

Abstract
We have shown recently that cholecystokinin octapeptide (CCK-8s) increases glutamate release from nerve terminals onto neurons of the nucleus tractus solitarius pars centralis (cNTS). The effects of CCK on gastrointestinal-related functions have, however, been attributed almost exclusively to its paracrine action on vagal afferent fibers. Because it has been reported that systemic or perivagal capsaicin pretreatment abolishes the effects of CCK, the aim of the present work was to investigate the response of cNTS neurons to CCK-8s in vagally deafferented rats. In surgically deafferented rats, intraperitoneal administration of 1 or 3 μg/kg CCK-8s increased c-Fos expression in cNTS neurons (139 and 251% of control, respectively), suggesting that CCK-8s’ effects are partially independent of vagal afferent fibers. Using whole cell patch-clamp techniques in thin brain stem slices, we observed that CCK-8s increased the frequency of spontaneous and miniature excitatory postsynaptic currents in 43% of the cNTS neurons via a presynaptic mechanism. In slices from deafferented rats, the percentage of cNTS neurons receiving glutamatergic inputs responding to CCK-8s decreased by ~50%, further suggesting that central terminals of vagal afferent fibers are not the sole site for the action of CCK-8s in the brain stem. Taken together, our data suggest that the sites of action of CCK-8s include the brain stem, and in cNTS, the actions of CCK-8s are not restricted to vagal central terminals but that nonvagal synapses are also involved.
Keywords: brain stem, electrophysiology, c-fos
Cholecystokinin, which is released from intestinal cells following ingestion of nutrients (34, 37, 47), has profound effects on gastrointestinal functions. In fact, it is well known that cholecystokinin, and, in particular, the cleaved octapeptide cholecystokinin-8s (CCK-8s), induces vagally mediated gastric relaxation, increases pancreatic exocrine secretion, and induces short-term satiety (36, 38, 41, 42, 48, 62).
Many studies have proposed that the vagally mediated effects of CCK-8s are almost exclusively due to a paracrine action of CCK-8s on peripheral, capsaicin-sensitive vagal afferent fibers (8, 14, 41). This postulate stems mainly from the observations that C-fiber ablation induced by perivagal capsaicin treatment greatly attenuates, or abolishes altogether, the effects of systemic administration of CCK-8s on gastric motility, gastric acid secretion, and pancreatic exocrine secretion (32, 33, 41, 58). One must keep in mind, however, that the localized perineural application involves the use of very high concentrations of capsaicin, usually 33 mM (i.e., 1% solution) (9, 23, 25, 41, 56, 63, 67). Such a high concentration of capsaicin raises the possibility that its observed effects are due to the toxic effects of massive calcium influx on fibers or neurons other than C-fibers due to the activation of VR-1 receptors. Although it is well known that the effects of capsaicin on ionic conductances are transient [reviewed in (7, 26)], it is also accepted that administration of elevated doses of capsaicin induces degenerative changes in CNS neurons and fibers, including vagal efferent fibers, that may not express VR1 receptors and even involves neuronal areas that do not receive sensory inputs from the periphery (24, 26, 49).
Furthermore, the very composition of the vagus, where more than 85% of the fibers are afferent, implies that even minor damages induced by perivagal application of capsaicin to the efferent fibers could not only be overlooked easily but would also have a potentially major impact on the vagal motor output, leading to an overestimation of the contribution of vagal afferent fibers to the CCK-8s-mediated gastrointestinal effects.
Finally, should the physiological effects of CCK on gastrointestinal and feeding-related circuits be due exclusively to a paracrine action of the hormone, then the response to systemic administration of CCK-8s falls short of explaining its mechanism of action. In fact, it is highly unlikely that systemically administered CCK-8s could cross the lamina propria at a concentration sufficient to induce a paracrine effect. It is very likely, therefore, that the actions of CCK are not limited to its paracrine effect on peripheral vagal afferent fibers, but other sites of action, including the second-order neurons of the NTS, should be taken into account. Indeed, we and others showed recently that CCK-8s increases glutamatergic excitatory inputs to NTS neurons of animals with intact vagal afferent fibers (2, 3). Because the activity of CCK is supposedly mediated in its entirety by vagal C-fibers (32, 33, 41, 58), chemical (i.e., perivagal capsaicin) or surgical (rhizotomy) deafferentation should prevent the response of brain stem vagal circuits to application of CCK.
We used the NTS subnucleus centralis (cNTS) as a convenient model for our studies because 1) the neuronal population of cNTS is quite homogeneous, since it comprises almost exclusively sensory neurons from the esophagus (1, 5); and, 2) its rostral location makes it highly unlikely that it receives a significant innervation from the controlateral NTS, unlike the more caudally located subnuclei medialis and commissuralis, which receive a robust input from the controlateral NTS (30).
In the present manuscript, we use different experimental approaches with the aim of characterizing the response of cNTS neurons to exogenous applications of CCK-8s in rats that underwent either chemical or surgical vagal sensory deafferentation.
METHODS
Research reported in the present manuscript conforms fully to National Institute of Health guidelines and was approved by the Pennington Biomedical Research Center-Louisiana State University System Institutional Animal Care and Use Committee.
Immunohistochemistry
Experiments were conducted on 24 Sprague-Dawley rats of either sex (200–250 g). One group of rats (n = 12) underwent a complete resection of the subdiaphragmatic anterior vagal trunk and surgical removal of the sensory rootlets of the right vagus (supranodose afferent rhizotomy; see below for surgical techniques). The other group of rats (n = 12) was used as control, i.e., no surgical procedures were performed. Three days later, all rats were injected with Fluorogold (Fluorochrome, 20 μg/ml saline ip; Englewood, CO) to label preganglionic neurons innervating the subdiaphragmatic viscera, allowing delineation of the boundaries of the dorsal motor nucleus of the vagus (DMV) and, in deafferented rats, to ascertain that the surgical procedures did not damage DMV neurons in those animals that underwent supranodose deafferentation (40, 66). Experiments were conducted 3–5 days after Fluorogold injection.
On the day of the experiment, the rats were injected either with saline, 1 or 3 μg/kg CCK-8s (0.5 ml ip). Ninety minutes later, the rats were anesthetized deeply (abolition of foot pinch withdrawal reflex) with isoflurane and perfused transcardially with chilled saline followed by Zamboni’s fixative (see below). After extraction, the brain stems were stored overnight in Zamboni’s fixative at 4°C. Following washout of the Zamboni’s solution, the brain stems were cut in 40-μm-thick coronal sections, and every third slice was mounted onto gelatin-coated coverslips. The slices were incubated at 37°C for 2 h with the primary antibody [rabbit-α-cFos; 1:5,000 in PBS containing 0.15% Triton-100 (TX) and 0.1% bovine serum albumin (BSA)]. The slices were rinsed with PBS-TX-BSA and incubated again at 37°C for 30 min with secondary antibody (goat-α-rabbit Texas red, 1:100 diluted in PBS-TX-BSA). The specimens were again rinsed with PBS-TX-BSA solution containing 1% goat antiserum, before being allowed to air dry, and mounted with Fluoromount-G (Southern Biotechnology Associates, Birmingham, AL). Specimens were coded and c-Fos immunoreactivity (IR) was counted manually using a calibrated grid by an investigator unaware of the treatment. In each rat, we counted, on average, 6.7 ± 1.2 slices from the caudal DVC, 6.5 ± 1.1 slices from the intermediate dorsal vagal complex (DVC) and 4.4 ± 0.9 slices from the rostral DVC, while 9.3 ± 1.7 slices were used to count the c-Fos expression in cNTS. Photomicrographs were taken using a Nikon E-400 microscope (×100 final magnification) equipped with tetramethylrhodamine isothiocyanate (to visualize cFos-IR) and UV (to visualize Fluorogold-IR) epifluorescent filters and a SPOT Insight camera and software (Diagnostic Instruments, Sterling Heights, MI) connected to a PC. Overlapping panels of the whole DVC area were digitally enhanced and montaged using ImageJ (developed at the U.S. National Institutes of Health and available from the Internet at http://rsb.info.nih.gov/ij) and Adobe Photoshop software (Adobe Systems, San Jose, CA).
