

Summary
The life span of C. elegans can be increased via reduced function of the mitochondria; however, the extent to which mitochondrial alteration in a single, distinct tissue may influence aging in the whole organism remains unknown. We addressed this question by asking whether manipulations to ETC function can modulate aging in a cell non-autonomous fashion. We report that the alteration of mitochondrial function in key tissues is essential for establishing and maintaining a pro-longevity cue. We find that regulators of mitochondrial stress responses are essential and specific genetic requirements for the electron transport chain (ETC) longevity pathway. Strikingly, we find that mitochondrial perturbation in one tissue is perceived and acted upon by the mitochondrial stress response pathway in a distal tissue. These results suggest that mitochondria may establish and perpetuate the rate of aging for the whole organism independent of cell-autonomous functions.
Introduction
An aging organism exhibits correlated and recognizable changes to its physiology over time. These changes occur coordinately across multiple tissues and organs, in concordance with theories that posit a strong role for the participation of the endocrine system in the regulation of age-related phenotypes (Russell and Kahn, 2007; Tatar et al., 2003). Within invertebrate model organisms such as C. elegans and Drosophila, evidence strongly suggests that tissue-specific manipulations of endocrine pathway components affect the aging process of the entire organism. These include alteration of signals from the somatic germline which control the aging of non-mitotic tissues (Arantes-Oliveira et al., 2002a; Hsin and Kenyon, 1999); restoration or reduction of insulin/IGF-1 signaling (IIS) in neuronal or fat tissues (Broughton et al., 2005; Hwangbo et al., 2004; Kapahi et al., 2004; Libina et al., 2003; Wolkow et al., 2000); and genetic manipulations to specific neurons which then alter the capacity for the entire animal to respond to dietary restriction (Bishop and Guarente, 2007). These systems have offered the simplicity of studying tissue-specific expression in organisms in which single-gene mutations can affect longevity, and have been extended to mammalian model systems (Bluher et al., 2003; Conboy et al., 2005; Taguchi et al., 2007). Such evidence indicates that there are key tissues that transmit longevity signals to modulate the aging process. Moreover, these adaptations may have evolved to provide the animal with a mechanism by which an environmental, extrinsic signal could be sensed and then amplified across the entire animal to coordinate the appropriate onset of reproduction, senescence and/or aging.
Life span can be increased by reduced function of the mitochondria. Mutation or reduced function in nuclear genes encoding electron transport chain (ETC) components in yeast, C. elegans, Drosophila, and mice delay the aging process (Copeland et al., 2009; Dell'Agnello et al., 2007; Dillin et al., 2002b; Feng et al., 2001; Hansen et al., 2008; Kirchman et al., 1999; Lapointe et al., 2009; Lee et al., 2002; Liu et al., 2005). In C. elegans, mitochondria undergo a period of dramatic proliferation during the L3/L4 stage (Tsang and Lemire, 2002). After this larval molt is completed, mitochondrial DNA proliferation in post-mitotic tissues is minimal (Tsang and Lemire, 2002). Intriguingly, the L3/L4 larval developmental period has proven to be a critical period in which the ETC modulates the aging process (Dillin et al., 2002b). Thus, the sensing and monitoring of key events during the L3/L4 transition by the ETC longevity pathway initiates and maintains the rate of aging of the animal for the rest of its life. Reduction of the ETC longevity pathway during adulthood can not result in increased longevity, even as ATP synthesis becomes impaired (Dillin et al., 2002a; Rea et al., 2007). How the mitochondrial signaling pathway modulates the aging process and the identity of the pathway constituents that transmit these longevity signals remain unknown.
One possible suggestion for the observation of increased longevity under conditions of reduced mitochondrial function originates from the “rate of living” theory of aging, in existence for over a hundred years, which suggests that the metabolic expenditures of an organism ultimately determine its life span (Pearl, 1928; Rubner, 1908). A modification to this theory has suggested alternatively that, because reactive oxygen species (ROS) are generated as a byproduct of the metabolic activity of the mitochondrial ETC during the production of ATP (Harman, 1956), a decrease in ROS production is the major contributing factor to the long-lived phenotypes of ETC mutants (Feng et al., 2001; Rea and Johnson, 2003). Recent evidence (Copeland et al., 2009; Gems and Doonan, 2008; Van Raamsdonk and Hekimi, 2009; Yang et al., 2007) does not support a linear relationship between ROS production and life span. With the increased skepticism towards the oxidative stress theory of aging comes the question: if not by manipulation of ROS in a cell-autonomous manner, then by what mechanism does reduction of mitochondrial function affect aging?
We attempted to address this question by asking whether manipulations to ETC function could modulate aging in a cell non-autonomous fashion in the nematode C. elegans. We asked whether key tissues could govern increases in longevity when components of the mitochondrial ETC are inactivated. We also reasoned that if we could identify the crucial tissues from which the ETC longevity pathway functions, we could identify the origin of the longevity signal and perhaps potential mediators of this signal.
Results
cco-1 Functions in Specific Tissues to Affect the Aging Process
To ascertain whether tissue-specific ETC knockdown could alter the life span of an organism, we created transgenic worms carrying an inverted repeat hairpin (HP) directed towards the nuclear-encoded cytochrome c oxidase-1 subunit Vb/COX4 (cco-1). cco-1 was chosen because knockdown of this gene results in intermediate phenotypes compared to knockdown of the other ETC genes by RNAi, allowing both positive and negative modulation of longevity to be identified (Dillin et al., 2002b; Lee et al., 2002; Rea et al., 2007). Furthermore, cco-1 RNAi does not result in the detrimental phenotypes observed when bacterial feeding RNAi against other components of the ETC is administered undiluted, such as severe developmental delay and lethality (Copeland et al., 2009; Gems and Doonan, 2008; Van Raamsdonk and Hekimi, 2009; Yang et al., 2007).
In worms and plants, RNAi can have a systemic effect due to spreading of the dsRNA molecules. For example, exposure of the intestine to bacterially expressed dsRNA results in the dsRNA entering through the intestinal lumen but eliciting knockdown in other cells, such as the muscle and hypodermis (Jose et al., 2009). To remove the systemic nature of RNAi from our experimental design, we used Systemic RNAi Deficient (sid-1(qt9)) mutant worms (Figure 1A). sid-1 encodes a transmembrane protein predicted to serve as a channel for dsRNA entry. While defective for systemic RNAi, the sid-1(qt9) mutants are fully functional for cell-autonomous RNAi (Winston et al., 2002). Lines were generated in the sid-1(qt9) mutant background using an inverted repeat of the cco-1 cDNA under the control of well-characterized promoters expressed in neurons (unc-119 and rab-3) (Maduro and Pilgrim, 1995; Nonet et al., 1997), intestine (ges-1) (Aamodt et al., 1991), and body-wall muscle cells (myo-3) (Miller et al., 1986; Okkema et al., 1993).
Figure 1.

Life-span analysis of cco-1 hairpin transgenic animals. A. Wild-type worms allow import of dsRNA from surrounding tissues, but sid-1(qt9) mutant worm can not import dsRNA and RNAi knockdown is no longer systemic but is maintained locally within the tissue in which the dsRNA is produced (Winston et al., 2002). B. Intestine-specific knockdown of cco-1 results in life-span extension. sid-1(qt9)/rol-6 control (black line, mean 18.8 +/- 0.7 days), ges-1p∷cco-1hairpin (green line, 23.9 +/- 0.8 days, p<.0001). C. Body wall muscle knockdown of cco-1 does not significantly affect life span. sid-1(qt9)/rol-6 control (black line, mean 18.6 +/-0.5 days), myo-3p∷cco-1 hairpin (blue line, mean 16.6+/- 0.5 days, p=0.0574). D. Neuronal knockdown of cco-1 extends life span. sid-1(qt9)/rol-6 control (black line, 18.2 +/-0.2 days), rab-3p∷cco-1 hairpin (purple line, 21.7 +/- 0.5 days, p<.0001). E. Neuronal knockdown of cco-1 driven by the unc-119 promoter also extends life span. sid-1(qt9)/rol-6 control (black line, mean 19.8 +/-0.7days), unc-119p∷cco-1 hairpin (red line, mean 23.8 +/-0.8 days, p=.0001). Please see Table S1 for all statistical analyses and also Figure S1 and Movie S1 for additional experiments.
Knockdown of cco-1 in the intestine using the ges-1 intestine-specific promoter driving a cco-1 hairpin construct significantly increased life span (Figure 1B, representative line of 13, Table S1), whereas the myo-3 muscle-specific promoter driving a cco-1 hairpin in the body-wall muscle either had no effect or even decreased life span (Figure 1C, representative line of 6, Table S1). The rab-3 neuron-specific promoter driving a cco-1 hairpin also increased life span (Figure 1D, representative line of 2, Table S1). Because the life-span extension in the neuronal promoter line was not as great as that observed in intestinal hairpin lines, we tested another neuronal promoter, the pan-neural unc-119 promoter. Consistent with the rab-3 promoter, we observed a moderate increase in life span across multiple unc-119 promoter transgenic lines (Figure 1E, representative line of 8, Table S1). The results of these experiments suggest a primary requirement for ETC knockdown in intestinal and neuronal tissues for increased longevity, albeit the neuronal derived ETC knockdown was consistently less robust compared to the intestinal knockdown.
We tested an alternative method for tissue-specific RNAi to verify our hairpin apporach. Tissue-specific RNAi can also be achieved by feeding dsRNA to rde-1 mutant animals in which the wild-type rde-1 gene has been rescued using tissue-specific promoters (Figure 2A) (Qadota et al., 2007). rde-1 encodes an essential component of the RNAi machinery encoding a member of the PIWI/STING/Argonaute family of proteins.
Figure 2.
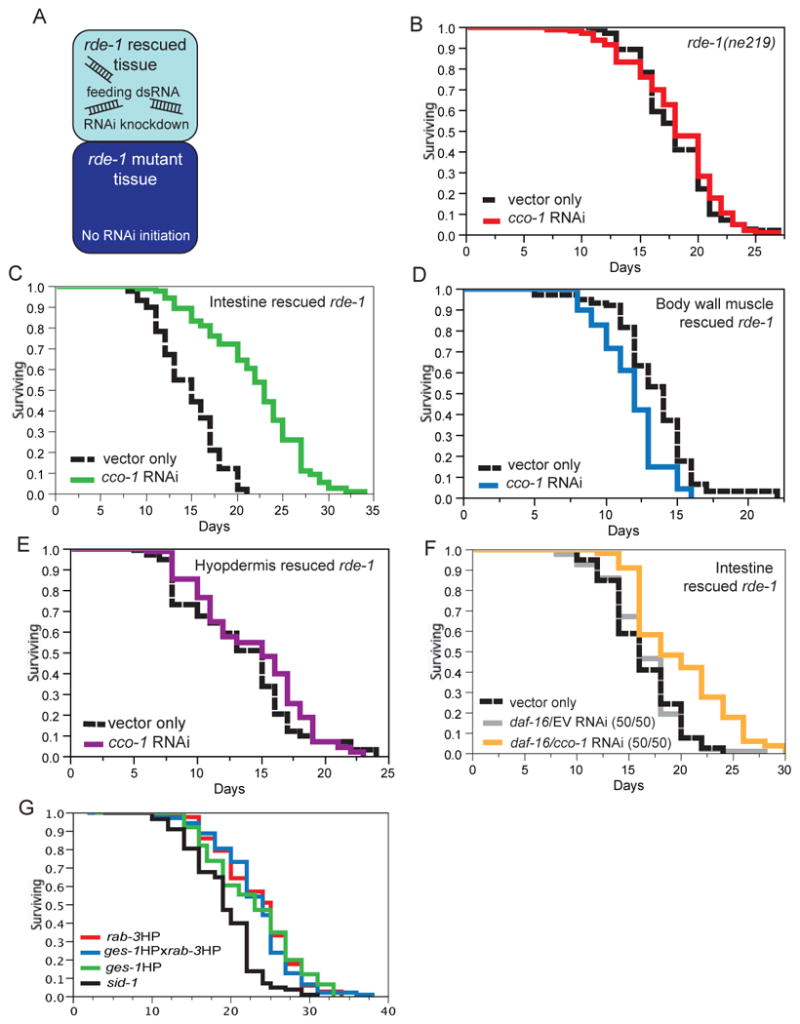
Life-span analysis of tissue-specific complementation of rde-1 with cco-1 feeding RNAi. A. Tissues exposed to dsRNA from feeding RNAi initiate knockdown if rde-1 has been rescued in the corresponding tissue. Neighboring tissues are unable to initiate RNAi if rde-1 is absent. B. rde-1(ne219) mutants do not respond to cco-1 feeding RNAi. Animals fed bacteria harboring an empty vector (black line, mean 18.0 +/- 0.3 days), cco-1 RNAi (red line, mean 18.16 +/-0.4 days, p<0.4043). C. rde-1 rescued in the intestine (VP303) extends life span when fed cco-1 dsRNA producing bacteria. Animals fed vector only bacteria (black line, mean 14.7 +/- 0.6 days), cco-1 RNAi (green line, mean 22.0 +/- 0.2 days, p<.0001). D. rde-1 rescued in the body wall muscle (NR350.5) decreases life span when fed cco-1 dsRNA bacteria. Animals fed bacteria harboring empty vector (black line, mean 13.5 +/- 0.3 days), cco-1 RNAi (blue line, mean 11.8 +/- 0.3 days, p<0.0002. E. rde-1 rescued in the hypodermis (NR222) has no effect on life span when fed cco-1 dsRNA producing bacteria. Vector only (black line, mean 13.5+/- 0.3days), cco-1 RNAi (purple line, mean 14.3 +/- 0.4 days, p=0.148). F. Life-span extension by cco-1 feeding RNAi in the intestine of ges-1:;rde-1 rescued animals is independent of daf-16. Intestinal rde-1 rescued worms were fed empty vector (black line), mean 16.4+/- 0.6 days, daf-16 RNAi diluted 50% with empty vector (gold line, mean 16.3+/- 0.4days) or daf-16 diluted 50% with cco-1 RNAi (green19.9+/- 0.5 days, p<.0001). Please see Table S1 for all statistical analysis. G. Double transgenic animals carrying rab-3:;cco-1HP and ges-1:cco-1HP (blue line, mean life span 23.4 +/-0.6 days) did not live longer than either the rab-3∷cco-1HP (red line, mean life span 23.2 +/-0.6 days, p =.64) or the the ges1∷cco-1HP (green line, mean life span 22.9+/-0.6 days, p=.75) animals. sid-1 control (black line, mean life span 19.2+/-0.5 days).
Life-span analyses were performed with rde-1(ne219) mutant animals in which rde-1 was restored by tissue-specific expression of wild-type rde-1 cDNA (Qadota et al., 2007). rde-1 was rescued in transgenic lines under the control of the lin-26 hypodermal promoter, the hlh-1 body wall muscle promoter, and the nhx-1 intestine expressing promoter (Qadota et al., 2007). These lines were then tested for their effects on life span when animals were fed cco-1 dsRNA producing bacteria.
As expected, feeding rde-1(ne219) mutant animals cco-1 dsRNA producing bacteria did not extend life span, since these animals fail to perform RNAi due to the lack of rde-1 (Figure 2B and Table S1). Consistent with the cco-1 hairpin approach, knockdown of cco-1 in the intestine, by restoring rde-1 using the intestine-specific nhx-1 promoter and feeding cco-1 dsRNA bacteria significantly increased life span. In fact, intestinal cco-1 dsRNA fed to rde-1(ne219);nhx-1p∷rde-1 worms was able to completely recapitulate the life-span extension generated by feeding cco-1 dsRNA to wild-type animals (Figure 2C and Table S1). Furthermore, cco-1 knockdown in the body wall muscle decreased life span (Figure 2D and Table S1), similar to results obtained from the muscle-specific cco-1 RNAi hairpin experiments. Hypodermal knockdown of cco-1 had no significant effect on life span (Figure 2E and Table S1). Consistent with cco-1 feeding RNAi increasing longevity in an insulin/IGF-1 pathway independent manner, we found the life-span extension of intestinal cco-1 RNAi animals to be daf-16 independent (Figure 2F and Table S1).