Electrophysiology
The methods of slicing the brain stem and the identification of cNTS neurons have been described previously (3, 4, 59). Briefly, 25- to 35-day-old Sprague-Dawley rats of either sex were anesthetized with isoflurane (abolition of the foot pinch withdrawal reflex) before being killed by severing the blood vessels in the chest. The brain stem was removed and glued to the platform of a vibratome, and three coronal slices (300-μm-thick) were cut starting from the posterior area postrema moving rostrally. The slices were stored at least 1 h in carboxygenated (95% O2-5% CO2) Krebs solution (see Solution composition) at 30°C before use. A single slice was then transferred to a perfusion chamber (volume 500 μl; Michigan Precision Instruments, Parma, MI), kept in place with a nylon mesh, and maintained at 35 ± 1°C by perfusion with warmed Krebs solution at a rate of 2.5–3.0 ml/min.
Whole cell recordings were conducted on putative cNTS neurons identified as per their location in close proximity (within 100 μm) to the tractus solitarius at a level from the posterior tip of the area postrema to ~0.5 mm rostral to the anterior portion of the area postrema. Recordings were made with patch pipettes (6–8 MΩ resistance) filled with a potassium gluconate solution (see Solution composition) by using an Axoclamp 2B amplifier (Axon Instruments, Union City, CA). Data were sampled at 10 kHz and filtered at 2 kHz, digitized via a Digidata 1322 interface (Axon Instruments), acquired with a PC utilizing pClamp8 software (Axon Instruments) and analyzed with MiniAnalysis software (Jaejin Software, Leonia, NJ). Rise and decay kinetics of the excitatory postsynaptic currents (EPSC) were calculated for each event using a subroutine of the MiniAnalysis software; the data were then averaged and their means were compared in the presence and absence of CCK-8s. Recordings were accepted only if the series resistance were <20 MΩ. In the experiments in which the events’ kinetics were analyzed, the series resistance was checked every 10 min, and the data were discarded if there were a variation of series resistance of more than 25%.
In some experiments (n = 19), a bipolar tungsten electrode was placed in the tractus solitarius. Pulses of direct current (up to 2 ms long) were delivered while recording from cNTS (HP = −50 mV) of rats that underwent vagal deafferentation (see Vagal deafferentation).
Drugs were applied to the bath via a series of manually operated valves at concentrations shown previously to be effective (3). Neurons were allowed to recover fully between additions of agonists (minimum washout period of 10 min). Cells were classified as CCK-8s responsive if perfusion with CCK-8s (100 nM) increased the frequency of spontaneous excitatory postsynaptic currents (sEPSC) by a minimum of 50% above baseline.
Vagal deafferentation
Both chemical and surgical methods were used to remove vagal sensory inputs to the brain stem (vagal nerve deafferentation). Chemical vagal deafferentation was conducted following methods described previously (23, 41). Briefly, 20-day-old rats of either sex were anesthetized with a mixture of ketamine, xylazine, and acepromazine (80, 1.6, 5 mg· ml−1 · kg−1 ip, respectively). Under aseptic conditions, a ventral midline incision on the neck exposed ~4 mm of the right cervical vagal trunk, which was isolated over a strip of Parafilm. A cotton pellet soaked in a 1% capsaicin solution (8:1:1 saline: DMSO: ethanol) was applied to the nerve for 30 min; the area was then dried, the Parafilm was removed, and the incision was closed with 5–0 suture. The electrophysiological experiments were performed 7 to 10 days after the perivagal capsaicin treatment.
Surgical vagal deafferentation was achieved by sectioning the vagal afferent nerve rootlets (supranodose afferent rhizotomy) using a technique similar to that described previously (61). Rats were anesthetized as above and placed in a stereotaxic frame. After a dorsolateral incision at the level of the occipital bone, muscle tissue was blunt dissected to expose the occipital bone and the first cervical vertebra; because the three supranodose vagal dorsal afferent rootlets are located ~1 mm medial to the occipital condyle, the occipital bone needed to be trimmed with a #6 dental drill to expose the vagal rootlets (32, 61). The dorsal rootlets are located beneath the caudal portion of the occipital bone. Once visualized, the supranodose dorsal rootlets on one of the vagal trunks were sectioned under microscopic guidance using a 27-gauge surgical needle. The complete resection of the rootlets was assessed and confirmed routinely by the person assisting the surgery. In one group of rats, 4 days after vagal deafferentation, the nodose ganglia were exposed by an incision in the neck using an aseptic technique (n = 3). The neck muscles were blunt dissected to expose the internal carotid artery, and the attached cervical vagus nerve, which was dissected carefully from the carotid artery in the rostral direction. Once exposed, a small (less than 0.3 mm) incision was made in the sheath surrounding the nodose ganglion to allow insertion of a micropipette containing a solution (5%) of rhodamine dextrane (lysine fixable, MW 3000). The tip of the pipette was guided through the incision in the sheath of the nodose ganglion, and pressure pulses were applied to inject the dye (total injected volume was ~0.1 μl). The pipette was withdrawn, the area was blotted dry, and the cervical incision was closed with 4–0 suture. Three to five 5 days later, the rats were anesthetized deeply (isoflurane 5%) and were perfused transcardially with chilled saline followed by Zamboni’s fixative. The brain stem was extracted and postfixed in Zamboni’s fixative overnight at 4°C. Following washout of the Zamboni’s solution, coronal sections (40 μm thick) containing the DVC were cut using a freezing microtome, and every third slice placed onto gelatin-coated coverslips and mounted with Fluoromount (Southern Biotechnology Associates, Birmingham, AL) to reduce fading. Confocal microscopic images were collected by using a Zeiss 510 confocal scanning laser microscope equipped with a Kr/Ar-ion laser and filters for the selective visualization of Texas red. Two Z-sections separated by 5 μm were taken (final magnification, ×100), and their projections were merged. Overlapping panels of the entire DVC area were digitally enhanced and montaged using ImageJ and Adobe Photoshop software (Fig. 1).
Fig. 1.
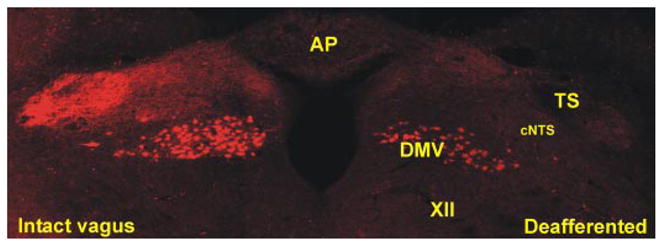
Anatomical proof of deafferentation. A coronal section (40 μm thick) at an intermediate level of the dorsal vagal complex (DVC). Note that on the deafferented side of the brain stem, rhodamine dextran-labeled vagal afferent nerve terminals are not present within the tractus solitarius (TS) and nucleus tractus solitarius pars centralis (cNTS), compared with the dense innervation within the vagally intact brain stem side. Note also that vagal preganglionic motoneurons are labeled in both sides of the brain stem, indicating the selectivity of the surgical deafferentation procedure. AP, area postrema; DMV, dorsal motor nucleus of the vagus; XII, nucleus of the hypoglossus; cNTS, nucleus tractus solitarius pars centralis.
When conducting immunocytochemistry experiments, contralateral subdiaphragmatic vagotomy was performed by removing 3–5 mm of the anterior branch of the vagus at a level rostral to the hepatic branch bifurcation or by removing 3–5 mm of the posterior vagal branch at midesophageal level. The incisions were closed using 5–0 suture, and the rats were allowed to recover for 5–10 days before experimentation.
Solution composition
The Krebs solution for electrophysiology had the following composition (in mM): 126 NaCl, 25 NaHCO3, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, and 11 dextrose, maintained at pH 7.4 by bubbling with 95% O2-5% CO2. The intracellular solution was composed of (in mM) 128 K gluconate, 10 KCl, 0.3 CaCl2, 1 MgCl2, 10 HEPES, 1 EGTA, 2 ATP, 0.25 GTP. Adjusted to pH 7.35 with KOH. Zamboni’s fixative was composed of (in mM) 1.6% (wt/vol) paraformaldehyde, 19 KH2PO4, and 100 Na2HPO4 · 7 H2O in 240 ml saturated picric acid-1,600 ml H2O; adjusted to pH 7.4 with HCl. PBS (in mM): 115 NaCl, 75 Na2HPO4 · 7 H2O, 7.5 KH2PO4, and 0.15% Triton-X.
Drugs and chemical
Capsaicin and TTX were purchased from TOCRIS Cookson (Ellisville, MO) and Alomone Laboratories (Jerusalem, Israel), respectively. Fluorogold was purchased from Fluorochrome, LLC (Denver, CO); c-Fos antibodies were purchased from Calbiochem (San Diego, CA); Alexa 568-conjugated goat-α-rabbit IgG was purchased from Molecular Probes (Eugene, OR). All other salts were purchased from Sigma (St. Louis, MO). Stock solutions were prepared freshly and diluted to the final concentration in Krebs-Ringer solution just before use.
Statistical analysis
Results are expressed as means ± SE, with significance defined as P <0.05. Results were compared before and after drug administration, with each neuron serving as its own control (Student’s paired t-test). Intergroup comparisons were conducted using the Student’s grouped t-test or the χ2-test.
RESULTS
CCK-8s increases c-Fos expression in cNTS of deafferented rats
c-Fos expression was measured in the cNTS following intraperitoneal administration of saline or CCK-8s. Rats were divided into two groups of 12 animals; one group was used as a control while the second group comprised rats that underwent surgical vagal deafferentation (i.e., afferent rhizotomy and contralateral subdiaphragmatic vagotomy). Both groups were subdivided in three subgroups consisting of four rats each that received the following treatments: 1) saline; 2) CCK-8s 1 μg/kg; and 3) CCK-8s 3 μg/kg.
Deafferentation did not affect the baseline c-Fos expression in NTS. In control animals that received saline injections, the whole NTS expressed 126 ± 50 c-Fos positive neurons, whereas the cNTS displayed 16 ± 7 c-Fos-positive neurons; in deafferented rats, after saline injection, there were 108 ± 42 and 16 ± 5 c-Fos-positive cells in the NTS and cNTS, respectively (P > 0.05 vs. control). After injection of 1 μg/kg CCK-8s in control rats, c-Fos expression increased to 229.5 ± 53.1% of control in cNTS (193.2 ± 44.5% for the whole NTS; P <0.05 vs. control). In deafferented animals, administration of 1 μg/kg CCK-8s increased c-Fos expression to 138.9 ± 30.2% of control in cNTS and 143.1 ± 28.3% for the whole NTS (P <0.05 vs. control; Fig. 2). Injection of 3 μg/kg CCK-8s in control rats increased c-Fos expression to 359 ± 56.7% of control in cNTS (275 ± 42.0% for the whole NTS; P <0.05 vs. control). In deafferented animals, administration of 3 μg/kg CCK-8s increased c-Fos expression to 251 ± 30.4% of control in cNTS and 281 ± 48.6% for the whole NTS (P <0.05 vs. control; Fig. 2). These data suggest that part of the c-fos expression in cNTS neurons induced by peripheral administration of a physiological (1 μg/kg) or a pharmacological (3 μg/kg) dose of CCK-8s occurs independently of vagal afferent fiber activation.
Fig. 2.
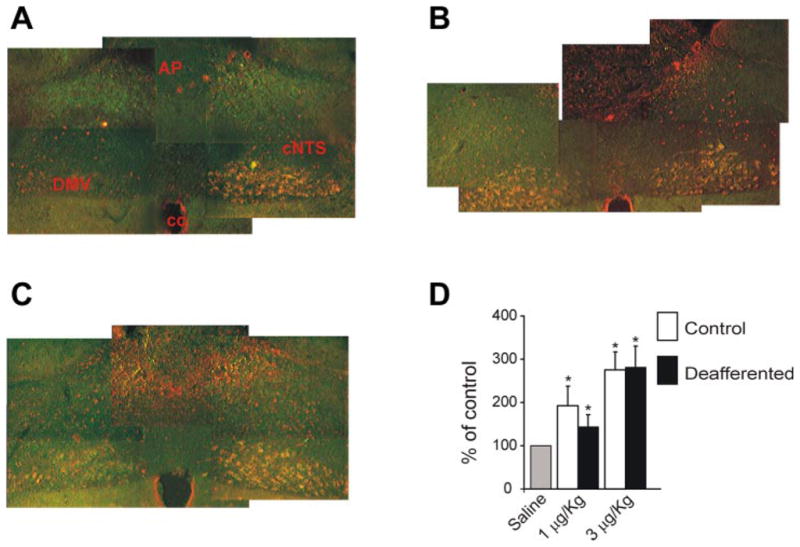
Cholecystokinin octapeptide (CCK-8s) increases c-fos expression in controls and deafferented rats. A. c-fos expression following injection of 0.5 ml saline in a deafferented rat. Note the prominent Fluorogold labeling of the left portion of the DMV and the almost complete absence of Fluorogold labeling in the right DMV. B: c-fos expression in a control rat following intraperitoneal administration of 1 μg/kg CCK-8s. Note that in this brain stem the intense Fluorogold labeling of both sides of the DMV. C: c-Fos expression in the NTS of a deafferented rat treated with 1 μg/kg CCK-8s intraperitoneally. D: Bar graph summarizing the c-fos expression evoked by CCK-8s intraperitoneally in the whole NTS of control and deafferented rats. cc, central canal. The micrographs were taken approximately at the same level, that is, +0.5 mm from the calamus scriptorium.
Differences were never observed in the CCK-8s-induced cFos expression on the right (rhizotomy) vs. the left (subdiaphragmatic vagotomy) side (Table 1). These data further suggest that the residual c-Fos expression observed in deafferented rats is determined by CCK-8s’ activation through routes other than the sectioned vagal afferent fibers or the projections from the controlateral NTS.
Table 1.
Summary of the effects on cFos expression of intraperitoneal injections of saline or CCK-8s
| Control |
Deafferented |
|||||
|---|---|---|---|---|---|---|
| Saline | CCK-8s, 1 μg/kg | CCK-8s, 3 μg/kg | Saline | CCK-8s, 1 μg/kg | CCK-8s, 3 μg/kg | |
| NTS | ||||||
| Rx | 100 | 215.6± 70.21* | 262.9± 26.40* | 100 | 155.8± 29.78* | 315.0± 62.95* |
| Lx | 100 | 213.3± 57.40 | 287.6± 55.78 | 100 | 141.1± 33.93 | 246.1± 41.70 |
| cNTS | ||||||
| Rx | 100 | 205.6± 37.49* | 318.1± 42.05* | 100 | 136.3± 41.70* | 308.5± 56.76* |
| Lx | 100 | 244.8± 59.84 | 364.1± 63.48 | 100 | 155.8± 29.32 | 213.5± 22.55 |
Values are expressed as means ± SE. CCK-8s, cholecystokinin octapeptide; NTS, nucleus tractus solitarius; cNTS, nucleus tractus solitarius pars centralis; Rx, right side (afferent rhizotomy); Lx, left side (subdiaphragmatic vagotomy).
P = not significant vs. Lx.
CCK-8 increases spontaneous and miniature postsynaptic current frequency in a subpopulation of cNTS cells
In neurons voltage clamped at −50 mV, 2–5 min perfusion with 100 nM CCK-8s increased both the frequency and amplitude of the inward sPSC from 2.08 ± 0.3 to 11.3 ± 1.4 events/s (P <0.05) and from 26.8 ± 0.8 pA to 35.4 ± 2 pA (P <0.05), respectively, in 45 out of 105 cNTS neurons (i.e., 43%; Fig. 3). The frequency and amplitude of the sPSC recovered to baseline upon washout. In the remaining 60 neurons, perfusion with CCK-8s changed neither the frequency nor the amplitude of the sPSC (1.6 ± 0.3 to 1.7 ± 0.3 events/s and 24.6 ± 0.7 to 23.7 ± 0.7 pA in control and CCK-8s, respectively).
Fig. 3.