Because cco-1 knockdown in two distinct tissues increased longevity, we tested whether combination of the intestinal and nervous system cco-1 knockdown could result in an even further increase in longevity compared to knockdown in each individual tissue. We crossed our long lived rab-3∷cco-1HP (neuronal) lines to the ges-1∷cco-1HP (intestinal) lines and tested the life span of the double transgenic animal. Animals with cco-1 knocked down in the nervous system and the intestine did not live longer than knockdown in either the nervous system or the intestine (Figure 2G and Table S1). To test this via a second method, we used worms in which a gly-19p∷sid-1 transgene could specifically restore the capacity for feeding RNAi within the intestine of sid-1 mutant animals. We again found that feeding worms bacteria expressing cco-1 dsRNA did not further extend the life span of our rab-3∷cco-1 HP animals (Figure S1A). Therefore, there does not appear to be synergy among the tissues in which cco-1 knockdown is required to extend longevity. This finding suggests that the nervous system and the intestinal cells communicate with each other to modulate aging in response to reduced cco-1 function.
Tissue-specific ETC Knockdown Uncouples Multiple Correlates of Longevity
Resistance to oxidative stress, UV damage, and heat stress is associated with multiple forms of increased longevity. We tested whether the increased longevity of the tissue-specific cco-1 RNAi animals was due to resistance to any of these stresses. Tissue-specific knockdown of cco-1 did not affect the response of animals to oxidative stress induced by paraquat in a manner correlated with their longevity phenotype (Table S2), consistent with recent results in worms and flies (Copeland et al., 2009; Doonan et al., 2008; Van Raamsdonk and Hekimi, 2009) (Lee et al., 2002). We next tested whether resistance to UV damage correlated with increased longevity. Again, none of the long-lived tissue-specific hairpin lines were more resistant to UV damage than wild type animals (Table S3). Finally, we tested whether the long-lived tissue-specific cco-1 hairpin lines were more resistant to heat stress than control animals and found that they were not (Table S4). Collectively, the increased longevity of tissue specific cco-1 hairpin animals did not correlate with the known stress resistance phenotypes associated with other pathways that modulate the aging process (Arantes-Oliveira et al., 2002b; Larsen et al., 1995; Lee et al., 1999; Lee et al., 2003; McElwee et al., 2004).
RNAi of cco-1 slows development, growth, movement and reduces fecundity (Dillin et al., 2002b). Through an RNAi dilution approach, many of these side effects could be uncoupled from longevity, an observation that suggested a quantitative model for ETC function upon these life history traits (Rea et al., 2007). We tested whether a qualitative difference among the mitochondrial ETC from different tissues could also explain these observed side effects. We found that many of these traits could be uncoupled from increased longevity conferred by simply reducing mitochondrial function in a particular tissue. For example, long lived animals in which cco-1 was reduced in neuronal cells produced worms of nearly identical length to their control counterparts (Figure S1B), reached adulthood at the similar rates (data not shown) and had similar numbers of progeny (Figure S1C). These results are consistent with ETC reduction in all tissues of Drosophila increased life span and decreased fertility, while knockdown in neurons increased longevity without affecting fertility (Copeland et al., 2009). Additionally, reduction of cco-1 in the intestine or nervous system did not result in slowed movement; however, reduction in the body wall muscles did (Movie S1). Therefore, in addition to the quantitative model proposed by Rea et al. to explain the developmental and behavioral deficits of ETC RNAi, contributions from specific tissues must also play an important role in these life history traits.
The Mitochondrial Unfolded Protein Response (UPRmt) is Required for ETC-Mediated Longevity
In response to a mitochondrial perturbation there exists a stress response mechanism that is communicated to the nucleus to increase the expression of mitochondrial associated protein chaperones, such as HSP-6 and HSP-60, referred to as the mitochondria-specific unfolded protein response (UPRmt) (Benedetti et al., 2006; Yoneda et al., 2004; Zhao et al., 2002). hsp-6 is the mitochondrial hsp70 heat-shock protein family member and hsp-60 is the mitochondrial GroE/hsp60/hsp10 chaperonin family member. The UPRmt) is activated upon different forms of mitochondrial stress including the misfolding of mitochondria-specific proteins or stoichiometric abnormalities of large multimeric complexes, such as ETC complexes (Yoneda et al., 2004). Disrupting subunits of ETC complexes by either RNAi or mutation activates the mitochondrial stress response (Benedetti et al., 2006; Yoneda et al., 2004). cco-1 RNAi is a potent inducer of hsp-6 and hsp-60 (Yoneda et al., 2004). Intrigued by this discovery, we tested whether the UPRmt might play a central and specific role in the increased longevity generated by ETC RNAi.
We tested whether other well-known pathways that regulate the aging process also induced the UPRmt. Unlike cco-1 RNAi treated animals (Figure 3A), animals treated with RNAi towards daf-2, the IIS receptor or, eat-2 mutant animals, a genetic surrogate for diet restriction induced longevity, did not induce the UPRmt (Figures 3B and 3C) even though each of these interventions increase longevity. Therefore, induction of the UPRmt appears specific to the ETC longevity pathway and not other longevity pathways.
Figure 3.
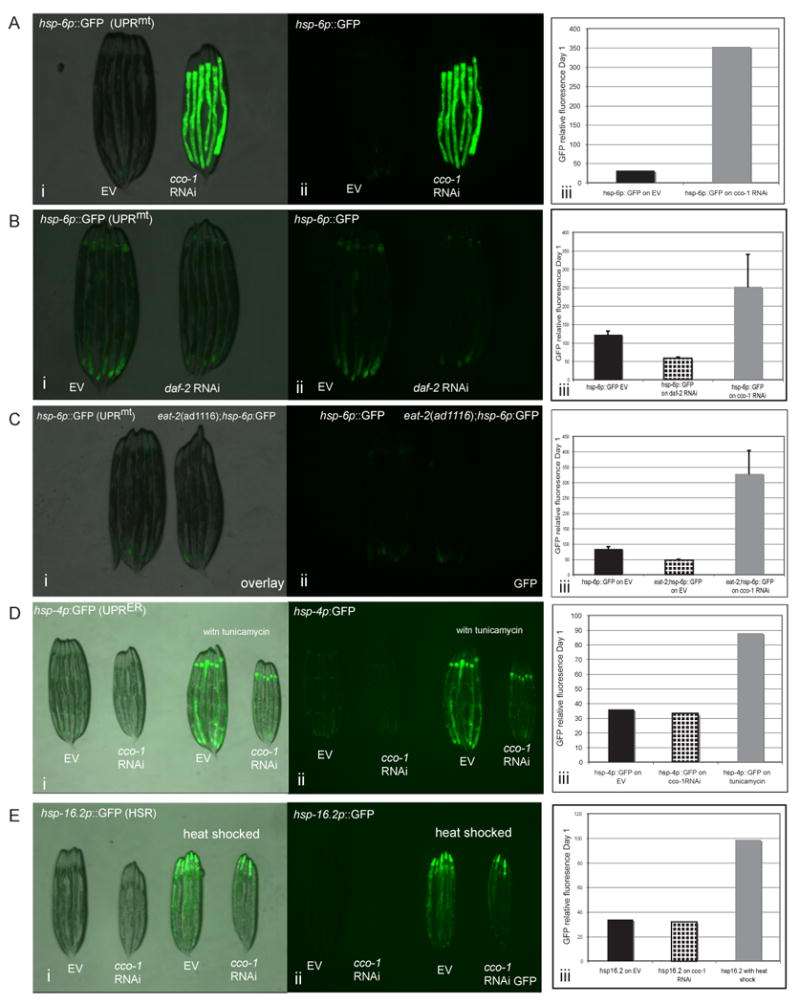
Induction of the UPRmt is specific to the ETC longevity pathway. A. hsp-6p∷GFP reporter worms fed empty vector (EV) containing bacteria have low levels of background GFP (i) overlay; (ii) GFP. hsp-6p∷GFP reporter worms fed cco-1 RNAi upregulate the UPRmt. Relative fluorescence was quantified using a fluorescence plate reader (iii). B. daf-2 RNAi does not induce hsp-6p∷GFP (i) overlay; (ii) GFP. hsp-6p∷GFP reporter worms were hatched on empty vector, cco-1, or daf-2 dsRNA expressing bacteria and allowed to grow to day 1 of adult hood. Relative fluorescence was quantified (iii). C. Dietary restricted eat-2(ad1116) mutant worms do not upregulate hsp-6p∷GFP reporter (i) overlay; (ii) GFP. Relative fluorescence was quantified (iii). D The UPRER is not induced by cco-1 RNAi, (i) overlay; (ii) GFP. hsp-4p∷GFP transgenic reporter worms were fed empty vector containing bacteria or cco-1 dsRNA bacteria. No fluorescence upregulation was detected (iii). Both EV and to a lesser extent cco-1 RNAi fed worms were able to upregulate the UPRER upon treatment with tunicamycin, (i and ii) which is known induce UPRER. Relative fluorescence of was quantified (iii). E. cco-1 RNAi does not induce a marker of cytosolic protein misfolding stress, (i) overlay; (ii) GFP. hsp16.2p∷GFP reporter worms were fed EV or cco-1 dsRNA bacteria. No fluorescence upregulation was detected (iii). As positive controls, heat shock for 6 hours at 31°C could induce the heat-shock response (HSR) and cco-1 RNAi did not block this response (i and ii). In all panels, error bars indicate standard deviations (SD).