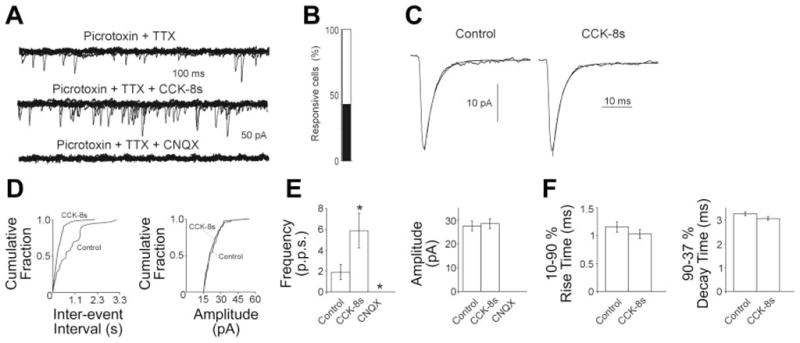
CCK-8s increases miniature excitatory postsynaptic currents (mEPSC) frequency via a presynaptic mechanism. A: representative traces showing that perfusion with 100 nM CCK-8s increases the frequency of glutamatergic mEPSC. Holding potential (HP) = −60 mV. B: bar graph showing the percentage of CCK-8s responsive cells in cNTS. C: averaged traces from the same cell in A in control (36 events) and in the presence of CCK-8s (104 events). The smooth line is one-exponential fitting of the mEPSC decay phase. D: frequency and amplitude histograms for the same cell in A. Note that perfusion with CCK-8s increases the frequency but not the amplitude of mEPSC. E: bar graphs showing the averaged results of frequency and amplitude for the mEPSC in control (40 μm picrotoxin + 0.5 μm TTX), 100 nM CCK-8s, and 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (n = 8 for all). Note that CNQX completely abolished the occurrence of mEPSC. *P <0.05 vs. control. F: bar graph summarizing the results for rise and decay time of mEPSC in control and in presence of 100 nM CCK-8s. Note that perfusion with CCK-8s does not affect the rise or the decay time of mEPSCs.
When voltage was clamped at −50 mV, perfusion of either picrotoxin (40 μM) or bicuculline (30 μM) with TTX (0.5 μM) did not eliminate the PSCs, which, instead, were abolished completely by perfusion with the non-NMDA antagonist CNQX (10 μM; n = 8; Fig. 3), suggesting that the PSC are due to glutamate receptor activation and can be classified as EPSCs.
In eight cells responsive to CCK-8s, in the presence of either picrotoxin (40 μM) or bicuculline (30 μM) with TTX (0.5 μM), CCK-8s increased the frequency of miniature EPSC (mEPSC) from 1.93 ± 0.74 to 5.8 ± 1.6 events/s (P <0.05) but not their amplitude (27.5 ± 2 and 28.5 ± 2.1 pA in control and following CCK-8s, respectively; P > 0.05; Fig. 3). Furthermore, perfusion with CCK-8s did not change either the 10–90% rise time (1.15 ± 0.09 and 1.02 ± 0.08 ms in control and following CCK-8s, respectively) or the 90–37% decay time (3.2 ± 0.37 and 3 ± 0.3 ms in control and following CCK-8s, respectively) of the mEPSC (Fig. 3).
Taken together, these results confirm our previous data (3) and suggest that in cNTS CCK-8s can increase the frequency of EPSCs independent of action potential generation. It must be noted, however, that CCK-8s induces a larger increase in sEPSC than in mEPSC (1,140.8 ± 341.3 and 357.9 ± 176.4%, respectively, P <0.05), suggesting that part of the CCK-8s’ effect is indeed due to action potential generation in the intrinsic circuitry that has been preserved in the slice.
CCK-8 increases sEPSC and mEPSC frequency in cNTS neurons from deafferented rats
In cNTS neurons from rats that underwent perivagal capsaicin treatment 7–10 days before experimentation, the frequency of sEPSC in cNTS was similar to that of untreated rats, 2.2 ± 0.4 events/s (n = 46) and 1.83 ± 0.2 events/s (n = 105) in capsaicin-treated and control, respectively. These data suggest that, in the slice preparation, the basal frequency of the sEPSC in cNTS neurons is not determined exclusively by the glutamate released from the central terminals of capsaicin-sensitive C-fibers.
After perivagal capsaicin treatment, perfusion with 100 nM CCK-8s increased the frequency of sEPSC in cNTS neurons from 2.6 ± 1.01 events/s to 10.6 ± 3.14 events/s and the amplitude from 24 ± 1.78 to 41 ± 8.6 pA in 9 out of 46 cells (P <0.05). Treatment with perivagal capsaicin reduced significantly the percentage of cells responsive to CCK-8s (20% compared with the 43% of control cells; P <0.05 χ2-test). These data suggest that ~50% of the CCK-8s-mediated excitatory response is due to actions on the terminals of capsaicin-sensitive vagal terminals.
Electrical stimulation of the tractus solitarius at current intensity and duration 1 order of magnitude higher than those previously shown as effective (64) did not evoke any current in cNTS neurons taken from rats that underwent surgical deafferentation 5–10 days before experimentation (n = 19). These data clearly show the completeness of the surgical deafferentation.
In cNTS neurons from rats that underwent surgical deafferentation 5–10 days before experimentation, the frequency of sEPSC in cNTS neurons was higher than that of untreated rats, 4.8 ± 1.26 events/s (n = 11) and 1.83 ± 0.2 events/s (n = 105) in surgically deafferented and control rats, respectively. These data suggest that, in the slice preparation, the basal frequency of the sEPSC in cNTS neurons is not determined exclusively by the glutamate released from the central terminals of vagal afferent fibers, which, rather appear to exert a tonic inhibitory influence, possibly via activation of local inhibitory circuits.
Following surgical deafferentation, perfusion with 100 nM CCK-8s increased the frequency of sEPSC in cNTS neurons from 4.8 ± 1.26 events/s to 11.4 ± 2.14 events/s in 11 out of 28 cells (P <0.05). Similarly, in seven neurons in which perfusion with CCK-8s increased the frequency of sEPSC, perfusion with 100 nM CCK-8s in the presence of 1 μM TTX increased the frequency of mEPSC from 2.8 ± 1.1 events/s to 5.1 ± 1.8 events/s in all of the cells (P <0.05; Fig. 4). Six neurons in which CCK-8s did not increase the frequency of sEPSCs were unresponsive, even when 100 nM CCK-8s was tested on mESPC (data not shown).
Fig. 4.
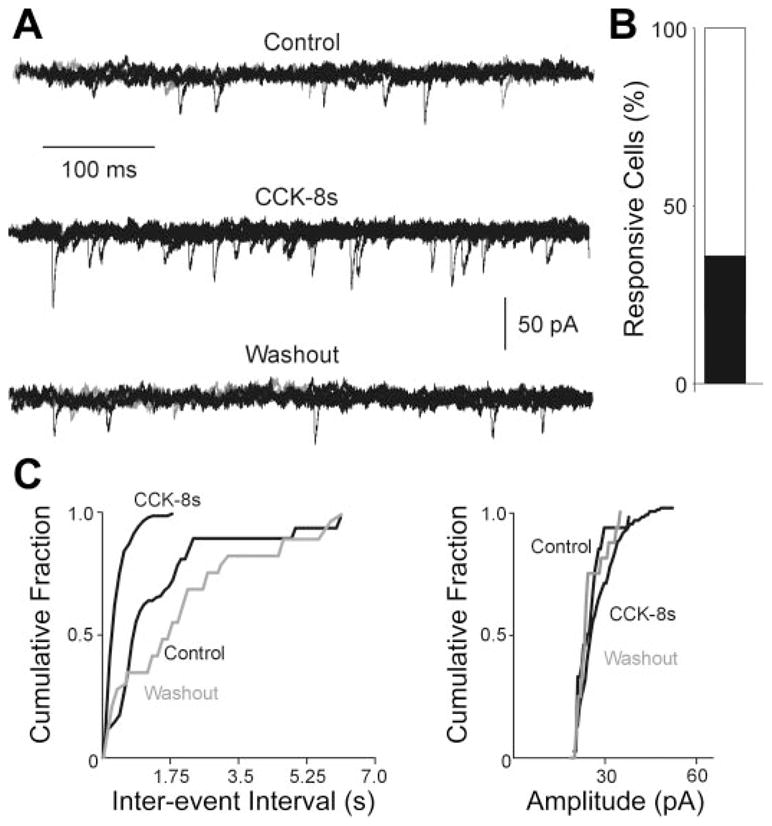
CCK-8s increases sEPSC frequency in rats that underwent vagal deafferentation. A: representative traces showing that perfusion with 100 nM CCK-8s increases the frequency of mEPSC. HP = −60 mV. B: bar graph showing the percentage of CCK-8s responsive cells in cNTS. C: frequency and amplitude histograms for the same cell in A. Note that perfusion with CCK-8s increases the frequency but not the amplitude of mEPSC.
These data suggest that CCK-8s does not act exclusively on the terminals of vagal afferent fibers.
DISCUSSION
In the present work, we have shown that: 1) surgical deafferentation reduces significantly, but does not eliminate, c-Fos expression in cNTS neurons following systemic CCK-8s; and 2) chemical or surgical vagal deafferentation does not abolish the ability of CCK-8s to increase the frequency of excitatory synaptic transmission to cNTS neurons.
Taken together, these data suggest that CCK-8s activates both capsaicin-sensitive and capsaicin-insensitive vagal sensory afferent terminals in the cNTS but imply that CCK-8s also excites cNTS neurons via a vagally independent mechanism.
The activation of brain stem vagal circuits by systemic administration of CCK-8s has been postulated as being determined exclusively by its paracrine effects, that is, CCK, released from intestinal cells in response to a meal, activates capsaicin-sensitive vagal afferent fibers, which then carry the information to brain stem vagal circuits that trigger the efferent vagal response (8, 14, 41). Anatomical evidence certainly supports the presence of CCK receptors on vagal afferent fibers (6), and this was proposed as the sole site of action of CCK because of the observations that, as with a complete vagotomy, C-fiber degeneration induced by perivagal capsaicin treatment greatly attenuates or even abolishes the effects of CCK-8s on gastric motility, gastric acid secretion and pancreatic exocrine secretion (21, 32, 33, 41, 58). One must be aware, however, that the high concentration of capsaicin used in the perivagal treatment (9, 23, 25, 41, 56, 63, 67) may have additional effects unrelated to its recognized actions on C-fibers or neurons receiving direct C-fiber-mediated inputs (24, 26, 49). In this regard, a monosynaptic connection between vagal afferent fibers and gastrointestinal-projecting DMV neurons has been described (44), raising the possibility that the lack of CCK-8s effects on gastrointestinal functions in systemically capsaicin-treated animals may be due to nonselective toxic effects of capsaicin on vagal motorneurons. In brief, major drawbacks exist in the use of capsaicin, even when administered perivagally. In consideration of this, we resolved to conduct, whenever possible, either chemical or surgical vagal deafferentation to assess the effects of CCK-8s in cNTS neurons.
The deafferented rats in the immunohistochemical portion of the present study underwent unilateral supranodose afferent rhizotomy and contralateral subdiaphragmatic vagotomy. The rationale for conducting this surgical approach resides in providing a complete deafferentation of the abdominal viscera prior to systemic administration of CCK-8s. If, as inferred, the activation of vagal brain stem circuits by CCK-8s is solely the result of the paracrine effect of the peptide, then complete surgical deafferentation should prevent c-Fos increase in the DVC. A postsurgical period of 5–10 days was sufficient to allow degeneration of vagal afferent fibers (rhizotomy side) and degeneration of both afferent fibers and motoneurons (controlateral vagotomy side) (28, 29). The Fluorogold staining [which is transported centrally by vagal fibers, among other pathways (40, 66)] confirmed that the supranodose afferent rhizotomy did not damage the efferent vagus, as DMV neurons were labeled only on the ipsilateral side. The contralateral side (both motor and sensory vagotomy) exhibited little, if any, Fluorogold staining, indicating the success of the subdiaphragmatic vagotomy.
The increase in c-Fos expression in vagal brain stem circuits following intraperitoneal administration of CCK-8s is well established (18, 20, 35, 45, 46). After surgical deafferentation, however, we show that systemic administration of CCK-8s still increased c-Fos expression in the NTS, including the cNTS. These data suggest that part of the c-fos expression in cNTS neurons induced by peripheral administration of CCK-8s at physiological (1 μg/kg) or pharmacological (3 μg/kg) doses occurs independently of the activation of peripheral vagal afferent fibers.
Although apparently at odds with previous studies conducted on vagotomized or capsaicin-treated animals (53), one should consider the technical differences between our study and the studies of Sayegh and Ritter (53), foremost the deafferentation procedures. In fact, Sayegh and Ritter did their experiments 8 wk after conducting a complete subdiaphragmatic vagotomy or after systemic administration of capsaicin; conversely, we conducted our experimental procedures 5–10 days after afferent rhizotomy and subdiaphragmatic vagotomy. The longer postsurgical recovery period allowed by these colleagues (i.e., 8 wk) before the experimentation may well have induced a degeneration of vagal afferent fibers, as well as NTS and DMV neurons, whereas our selective surgery and relatively short recovery period are likely to have affected only vagal afferent terminals. Sayegh and Ritter (53) reported that the c-Fos expression elicited by CCK-8s was decreased in vagotomized vs. control rats [see, for example, columns 1 (control saline), vs. column 2 (control CCK-8s), vs. column 5 (vagotomized CCK-8s), in Table 1 of Ref. 53]. They did not compare, however, c-Fos expression in response to CCK-8s or saline in vagotomized rats [see, for example, columns 4 (vagotomized saline), vs. column 5 (vagotomized CCK-8s) in Table 1 of Ref. 53]. Indeed, similar to our data, systemic CCK-8s administration induced a (possibly significant) increase in cFos expression also in the vagotomized rats reported in their study (53).
In a more recent report, van de Wall and colleagues (60) demonstrated that while c-Fos expression in response to CCK administration is markedly reduced in capsaicin-treated rats, CCK increases c-Fos expression induced by gastric distention. These data suggest that CCK activates capsaicin-sensitive fibers directly but also enhances the vagally mediated responses of distention-activated, capsaicin-insensitive fibers that synapse in the NTS.
Our electrophysiological results confirm the excitatory effect of CCK-8s on glutamatergic terminals apposing cNTS neurons reported previously (2, 3). The increase in mEPSC frequency, but not amplitude, and the lack of effect on the EPSC kinetics indicate a presynaptic site of action and suggests that CCK-8s has a profound modulatory role of the glutamatergic inputs to cNST.
In accordance with our cFos data, we observed that perfusion with CCK-8s increased glutamatergic transmission to cNTS neurons from rats that underwent chemical (perivagal capsaicin) or surgical (rhizotomy) deafferentation. These data were somewhat surprising since, as mentioned before, the excitatory effects of CCK-8s on NTS neurons have been ascribed almost exclusively to a paracrine effect of the peptide on capsaicin-sensitive fibers. Certainly, the lower percentage of CCK-8s responsive cNTS cells observed in slices from chemically deafferented rats confirms rather than negates the paracrine effects of CCK-8s but, at the same time, indicates the involvement of other routes of NTS neuronal activation by CCK-8s. This postulate is reinforced by the data showing that glutamatergic transmission to cNTS is still enhanced when CCK-8s is applied onto cNTS neurons from surgically deafferented rats.
Recent evidence indicates that CCK-8s might also have an effect at sites other than vagal afferent fibers. In fact, the satiating effects of CCK-8s are attenuated by CCK receptor antagonists that cross the blood brain barrier but not by CNS-impermeable antagonists (43). Similarly, subdiaphragmatic vagotomy does not attenuate the reduction of food intake induced by pharmacological doses (8 nmol/kg) of exogenous CCK-8s, but it completely abolishes the reduction of food intake by more physiological doses of CCK-8s (1–2 nmol/kg) (42, 43). These reports suggest that higher levels of circulating CCK, which could possibly be attained by prolonged secretion of intestinal CCK, may act at a site outside the abdominal cavity, including the brain. Indeed, electrophysiological studies have also shown that CCK-8s has profound effects on gastric-projecting dorsal vagal motorneurons (65), on NTS POMC neurons in the caudal brain stem (2), on esophageal sensory neurons of the cNTS (3), on isolated cultured sensory neurons of the nodose ganglion (54), as well as in nonidentified vagal brain stem neurons (11, 12, 39). In all instances, the threshold concentration of CCK-8s (~1 nM), is close to the postprandial levels of plasma CCK (in the picomolar range) (55), and gastric relaxation is observed to occur after systemic administration of CCK-8s at low nanomolar concentrations (58). It is well accepted that, in vitro, the concentration-response curve to agonists is shifted to the right by two or more orders of magnitude (17), thus placing the observed threshold for the response to CCK-8s in the physiological range.