In addition to the UPRmt, the unfolded protein response in the endoplasmic reticulum (UPRER) is also induced under conditions of protein misfolding, although confined to the ER (Ron and Walter, 2007). We tested whether mitochondrial reduction resulted in a general up regulation of all protein misfolding pathways by treating hsp-4p∷GFP reporter worm strains (Calfon et al., 2002) with cco-1 RNAi. HSP-4 is the worm orthologue of the ER chaperone, BiP, which is transcriptionally induced by the UPRER. Unlike the UPRmt, cco-1 RNAi did not induce expression of the UPRER (Figure 3D), although ER stress induced by tunicamycin did. Furthermore, cco-1 RNAi did not inhibit the ability of cells to induce the UPRER by treatment with tunicamycin. We also tested if cco-1 RNAi induced a marker of cytosolic protein misfolding by treating animals containing the hsp-16.2p∷GFP reporter strain (Link et al., 1999) with cco-1 RNAi. HSP-16.2 is a small heat-shock protein of the hsp20/alpha-B crystallin family and is under transcriptional control of the Heat-Shock Response (HSR) regulated by HSF-1. Much like the UPRER, cco-1 RNAi was unable to induce this reporter associated with cytosolic misfolding (Figure 3E). As positive controls, heat shock could induce the HSR reporter and cco-1 RNAi did not block this response. Thus, it appears that knockdown of cco-1 specifically induces the UPRmt, and not other protein misfolding pathways.
The UPRmt is a Potent Transducer of the ETC-Longevity Pathway
We tested whether the UPRmt is a key component of the ETC longevity pathway since there appeared to be a positive and specific correlation of induction of the UPRmt and ETC mediated longevity. If the UPRmt is indeed a regulator of the ETC longevity pathway, we predicted that loss of the UPRmt would specifically suppress the extended longevity of ETC reduced animals and not other longevity pathways.
The UPRmt consists of a signaling cascade that results in upregulation of nuclear-encoded genes to alleviate the stress sensed in the mitochondria. Perception of misfolding in the mitochondria requires the nuclear localized ubiquitin-like protein UBL-5, which acts as an essential and specific coactivator of the homeodomain transcription factor, DVE-1. Together, UBL-5 and DVE-1 respond to mitochondrial perturbation to increase expression of mitochondrial chaperones, including hsp-6 and hsp-60 (Benedetti et al., 2006). ClpP is the homolog of the E.coli ClpP protease located in the mitochondria that plays a role in generating the mitochondrial derived signal to activate DVE-1/UBL-5 stress responsive genes (Haynes et al., 2007).
We treated long-lived ETC mutant animals with RNAi directed towards the known pathway components of the UPRmt and tested the resulting life span. Because cco-1 RNAi is extremely sensitive to dilution and is not efficiently knocked down in combination with a second RNAi (Figures S2A and S2B), we first chose to examine the requirement for UPRmt genes on several long-lived mitochondrial mutants. RNAi of ubl-5, the dve-1 transcriptional co-regulator, specifically blocked the extended life span of the mitochondrial mutants, isp-1 (qm150) and clk-1(e2519) (Figures 4A and S2C; Table S1) compared to the life span of wild-type animals. RNAi of ubl-5 did not suppress the extended life span of long-lived daf-2 or eat-2 mutant animals (Figures 4B and 4C; Table S1). Furthermore, ubl-5 RNAi did not shorten the life span of wild type animals (Figure 4D and Table S1). Taken together, ubl-5 appears essential and specific for the extended longevity of mitochondrial mutants.
Figure 4.

ubl-5 is necessary and specific for ETC mediated longevity. A. The long life span of isp-1(qm150) mutant animals is dependent upon ubl-5. isp-1(qm150) (empty vector, black line, mean 25.8 +/- 1.0 days), isp-1(qm150) fed ubl-5 dsRNA bacteria (orange line, mean 15.5 +/-0.7 days, p<.0001), N2 wild-type (grey line, mean 19 +/- 0.5 days). B. daf-2(e1370) mutant life span is unaffected by ubl-5 knockdown. daf-2(e1370) mutant animals grown on empty vector bacteria (black line, mean 40.1+/- 1.2 days), daf-2(e1370) fed ubl-5 dsRNA bacteria (orange line, mean 39.9 +/-1.2 days, p=.327). C. Dietary restricted eat-2(ad1116) mutant life span is not dependent upon ubl-5. N2 on empty vector (grey line, mean life span 18.2+/-0.4 days); eat-2(ad1116) on empty vector (black line, mean 26.4+/-0.6 day)s; eat-2(ad1116) fed ubl-5 dsRNA bacteria (orange line, mean 23.3+/-0.7 days, p<0.0004). D. N2 wild-type life span is unaffected by ubl-5 knockdown. N2 grown on empty vector bacteria (black line, mean 18.2 +/-0.4 days), N2 fed ubl-5 dsRNA bacteria (orange line, mean 20.3 +/- 0.4 days, p=0.0834). All statistical data can be found in Table S1. See also Figure S2 for additional experiments.
RNAi of dve-1 suppressed the life span of all long-lived animals and shortened the life span of wild type animals (Figure S2D-S2G). This result is not surprising given the roles of dve-1 in growth and development and the embryonic lethality observed for homozygous dve-1 mutant animals (Burglin and Cassata, 2002; Haynes et al., 2007). Furthermore, RNAi of hsp-6, hsp-60 or clpp-1 suppressed longevity in the same manner as dve-1, suggesting that these RNAi treatments were pleiotropic and simply made the animals sick (data not shown). Thus, our results indicate that ubl-5 is specific for the longevity response, possibly by specifying the transcriptional activity of DVE-1, to mitochondrial ETC mediated longevity and dve-1, hsp-6, hsp-60 and clpp-1 have more broad roles in development making their specific roles in mitochondrial ETC mediated longevity difficult to discern at this time.
The Temporal Requirements of the UPRmt and ETC-Mediated Longevity Overlap
The life-span extension by ETC RNAi has a distinct temporal requirement during the L3/L4 stages of larval development (Dillin et al., 2002b; Rea et al., 2007). Furthermore, markers of the UPRmt, namely hsp-6p∷GFP have their greatest activation late in larval development at the L4 stage when challenged with mitochondrial stress (Yoneda et al., 2004). We verified these findings by following the activation of the UPRmt of animals treated with cco-1 RNAi and tested whether the timing requirement of cco-1 mediated longevity could be uncoupled from the induction of the UPRmt. Worms carrying the hsp-6p∷GFP UPRmt reporter were transferred onto bacteria expressing cco-1 dsRNA at every developmental stage from embryo to day 2 of adulthood (Figure 5A-5C). Worms transferred to cco-1 dsRNA at the L1 stage induced hsp-6p∷GFP throughout development, and this signal perpetuated itself into adulthood (Figure S3). Worms could induce the UPRmt if transferred to the cco-1 RNAi treatment before the L4 larval stage (Figure 5B and 5C). After the L4 larval stage, worms transferred to bacteria expressing cco-1 dsRNA were unable to induce the hsp-6p∷GFP marker (Figure 5B and 5C) and were not long lived (Dillin et al., 2002b; Rea et al., 2007). Thus, inactivation of cco-1 must be instituted before the L3/L4 larval stage to initiate induction of the UPRmt. Inactivation in adulthood does not induce the UPRmt and does not result in increased longevity.