Particular mention must be made regarding a relatively recent manuscript (54) examining, with electrophysiological tools, the response to both capsaicin and CCK-8s in isolated, identified type A and/or type C nodose ganglion neurons. Both substances were reported to induce a depolarization in nodose neurons. Of particular interest and relevance are the observations that 1) CCK-8s depolarizes similar proportions of A- and C-type neurons; and 2) only C-type neurons were responsive to administration of low concentrations of capsaicin. Because capsaicin is supposedly selective for C-fibers, these observations, then, fall short of explaining the in vivo results showing that capsaicin, whether administered systemically or perivagally, antagonizes completely the effects of systemic CCK-8s. The results of Simasko and Ritter, however, would support the case of a nonselective action of capsaicin that, by inducing degeneration of fibers other than C-fibers, occludes or attenuates significantly the vagal efferent response, thus leading previous investigators to conclude that vagal afferent fibers only were responsive for the physiological actions of systemic CCK-8s. These conclusions are supported by other reported observations, including our published results, showing that CCK-8s has effects on neurons of the DVC (2, 3, 11, 12, 39, 65) and are strengthened by the immunohistochemical and electrophysiological data reported herein.
Our data then raise the question as to the whereabouts of this source of CCK acting in the NTS. One possibility is that CCK originates from local peptide-containing interneurons; indeed, CCK is one the most widespread neuropeptides in the brain, and there is abundant neurochemical evidence showing the presence of both CCK receptors, as well as CCK-containing neurons and terminals in the caudal brain stem (15, 16, 22, 31, 57, 65), suggesting that local neurons containing CCK may also contribute to its effects in vagal brain stem circuits.
Another possibility is that CCK reaches the NTS via its plasma transport. Although it has been suggested that CCK does not cross the blood-brain barrier (BBB), we have to consider that the NTS is a circumventricular organ with a leaky BBB, fenestrated capillaries, and enlarged perivascular space that allows the passage of large molecules (13, 19, 50, 51), raising the possibility that circulating CCK might also reach these neuronal circuits. Additionally, the adjacent area post-rema, which lies entirely outside the BBB, has a series of short, communicating vessels that potentially send postremal venous drainage to the NTS (52). These morphological characteristics raise the possibility that NTS neuronal activity can be modulated by circulating molecules, including CCK. In fact, there is functional evidence that CCK can cross the BBB to activate a solitariusnigral pathway (27), to phosphorylate CCK-A receptors in the DVC (65) and to induce short-term satiety (10).
The reader, however, has to keep in mind that we do not intend to dispute the powerful and well-documented paracrine effects of CCK on peripheral vagal afferent fibers. We should like, instead, to suggest that the effects of CCK-8s are due to actions not only on those fibers, but actions at other sites, including NTS neurons, and must be taken into account. The cellular vs. “in vivo” mismatch reported herein could be due to the different sensitivity of the experimental approaches. In fact, the cellular approaches we used are capable of discerning the minute changes determined by CCK-8s effects on the membrane of a single neuron. Conversely, the “in vivo” approach, by measuring the overall impact of a treatment, has a lower capability of observing minute variations. One should also keep in mind that the very composition of the vagus, where the vast majority of the fibers are afferent, implies that even minor damage to the efferent fibers induced by perivagal application of capsaicin could not only be overlooked easily but would also have a potentially major impact on the vagal motor output, leading to an overestimation of the contribution of vagal afferent fibers to the CCK mediated gastrointestinal effects.
In conclusion, our data suggest that, in addition to its paracrine effects on peripheral vagal afferent fibers, CCK also activates vagal sensory afferent terminals in the cNTS but also imply that CCK-8s excites cNTS neurons via a vagally independent mechanism. It is then possible that the site of action through which CCK influences gastric and pancreatic functions is not limited to its paracrine effect on peripheral vagal afferent fibers but involves also other areas, including NTS neurons.
Acknowledgments
We thank Cesare M. Travagli and W. Nairn Browning for support and encouragement. We also thank Dr. Zhongling Zheng for help with the confocal microscopy and Dr. Huiyuan Zhang for demonstrating the nodose injection procedures.
GRANTS
This work was supported by National Institutes of Health Grants DK-55530 and DK-56373.
References
- 1.Altschuler SM, Bao X, Bieger D, Hopkins DA, Miselis RR. Viscerotopic representation of the upper alimentary tract in the rat: sensory ganglia and nuclei of the solitary and spinal trigeminal tracts. J Comp Neurol. 1989;283:248–268. doi: 10.1002/cne.902830207. [DOI] [PubMed] [Google Scholar]
- 2.Appleyard SM, Bailey TW, Doyle MW, Jin YH, Smart JL, Low MJ, Andresen MC. Proopiomelanocortin neurons in nucleus tractus solitarius are activated by visceral afferents: regulation by cholecystokinin and opioids. J Neurosci. 2005;25:3578–3585. doi: 10.1523/JNEUROSCI.4177-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baptista V, Zheng Z, Coleman FH, Rogers RC, Travagli RA. Cholecystokinin octapeptide increases spontaneous glutamatergic synaptic transmission to neurons of the nucleus tractus solitarius centralis. J Neurophysiol. 2005;94:2763–2771. doi: 10.1152/jn.00351.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baptista V, Zheng ZL, Coleman FH, Rogers RC, Travagli RA. Characterization of neurons of the nucleus tractus solitarius pars centralis. Brain Res. 2005;1052:139–146. doi: 10.1016/j.brainres.2005.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barraco R, El-Ridi M, Parizon M, Bradley D. An atlas of the rat subpostremal nucleus tractus solitarius. Brain Res Bull. 1992;29:703–765. doi: 10.1016/0361-9230(92)90143-l. [DOI] [PubMed] [Google Scholar]
- 6.Berthoud HR, Patterson LM. Anatomical relationship between vagal afferent fibers and CCK-immunoreactive entero-endocrine cells in the rat small intestinal mucosa. Acta Anat (Basel) 1996;156:123–131. doi: 10.1159/000147837. [DOI] [PubMed] [Google Scholar]
- 7.Bevan S, Szolcsanyi J. Sensory neuron-specific actions of capsaicin: mechanisms and applications. Trends Pharmacol Sci. 1990;11:330–333. doi: 10.1016/0165-6147(90)90237-3. [DOI] [PubMed] [Google Scholar]
- 8.Blackshaw LA, Grundy D. Effects of cholecystokinin (CCK-8) on two classes of gastroduodenal vagal afferent fibre. J Auton Nerv Syst. 1990;31:191–202. doi: 10.1016/0165-1838(90)90185-l. [DOI] [PubMed] [Google Scholar]