Figure 5.
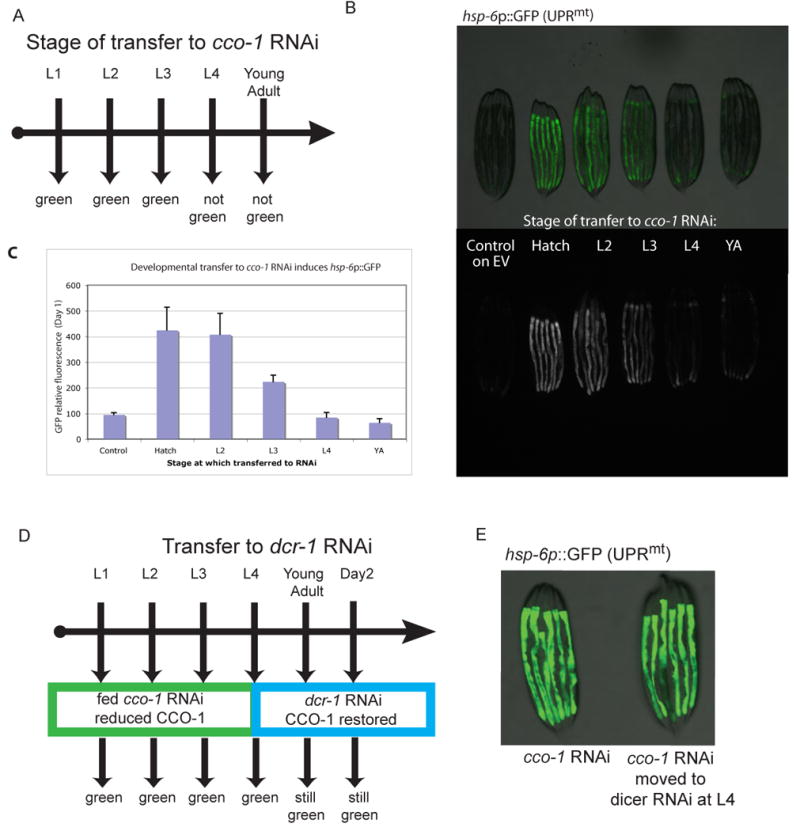
The temporal activation of ETC generated longevity signal is coincident with induction of the UPRmt.
A. hsp-6p∷GFP reporter worms were transferred to cco-1 RNAi at each larval developmental stage and early adulthood. GFP fluorescent measurements were taken 16 hours after reaching young adulthood in all cases. B. hsp-6p∷GFP is upregulated if transfer occurs before the L4 stage of development. C. Quantification of hsp-6p∷GFP in (A); error bars represent standard deviation (SD). D. cco-1 knockdown during larval development is sufficient to induce the hsp-6p∷GFP reporter in adulthood. hsp-6p∷GFP reporter worms we grown on cco-1 dsRNA bacteria during development and then moved to dcr-1 dsRNA producing bacteria at the L4 larval stage, to disrupt the RNAi machinery allowing CCO-1 levels to return to normal (Dillin et al., 2002). UPRmt remains induced. E. hsp-6p∷GFP fluorescence 48 hours after transfer to dcr-1 RNAi as described by schematic D. See also Figure S3.
Inactivation of ETC components during larval development is sufficient to confer increased longevity on adult animals even though the knocked-down ETC component can be restored in adulthood (Dillin et al., 2002b; Rea et al., 2007). We tested whether developmental inactivation of cco-1 could not only induce, but whether it could also maintain activation of the UPRmt during adulthood, even though adult inactivation of cco-1 was unable to induce the UPRmt. Worms treated with cco-1 RNAi during larval development and then moved to dicer (dcr-1) RNAi (a key component of the RNAi machinery) to block further RNAi activity on day 1 of adulthood have an extended life span (Dillin et al., 2002b). Similarly, hsp-6p∷GFP worms treated with cco-1 RNAi during larval development and moved onto dcr-1 RNAi maintained the induced response of the UPRmt (Figures 5D and 5E). Therefore, inactivation during larval development of cco-1 is sufficient to initiate and maintain a signal to increase longevity and induce the UPRmt in adult animals. The results of these experiments match the timing requirements of the life-span extension for ETC RNAi treated worms and support the idea that the signals for increased longevity and induction/maintenance of the UPRmt are not separable.
The UPRmt Responds to Cell Non-Autonomous Cues from ETC knockdown
Intrigued by the tissue-specific nature by which cco-1 depletion can modulate the aging process of the entire animal, the specific role of the UPRmt in the longevity response in ETC mutant animals and the overlapping timing requirements for both ETC RNAi and induction of the UPRmt, we hypothesized that the induction of the UPRmt may be able to act cell non-autonomously in a multicellular organism. If so, we reasoned that induction of the UPRmt in one tissue by cco-1 reduction might lead to the UPRmt being upregulated in a distal tissue that has not experienced cco-1 reduction (Figure 6A). Consistent with this hypothesis, transgenic worms with the cco-1 hairpin expressed in all neurons (either the rab-3 or the unc-119 promoter driven cco-1 hairpin) were able to induce hsp-6p:GFP expression in the intestine (Figures 6Bii and 6Biii). In fact, neuronal RNAi of cco-1 induced the UPRmt reporter to the same extent as animals with intestinal cco-1 RNAi (Figures 6B and 6C). We were unable to ascertain whether mitochondrial ETC knockdown in the intestine could signal to the nervous system to induce the UPRmt due to the low expression of the hsp-6p∷GFP reporter in neuronal cells.
Figure 6.
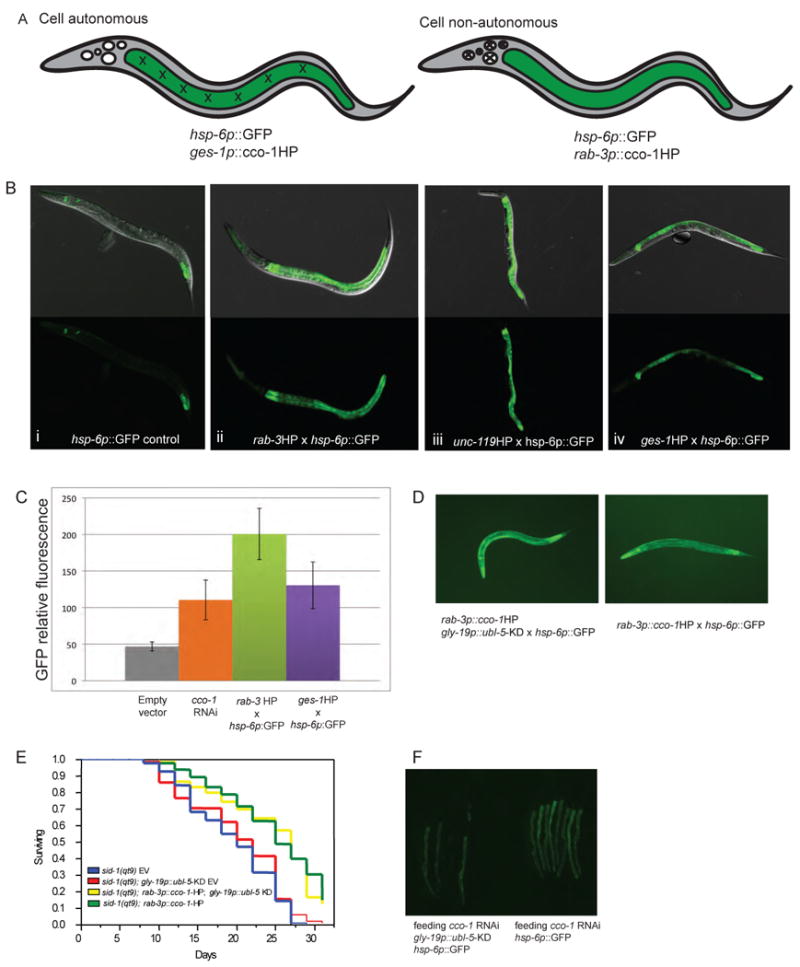
Cell non-autonomous upregulation of the UPRmt.
A. Representation of cell-autonomous and non-autonomous upregulation of UPRmt. “X's” depict tissue where cco-1 is knocked down (intestine or neurons). Green indicates location of upregulation of hsp-6p∷GFP reporter (intestine upon knockdown in intestine or neurons). B. hsp-6p∷GFP reporter worms were crossed to tissue-specific cco-1 hairpin lines. Control hsp-6p∷GFP shows only background GFP (i). Neuron-specific cco-1 hairpin results in upregulation of hsp-6p∷GFP in the intestine (rab-3 (ii) and unc-119 (iii) lines shown). Intestine-specific ges-1p∷cco-1 hairpin (iv) also results in upregulation of the hsp-6p∷GFP reporter in the intestine. C. Fluorescent quantification of B; error bars represent standard deviation (SD). D. Intestinal knockdown of ubl-5 does not block UPRmt induction caused by neuronal cco-1 reduction. Worm strains were created with rab-3∷cco-1HP; hsp-6p∷gfp with gly-19p∷ubl-5HP. E. Intestinal reduction of ubl-5 does not block the life-span extensions of rab-3∷cco-1HP animals. N2 fed empty vector (black line, mean=19.1+/-0.4); sid-1(qt9) (blue line, mean=19.4+/-0.6 days); sid-1(qt9); gly-19p∷ubl-5-KD (red line, mean=20.1+/-0.7 +/-0.7days); rab-3p∷cco-1-HP; gly-19p∷ubl-5-KD (yellow line, mean= 24.1+/-0.7days); rab-3p∷cco-1HP (green line, mean= 24.5+/-0.7days); N2 on cco-1 RNAi (mean= 27.3+/-0.6days. p>0.66 (green vs. yellow)) F. Intestinal reduction of ubl-5 (gly-19p∷ubl-5HP) blocks UPRmt induction of animals fed bacteria expressing cco-1 dsRNA. See also Figure S4.
Because ubl-5 RNAi could block the long life span of mitochondrial mutants and ubl-5 is required for induction of the UPRmt, we tested whether reduction of ubl-5 in the intestinal cells could block the induction of the cell non-autonomous signal for the UPRmt that originated from the nervous system. We created lines expressing the rab-3p∷cco-1HP in conjunction with a gly-19p∷ubl-5HP containing the hsp-6p∷GFP reporter. Surprisingly, we found that ubl-5 reduction in the intestinal cells could not block the induction of the UPRmt from signals generated in the nervous system (Figure 6D). Furthermore, intestine-specific depletion of ubl-5 was not sufficient to block the long life span of animals with reduced cco-1 expression in the nervous system (Figure 6E). However, intestine-specific knockdown of ubl-5 did block induction of the UPRmt in the intestinal cells of animals fed bacteria expressing dsRNA of cco-1 (Figure 6F and Figure S4A). Neuronal cells are not able to induce a RNAi response to feeding dsRNA, indicating that ubl-5 is required for induction of the UPRmt in response to cco-1 reduction in non-neuronal cells, but other, yet to be defined factors, are required to respond to the cell non-autonomous signals from the nervous system to induce the UPRmt.
Discussion
This work identified key tissues, genes essential and specific for mitochondrial longevity and at least one mechanism necessary for increased longevity in response to altered mitochondrial function in a metazoan. The UPRmt was essential for the extended longevity of ETC mutant animals and has been previously reported to be upregulated in response to RNAi of cco-1 (Yoneda et al., 2004). Consistent with the temporal requirements of the ETC to modulate longevity during the L3/L4 larval stages, the UPRmt could only be induced when cco-1 RNAi was administered before the L3/L4 larval stage, but not in adulthood. Therefore, induction of the UPRmt mirrored the temporal requirements of the ETC to promote longevity when reduced. More importantly, the fact that induction of the UPRmt can be maintained long into adulthood, well after the mitochondrial insult had been given in larval development, indicates that the animal might possess an epigenetic mechanism to ensure increased resistance to future mitochondrial perturbations.
One of the most surprising findings is the UPRmt can be activated in a cell non-autonomous manner. Because the hsp-6p∷GFP reporter is primarily limited to expression in the intestine, we were well poised to ask if perturbation of cco-1 in the nervous system could induce the UPRmt in the intestine. Therefore, neuronal limited knockdown of cco-1 could profoundly induce the hsp-6 reporter indicates that a cue from the nervous system must travel to the intestine to induce the UPRmt (Figure 7). It is not clear whether the factor is proteinacious, nucleic acid based or a small molecule, but it is clear that its production in a limited number of cells can profoundly influence the survival of the entire organism. Because this signal is the product of perceived mitochondrial stress that results in increased survival, we have termed this cell non-autonomous signal a “mitokine”.
Figure 7.

Model for the cell non-autonomous nature of the UPRmt.
Cells experiencing mitochondrial stress, in this scenario neuronal cells (circles) marked within the yellow box, produce a signal that is transmitted from the mitochondria to the nucleus to regulate the expression of genes regulated by UBL-5 and possibly DVE-1. These cells serve as sending cells and produce an extracellular signal (mitokine) that can be transmitted to distal, receiving cells, in this case intestinal cells marked in the green box. Receiving cells perceive the mitokine and induce the mitochondrial stress response. See also Figure S5.
While many of these perturbations have pleiotropic effects that result in their short life span, their ability to upregulate the UPRmt is not sufficient to overcome these potentially harmful side effects. We found that muscle-specific cco-1 RNAi could also induce the intestinal hsp-6p∷GFP reporter, yet these animals were not long lived (Figure S4B). We also find that short-lived mev-1 mutant animals also induce the UPRmt (data not shown). Finally, many of the nuclear-encoded mitochondrial genes discovered to induce the UPRmt when inactivated using RNAi (Yoneda et al., 2004) are not long lived (data not shown). Therefore, ectopic induction of the UPRmt is required but is not sufficient in the establishment of the pro-longevity cue from mitochondria in these settings.
Of the currently identified UPRmt pathway members, the ubiquitin like protein, UBL-5, which provides transcriptional specificity for the homeobox transcription factor DVE-1 in response to unfolded proteins in the mitochondria, is essential for the increased longevity of ETC mutant animals. Knockdown of ubl-5 specifically in the intestine was not sufficient to block mitokine signaling from the nervous system, but was able to block induction of animals fed dsRNA of cco-1, which can not reduce cco-1 in the nervous system. Therefore, it appears that ubl-5 either functions exclusively in a cell-autonomous fashion, or the signals generated to induce the UPRmt in the nervous system are very different from the signal generated in other tissues as ubl-5 was not required for neuronal induction.
It is intriguing to speculate why reduced mitochondrial ETC in only a few tissues are able to send a pro-longevity cue, or mitokine, but others do not. Because the intestine and the sensory neurons (amphids and phasmids) are the only cells that are in direct contact with the worm's environment (the hypodermis/skin is wrapped in a protective, dense cuticle), perhaps these cells are fine-tuned to perceive mitochondrial insults that might be present in the environment. Alternatively, mitochondrial metabolism in the nervous system and intestine might have different requirements than other tissues making disruptions in these tissues more susceptible to perturbation and subsequent UPRmt upregulation. As another possibility, there is a growing body of research emphasizing the importance of ROS, not as damaging agents, but as crucial components of cell signaling. It remains a possibility that ROS may act as signaling molecules and potentially serve as the mitokine or intermediary to elicit a nuclear response. However, animals treated with high doses of the antioxidants N-acetyl-L-cysteine (NAC) or ascorbic acid (vitamin C) were not able to block mitokine signaling (Figure S5), a thorough investigation of mitochondrial function from each tissue will be essential to test these hypotheses.