- 9.Blackshaw LA, Page AJ, Partosoedarso ER. Acute effects of capsaicin on gastrointestinal vagal afferents. Neuroscience. 2000;96:407–416. doi: 10.1016/s0306-4522(99)00547-3. [DOI] [PubMed] [Google Scholar]
- 10.Blevins JE, Stanley BG, Reidelberger RD. Brain regions where cholecystokinin suppresses feeding in rats. Brain Res. 2000;860:1–10. doi: 10.1016/s0006-8993(99)02477-4. [DOI] [PubMed] [Google Scholar]
- 11.Branchereau P, Bohme GA, Champagnat J, Morin-Surun MP, Durieux C, Blanchard JC, Roques BP, Denavit-Saubie M. CholecystokininA and cholecystokininB receptors in neurons of the brainstem solitary complex of the rat: pharmacological identification. J Pharmacol Exp Ther. 1992;260:1433–1440. [PubMed] [Google Scholar]
- 12.Branchereau P, Champagnat J, Denavit-Saubie M. Cholecystokinin-gated currents in neurons of the rat solitary complex in vitro. J Neurophysiol. 1993;70:2584–2595. doi: 10.1152/jn.1993.70.6.2584. [DOI] [PubMed] [Google Scholar]
- 13.Cottrell GT, Ferguson AV. Sensory circumventricular organs: central roles in integrated autonomic regulation. Regul Pept. 2004;117:11–23. doi: 10.1016/j.regpep.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 14.Davison JS, Clarke GD. Mechanical properties and sensitivity to CCK of vagal gastric slowly adapting mechanoreceptors. Am J Physiol Gastroin-test Liver Physiol. 1988;255:G55–G61. doi: 10.1152/ajpgi.1988.255.1.G55. [DOI] [PubMed] [Google Scholar]
- 15.Dockray GJ. Immunochemical evidence of cholecystokinin-like peptides in brain. Nature. 1976;264:568–570. doi: 10.1038/264568a0. [DOI] [PubMed] [Google Scholar]
- 16.Fallon JH, Seroogy KB. The distribution and some connections of cholecystokinin neurons in the rat brain. Ann NY Acad Sci. 1985;448:121–132. doi: 10.1111/j.1749-6632.1985.tb29912.x. [DOI] [PubMed] [Google Scholar]
- 17.Fatt P, Katz B. An analysis of the end plate potential recorded with an intracellular electrode. J Physiol. 1952;115:320–370. doi: 10.1113/jphysiol.1951.sp004675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glatzle J, Kreis ME, Kawano K, Raybould HE, Zittel TT. Postprandial neuronal activation in the nucleus of the solitary tract is partly mediated by CCK-A receptors. Am J Physiol Regul Integr Comp Physiol. 2001;281:R222–R229. doi: 10.1152/ajpregu.2001.281.1.R222. [DOI] [PubMed] [Google Scholar]
- 19.Gross PM, Wall KM, Pang JJ, Shaver SW, Wainman DS. Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol. 1990;259:R1131–R1138. doi: 10.1152/ajpregu.1990.259.6.R1131. [DOI] [PubMed] [Google Scholar]
- 20.Gulley S, Sharma SK, Moran TH, Sayegh AI. Cholecystokinin-8 increases Fos-like immunoreactivity in the brainstem and myenteric neurons of rats through CCK1 receptors. Peptides. 2005;26:1617–1622. doi: 10.1016/j.peptides.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 21.Helke CJ, Niederer AJ. Studies on the coexistence of Substance P with other putative transmitters in the nodose and petrosal ganglia (Abstract) Synapse. 1990;5:144–151. doi: 10.1002/syn.890050209. [DOI] [PubMed] [Google Scholar]
- 22.Herbert H, Saper CB. Cholecystokinin-, galanin-, and corticotropin-releasing factor-like immunoreactive projections from the nucleus of the solitary tract to the parabrachial nucleus in the rat. J Comp Neurol. 1990;293:581–598. doi: 10.1002/cne.902930405. [DOI] [PubMed] [Google Scholar]
- 23.Holzer HH, Turkelson CM, Solomon TE, Raybould HE. Intestinal lipid inhibits gastric emptying via CCK and a vagal capsaicin-sensitive afferent pathway in rats. Am J Physiol Gastrointest Liver Physiol. 1994;267:G625–G629. doi: 10.1152/ajpgi.1994.267.4.G625. [DOI] [PubMed] [Google Scholar]
- 24.Holzer P. Neural injury, repair, and adaptation in the GI tract. II. The elusive action of capsaicin on the vagus nerve. Am J Physiol Gastrointest Liver Physiol. 1998;275:G8–G13. doi: 10.1152/ajpgi.1998.275.1.G8. [DOI] [PubMed] [Google Scholar]
- 25.Holzer P. Capsaicin-sensitive afferent neurones and gastrointestinal propulsion in the rat. Arch Pharm (Weinheim) 1986;332:62–65. doi: 10.1007/BF00633198. [DOI] [PubMed] [Google Scholar]
- 26.Holzer P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol Rev. 1991;43:143–201. [PubMed] [Google Scholar]
- 27.Hommer DW, Palkovits M, Crawley JN, Paul SM, Skirboll LR. Cholecystokinin-induced excitation in the substantia nigra: evidence for peripheral and central components. J Neurosci. 1985;5:1387–1392. doi: 10.1523/JNEUROSCI.05-06-01387.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji J, Dheen ST, Wah Tay SS. Molecular analysis of the vagal motoneuronal degeneration after right vagotomy. J Neurosci Res. 2006;69:406–417. doi: 10.1002/jnr.10300. [DOI] [PubMed] [Google Scholar]
- 29.Jia YS, Wang XA, Ju G. Nitric oxide synthase expression in vagal complex following vagotomy in the rat. Neuroreport. 1994;5:793–796. doi: 10.1097/00001756-199403000-00014. [DOI] [PubMed] [Google Scholar]
- 30.Kalia M, Sullivan JM. Brainstem projections of sensory and motor components of the vagus nerve in the rat. J Comp Neurol. 1982;211:248–264. doi: 10.1002/cne.902110304. [DOI] [PubMed] [Google Scholar]
- 31.Kubota Y, Inagaki S, Shiosaka S, Cho HJ, Tateishi K, Hashimura E, Hamahoka T, Tohyama M. The distribution of cholecystokinin octapeptide-like structures in the lower brain stem of the rat: An immunohistochemical analysis. Neuroscience. 1983;9:587–604. doi: 10.1016/0306-4522(83)90176-8. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Owyang C. Vagal afferent pathway mediates physiological action of cholecystokinin on pancreatic enzyme secretion. J Clin Invest. 1993;92:418–424. doi: 10.1172/JCI116583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Owyang C. Endogenous cholecystokinin stimulates pancreatic enzyme secretion via vagal afferent pathway in rats. Gastroenterology. 1994;107:525–531. doi: 10.1016/0016-5085(94)90180-5. [DOI] [PubMed] [Google Scholar]
- 34.Liddle RA. Regulation of cholecystokinin synthesis and secretion in rat intestine. J Nutr. 1994;124:1308S–1314S. doi: 10.1093/jn/124.suppl_8.1308S. [DOI] [PubMed] [Google Scholar]
- 35.Mönnikes H, Lauer G, Arnold R. Peripheral administration of cholecystokinin activates c-fos expression in the locus coeruleus/subcoeruleus nucleus, dorsal vagal complex and paraventricular nucleus via capsaicin-sensitive vagal afferents and CCK-A receptors in the rat. Brain Res. 1997;770:277–288. doi: 10.1016/s0006-8993(97)00865-2. [DOI] [PubMed] [Google Scholar]
- 36.Moran TH, Kinzig KP. Gastrointestinal satiety signals II. Cholecystokinin. Am J Physiol Gastrointest Liver Physiol. 2004;286:G183–G188. doi: 10.1152/ajpgi.00434.2003. [DOI] [PubMed] [Google Scholar]
- 37.Moran TH, Ladenheim EE, Schwartz GJ. Within-meal gut feedback signaling. Int J Obes Relat Metab Disord. 2001;25(Suppl 5):S39–S41. doi: 10.1038/sj.ijo.0801910. [DOI] [PubMed] [Google Scholar]
- 38.Owyang C, Logsdon CD. New insights into neurohormonal regulation of pancreatic secretion. Gastroenterology. 2004;127:957–969. doi: 10.1053/j.gastro.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 39.Plata-Salaman CR, Fukuda A, Oomura Y, Minami T. Effects of sulphated cholecystokinin octapeptide (CCK-8) on the dorsal motor nucleus of the vagus. Brain Res Bull. 1988;21:839–842. doi: 10.1016/0361-9230(88)90054-8. [DOI] [PubMed] [Google Scholar]
- 40.Powley TL, Fox EA, Berthoud HR. Retrograde tracer technique for assessment of selective and total subdiaphragmatic vagotomies. Am J Physiol Regul Integr Comp Physiol. 1987;253:R361–R370. doi: 10.1152/ajpregu.1987.253.2.R361. [DOI] [PubMed] [Google Scholar]
- 41.Raybould HE, Tache Y. Cholecystokinin inhibits gastric motility and emptying via a capsaicin-sensitive vagal pathway in rats. Am J Physiol Gastrointest Liver Physiol. 1988;255:G242–G246. doi: 10.1152/ajpgi.1988.255.2.G242. [DOI] [PubMed] [Google Scholar]
- 42.Reidelberger RD. Cholecystokinin and control of food intake. J Nutr. 1994;124:1327S–1333S. doi: 10.1093/jn/124.suppl_8.1327S. [DOI] [PubMed] [Google Scholar]
- 43.Reidelberger RD, Hernandez J, Fritzsch B, Hulce M. Abdominal vagal mediation of the satiety effects of CCK in rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1005–R1012. doi: 10.1152/ajpregu.00646.2003. [DOI] [PubMed] [Google Scholar]
- 44.Rinaman L, Card JP, Schwaber JS, Miselis RR. Ultrastructural demonstration of a gastric monosynaptic vagal circuit in the nucleus of the solitary tract in rat. J Neurosci. 1989;9:1985–1996. doi: 10.1523/JNEUROSCI.09-06-01985.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rinaman L, Hoffman GE, Stricker EM, Verbalis JG. Exogenous cholecystokinin activates cFos expression in medullary but not hypothalamic neurons in neonatal rats. Dev Brain Res. 1994;77:140–145. doi: 10.1016/0165-3806(94)90222-4. [DOI] [PubMed] [Google Scholar]
- 46.Rinaman L, Verbalis JG, Stricker EM, Hoffman GE. Distribution and neurochemical phenotypes of caudal medullary neurons activated to express cFos following peripheral administration of cholecystokinin. J Comp Neurol. 1993;338:475–490. doi: 10.1002/cne.903380402. [DOI] [PubMed] [Google Scholar]
- 47.Ritter RC. Gastrointestinal mechanisms of satiation for food. Physiol Behav. 2004;81:249–273. doi: 10.1016/j.physbeh.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 48.Ritter RC. Increased food intake and CCK receptor antagonists: beyond abdominal vagal afferents. Am J Physiol Regul Integr Comp Physiol. 2004;286:R991–R993. doi: 10.1152/ajpregu.00116.2004. [DOI] [PubMed] [Google Scholar]
- 49.Ritter S, Dinh TT. Age-related changes in capsaicin-induced degeneration in rat brain. J Comp Neurol. 1992;318:103–116. doi: 10.1002/cne.903180108. [DOI] [PubMed] [Google Scholar]
- 50.Rogers RC, Hermann GE, Travagli RA. Brainstem control of gastric function. In: Johnson LR, editor. Physiology of the Gastrointestinal Tract. San Diego, CA: Elsevier; 2005. [Google Scholar]
- 51.Rogers RC, McTigue DM, Hermann GE. Vagovagal reflex control of digestion: afferent modulation by neural and “endoneurocrine” factors. Am J Physiol Gastrointest Liver Physiol. 1995;268:G1–G10. doi: 10.1152/ajpgi.1995.268.1.G1. [DOI] [PubMed] [Google Scholar]
- 52.Roth GI, Yamamoto WS. The microcirculation of the area postrema of the rat. J Comp Neurol. 1968;133:329–340. doi: 10.1002/cne.901330304. [DOI] [PubMed] [Google Scholar]
- 53.Sayegh AI, Ritter RC. Vagus nerve participates in CCK-induced Fos expression in hind brain but not myenteric plexus. Brain Res. 2000;878:155–162. doi: 10.1016/s0006-8993(00)02731-1. [DOI] [PubMed] [Google Scholar]
- 54.Simasko SM, Ritter RC. Cholecystokinin activates both A- and C-type vagal afferent neurons. Am J Physiol Gastrointest Liver Physiol. 2003;285:G1204–G1213. doi: 10.1152/ajpgi.00132.2003. [DOI] [PubMed] [Google Scholar]
- 55.Soudah HC, Lu Y, Hasler WL, Owyang C. Cholecystokinin at physiological levels evokes pancreatic enzyme secretion via a cholinergic pathway. Am J Physiol Gastrointest Liver Physiol. 1992;263:G102–G107. doi: 10.1152/ajpgi.1992.263.1.G102. [DOI] [PubMed] [Google Scholar]
- 56.South EH, Ritter RC. Capsaicin application to central or peripheral vagal fibers attenuates CCK satiety. Peptides. 1988;9:601–612. doi: 10.1016/0196-9781(88)90171-4. [DOI] [PubMed] [Google Scholar]
- 57.Takagi H, Kubota Y, Mori S, Tateishi K, Hamaoka T, Tohyama M. Fine structural studies of cholecystokinin-8-like immunoreactive neurons and axon terminals in the nucleus of tractus solitarius of the rat. J Comp Neurol. 1984;227:369–379. doi: 10.1002/cne.902270307. [DOI] [PubMed] [Google Scholar]
- 58.Takahashi T, Owyang C. Mechanism of cholecystokinin-induced relaxation of the rat stomach. J Auton Nerv Syst. 1999;75:123–130. doi: 10.1016/s0165-1838(98)00181-7. [DOI] [PubMed] [Google Scholar]
- 59.Travagli RA, Gillis RA, Rossiter CD, Vicini S. Glutamate and GABA-mediated synaptic currents in neurons of the rat dorsal motor nucleus of the vagus. Am J Physiol Gastrointest Liver Physiol. 1991;260:G531–G536. doi: 10.1152/ajpgi.1991.260.3.G531. [DOI] [PubMed] [Google Scholar]
- 60.van de Wall EH, Duffy P, Ritter RC. CCK enhances response to gastric distension by acting on capsaicin-insensitive vagal afferents. Am J Physiol Regul Integr Comp Physiol. 2005;289:R695–R703. doi: 10.1152/ajpregu.00809.2004. [DOI] [PubMed] [Google Scholar]
- 61.Walls EK, Wang FB, Holst MC, Phillips RJ, Voreis JS, Perkins AR, Pollard LE, Powley TL. Selective vagal rhizotomies: a new dorsal surgical approach used for intestinal deafferentations. Am J Physiol Regul Integr Comp Physiol. 1995;269:R1279–R1288. doi: 10.1152/ajpregu.1995.269.5.R1279. [DOI] [PubMed] [Google Scholar]
- 62.Woods SC. Gastrointestinal satiety signals. I. An overview of gastrointestinal signals that influence food intake. Am J Physiol Gastrointest Liver Physiol. 2004;286:G7–G13. doi: 10.1152/ajpgi.00448.2003. [DOI] [PubMed] [Google Scholar]
- 63.Zafra MA, Molina F, Puerto A. Effects of perivagal administration of capsaicin on post-surgical food intake. Auton Neurosci. 2003;107:37–44. doi: 10.1016/S1566-0702(03)00128-0. [DOI] [PubMed] [Google Scholar]
- 64.Zheng H, Patterson LM, Morrison C, Banfield BW, Randall JA, Browning KN, Travagli RA, Berthoud HR. Melanin concentrating hormone innervation of caudal brainstem areas involved in gastrointestinal functions and energy balance. Neuroscience. 2005;135:611–625. doi: 10.1016/j.neuroscience.2005.06.055. [DOI] [PubMed] [Google Scholar]
- 65.Zheng Z, Lewis MW, Travagli RA. In vitro analysis of the effects of cholecystokinin (CCK) on rat brainstem motorneurons. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1066–G1073. doi: 10.1152/ajpgi.00497.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng ZL, Rogers RC, Travagli RA. Selective gastric projections of nitric oxide synthase-containing vagal brainstem neurons. Neuroscience. 1999;90:685–694. doi: 10.1016/s0306-4522(98)00586-7. [DOI] [PubMed] [Google Scholar]
- 67.Zittel TT, Rothenhofer I, Meyer JH, Raybould HE. Small intestinal capsaicin-sensitive afferents mediate feedback inhibition of gastric emptying in rats. Am J Physiol Gastrointest Liver Physiol. 1994;267:G1142–G1145. doi: 10.1152/ajpgi.1994.267.6.G1142. [DOI] [PubMed] [Google Scholar]