In the future it will be important to understand how mitochondrial stress initiates the UPRmt in a cell-autonomous fashion and how this stress is then transmitted throughout the organism to induce the UPRmt in cells that have yet to possess mitochondrial stress (i.e. a cell non-autonomous fashion). Furthermore, the identity and mode of action of the mitokine will provide an avenue to explore treatment of mitochondrial diseases in a tissue and cell-type specific manner if conserved from worm to man.
Experimental Procedures
Strains
HC114 (sid-1(qt9)), MQ887 (isp-1(qm150)), CB4876 (clk-1(e2519)), CF1041 (daf-2(e1370)), TK22 (mev-1(kn1)III), WB27 (rde-1(ne219)), NR222 (rde-1(ne219)V; kzIs9, NR350 (rde-1(ne219) V; kzIs20), SJ4100 (zcIs13[hsp-6p∷GFP]), SJ4058 (zcIs9[hsp-60p∷GFP]), CL2070 (dvIs[hsp-16.2∷GFP]) and N2 wild-type were obtained from the Caenorhabditis Genetics Center. VP303 was a generous gift from the Strange lab.
The myo-3 promoter hairpin RNAi transgene was created by inserting PCR amplified cco-1 cDNA with no stop codon into pPD97.86 (Addgene). The reverse complement cco-1 cDNA was inserted into pGEX2T after the GST linker to be used as the hairpin loop as described (Tavernakis 2001). PCR amplifications were used to add an AgeI site to the 3′ end of the cco-1 cDNA and NgoMIV (compatible and non-recleavable with AgeI) to the 5′ end of the GST linker. Ligation of the PCR products in the presence of AgeI enzyme and NgoMIV were followed by gel extraction of the promoter hairpin fragment as described (Hobert 2002). The ges-1 and unc-119 promoters were PCR amplified from genomic DNA and cloned in place of the myo-3 promoter driving cco-1. The rab-3 promoter was a gift from Kang Shen, Stanford University, and sequence verified. Transgenic tissue-specific RNAi hairpin expressing strains were generated by microinjecting gel extracted hairpin RNAi constructs (40-60ng/μl) mixed with an equal concentration of pRF4(rol-6) co-injection marker or myo-2∷GFP into sid-1(qt9) worms. Control lines were generated by injecting sid-1(qt9) with 50ng/μl pRF4(rol-6). Extrachromosomal arrays were integrated and backcrossed 5 times as described (Hope 1999).
The gly-19 intestinal promoter driving wild-type sid-1 was injected into rab-3p∷cco-1HP transgenic worms to enable knockdown of cco-1 in the neurons by the hairpin transgene and in the intestine by feeding RNAi.
ubl-5 knockdown strains were generated with constructs as described (Esposito et al., 2007; Hobert, 2002). Forward and reverse orientation ubl-5 constructs were driven by the gly-19 intestinal promoter and co-injected with the myo-3p∷tdTomato marker.
Life-span Analyses
Life-span analyses were performed as described previously (Dillin et al., 2002a). 80-100 animals were used per condition and scored every day or every other day. All life-span analyses were conducted at 20°C and repeated at least twice. JMP IN 8 software was used for statistical analysis. In all cases, P-values were calculated using the log-rank (Mantel–Cox) method.
GFP Expression and Quantification
SJ4100 hsp-6∷GFP were bleached to collect synchronous eggs and grown on cco-1 RNAi. At each stage from larval stage 1 to Day 1 of adulthood, worms were assayed for GFP expression. Alternatively, SJ4100 worms were grown on empty vector and transferred to cco-1 RNAi at each developmental stage at which time GFP was assayed at Day 1 or 2 of adulthood.
Integrated hairpin RNAi worm lines were crossed to SJ4100 hsp-6p∷GFP reporter lines. GFP was monitored in Day 1 adults. Fluorimetry assays were performed using a Tecan fluorescence plate reader. 100 roller worms were picked at random (25 into 4 wells of a black walled 96-well plate) and each well was read three times and averaged. Each experiment was repeated three times.
Supplementary Material
Table S1, Statistics for Life spans of Figures 1,2, 4 and 6.
Table S2, Statistics for Paraquat Experiments
Table S3, Statistics for UV Experiments
Table S4, Statistics for Heat-Shock Experiments
Movie S1, related to Figure 1. Effect of tissue-specific cco-1 RNAis on movement.
Figure S1, related to Figure 1. Effect of tissue-specific cco-1 RNAis on size and progeny.
Figure S2, related to Figure 4. Effects of ubl-5 and dve-1 RNAi on ETC regulated longevity.
Figure S3, related to Figure 5. hsp-6p∷GFP activation by cco-1 feeding RNAi from hatch is constantly expressed throughout all larval stages.
Figure S4, related to Figure 6. Upregualtion of hsp-6p∷GFP by muscle-specific cco-1 RNAi.
Figure S5, related to Figure 7. Exploration of ROS as a potential Signaling Molecule.
Acknowledgments
We thank Drs. W. Mair, S. Panowski, M. Raices, P. Douglas, and N. Baird for thoughtful editing and scientific insight. We thank Z. Liu for cloning and strain integration. We are grateful to Drs. C. Hunter, C. Haynes, D. Ron and K. Shen for strains and reagents and anonymous reviewers for their insight. This work was supported by NIA R01 AG024365, NIH Developmental Biology Training Grant, and HHMI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aamodt EJ, Chung MA, McGhee JD. Spatial control of gut-specific gene expression during Caenorhabditis elegans development. Science (New York, NY. 1991;252:579–582. doi: 10.1126/science.2020855. [DOI] [PubMed] [Google Scholar]
- Apfeld J, Kenyon C. Cell nonautonomy of C. elegans daf-2 function in the regulation of diapause and life span. Cell. 1998;95:199–210. doi: 10.1016/s0092-8674(00)81751-1. [DOI] [PubMed] [Google Scholar]
- Arantes-Oliveira N, Apfeld J, Dillin A, Kenyon C. Regulation of lifespan by germ-line stem cells in Caenorhabditis elegans. Science (New York, NY. 2002a;295:502–505. doi: 10.1126/science.1065768. [DOI] [PubMed] [Google Scholar]
- Arantes-Oliveira N, Apfeld J, Dillin A, Kenyon C. Regulation of Life-Span by Germ-Line Stem Cells in Caenorhabditis elegans. Science. 2002b;295:502–505. doi: 10.1126/science.1065768. [DOI] [PubMed] [Google Scholar]
- Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174:229. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop NA, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007;447:545–549. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- Bluher M, Kahn BB, Kahn CR. Extended Longevity in Mice Lacking the Insulin Receptor in Adipose Tissue. Science (New York, NY. 2003;299:572–574. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- Broughton SJ, Piper MDW, Ikeya T, Bass TM, Jacobson J, Driege Y, Martinez P, Hafen E, Withers DJ, Leevers SJ. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proceedings of the National Academy of Sciences. 2005;102:3105. doi: 10.1073/pnas.0405775102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burglin TR, Cassata G. Loss and gain of domains during evolution of cut superclass homeobox genes. International Journal of Developmental Biology. 2002;46:115–124. [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. Extension of Drosophila Life Span by RNAi of the Mitochondrial Respiratory Chain. Current Biology. 2009 doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Dell'Agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, Zeviani M. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Human Molecular Genetics. 2007;16:431. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- Dillin A, Crawford DK, Kenyon C. Timing Requirements for Insulin/IGF-1 Signaling in C. elegans. Science (New York, NY. 2002a;298:830–834. doi: 10.1126/science.1074240. [DOI] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of Behavior and Aging Specified by Mitochondrial Function During Development. Science (New York, NY. 2002b;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P, Matscheski A, Vanfleteren JR, Gems D. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes & Development. 2008;22:3236. doi: 10.1101/gad.504808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Di Schiavi E, Bergamasco C, Bazzicalupo P. Efficient and cell specific knock-down of gene function in targeted C. elegans neurons. Gene. 2007;395:170–176. doi: 10.1016/j.gene.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Feng J, BussiËre F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Developmental Cell. 2001;1:633–644. doi: 10.1016/s1534-5807(01)00071-5. [DOI] [PubMed] [Google Scholar]
- Gems D, Doonan R. Oxidative Stress and Aging in the Nematode Caenorhabditis elegans. Oxidative Stress in Aging: From Model Systems to Human Diseases. 2008:81. [Google Scholar]
- Golden TR, Beckman KB, Lee AHJ, Dudek N, Hubbard A, Samper E, Melov S. Dramatic age-related changes in nuclear and genome copy number in the nematode Caenorhabditis elegans. Aging Cell. 2007;6:179. doi: 10.1111/j.1474-9726.2007.00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008;4:e24. doi: 10.1371/journal.pgen.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Developmental Cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, Driscoll M. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419:808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- Hobert O. PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans. Biotechniques. 2002;32:728–730. doi: 10.2144/02324bm01. [DOI] [PubMed] [Google Scholar]
- Hsin H, Kenyon C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 1999;399:362–366. doi: 10.1038/20694. [DOI] [PubMed] [Google Scholar]
- Hwangbo DS, Gersham B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 2004;429:562–566. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- Iser WB, Gami MS, Wolkow CA. Insulin signaling in Caenorhabditis elegans regulates both endocrine-like and cell-autonomous outputs. Developmental biology. 2007;303:434–447. doi: 10.1016/j.ydbio.2006.04.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose AM, Smith JJ, Hunter CP. Export of RNA silencing from C. elegans tissues does not require the RNA channel SID-1. Proceedings of the National Academy of Sciences. 2009;106:2283. doi: 10.1073/pnas.0809760106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Current Biology. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchman PA, Kim S, Lai CY, Jazwinski SM. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics. 1999;152:179–190. doi: 10.1093/genetics/152.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe J, Stepanyan Z, Bigras E, Hekimi S. Reversal of the mitochondrial phenotype and slow development of oxidative biomarkers of aging in long-lived Mclk1+/-mice. Journal of Biological Chemistry. 2009:M109. doi: 10.1074/jbc.M109.006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PL, Albert PS, Riddle DL. Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics. 1995;139:1567–1583. doi: 10.1093/genetics/139.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science (New York, NY. 1999;285:1390. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- Lee SS, Kennedy S, Tolonen AC, Ruvkun G. DAF-16 Target Genes That Control C. elegans Life-Span and Metabolism. Science (New York, NY. 2003;300:644–647. doi: 10.1126/science.1083614. [DOI] [PubMed] [Google Scholar]
- Lee SS, Lee RYN, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. nature genetics. 2002;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003;115:489–502. doi: 10.1016/s0092-8674(03)00889-4. [DOI] [PubMed] [Google Scholar]
- Link CD, Cypser JR, Johnson CJ, Johnson TE. Direct observation of stress response in Caenorhabditis elegans using a reporter transgene. Cell stress & chaperones. 1999;4:235–242. doi: 10.1379/1466-1268(1999)004<0235:doosri>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes & development. 2005;19:2424–2434. doi: 10.1101/gad.1352905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro M, Pilgrim D. Identification and Cloning of unc-119, a Gene Expressed in the Caenorhabditis elegans Nervous System. Genetics. 1995;141:977–988. doi: 10.1093/genetics/141.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElwee JJ, Schuster E, Blanc E, Thomas JH, Gems D. Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. Journal of Biological Chemistry. 2004;279:44533. doi: 10.1074/jbc.M406207200. [DOI] [PubMed] [Google Scholar]
- Miller DM, Stockdale FE, Karn J. Immunological identification of the genes encoding the four myosin heavy chain isoforms of Caenorhabditis elegans. Proceedings of the National Academy of Sciences. 1986;83:2305–2309. doi: 10.1073/pnas.83.8.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, Meyer BJ. Caenorhabditis elegans rab-3 Mutant Synapses Exhibit Impaired Function and Are Partially Depleted of Vesicles. J Neurosci. 1997;17:8061–8073. doi: 10.1523/JNEUROSCI.17-21-08061.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A. Sequence Requirements for Myosin Gene Expression and Regulation in Caenorhabditis elegans. Genetics. 1993;135:385–404. doi: 10.1093/genetics/135.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl R. The Rate of Living. London, UK: University of London Press; 1928. [Google Scholar]
- Qadota H, Inoue M, Hikita T, Köppen M, Hardin JD, Amano M, Moerman DG, Kaibuchi K. Establishment of a tissue-specific RNAi system in C. elegans. Gene. 2007;400:166–173. doi: 10.1016/j.gene.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S, Johnson TE. A metabolic model for life span determination in Caenorhabditis elegans. Developmental cell. 2003;5:197–203. doi: 10.1016/s1534-5807(03)00242-9. [DOI] [PubMed] [Google Scholar]
- Rea SL, Ventura N, Johnson TE. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007;5:e259. doi: 10.1371/journal.pbio.0050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rubner M. Das Problem der Lebensdauer und seine Beziehungen zu Wachstum und ErnaÈ hrung. Muenchen, Germany: R. Oldenburg; 1908. [Google Scholar]
- Russell SJ, Kahn CR. Endocrine regulation of ageing. Nature Reviews Molecular Cell Biology. 2007;8:681–691. doi: 10.1038/nrm2234. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science (New York, NY. 2007;317:369. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- Tatar M, Bartke A, Antebi A. The Endocrine Regulation of Aging by Insulin-like Signals. Science (New York, NY. 2003;299:1346–1351. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- Tsang WY, Lemire BD. Mitochondrial genome content is regulated during nematode development. Biochemical and biophysical research communications. 2002;291:8–16. doi: 10.1006/bbrc.2002.6394. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Hekimi S. Deletion of the Mitochondrial Superoxide Dismutase sod-2 Extends Lifespan in Caenorhabditis elegans. PLoS Genetics. 2009;5 doi: 10.1371/journal.pgen.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessells RJ, Fitzgerald E, Cypser JR, Tatar M, Bodmer R. Insulin regulation of heart function in aging fruit flies. Nature genetics. 2004a;36:1275–1281. doi: 10.1038/ng1476. [DOI] [PubMed] [Google Scholar]
- Wessells RJ, Fitzgerald E, Cypser JR, Tatar M, Bodmer R. Insulin regulation of heart function in aging fruit flies. Nat Genet. 2004b;36:1275–1281. doi: 10.1038/ng1476. [DOI] [PubMed] [Google Scholar]
- Winston WM, Molodowitch C, Hunter CP. Systemic RNAi in C. elegans Requires the Putative Transmembrane Protein SID-1. Science (New York, NY. 2002;295:2456–2459. doi: 10.1126/science.1068836. [DOI] [PubMed] [Google Scholar]
- Wolkow CA, Kimura KD, Lee MS, Ruvkun G. Regulation of C. elegans Life-Span by Insulinlike Signaling in the Nervous System. Science (New York, NY. 2000;290:147–150. doi: 10.1126/science.290.5489.147. [DOI] [PubMed] [Google Scholar]
- Yang W, Li J, Hekimi S. A Measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans. Genetics. 2007;177:2063–2074. doi: 10.1534/genetics.107.080788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117:4055–4066. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. The EMBO Journal. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, Statistics for Life spans of Figures 1,2, 4 and 6.
Table S2, Statistics for Paraquat Experiments
Table S3, Statistics for UV Experiments
Table S4, Statistics for Heat-Shock Experiments
Movie S1, related to Figure 1. Effect of tissue-specific cco-1 RNAis on movement.
Figure S1, related to Figure 1. Effect of tissue-specific cco-1 RNAis on size and progeny.
Figure S2, related to Figure 4. Effects of ubl-5 and dve-1 RNAi on ETC regulated longevity.
Figure S3, related to Figure 5. hsp-6p∷GFP activation by cco-1 feeding RNAi from hatch is constantly expressed throughout all larval stages.
Figure S4, related to Figure 6. Upregualtion of hsp-6p∷GFP by muscle-specific cco-1 RNAi.
Figure S5, related to Figure 7. Exploration of ROS as a potential Signaling Molecule.