

Abstract
The fruit fly, Drosophila melanogaster, has a long and rich history as an important model organism for biologists. In particular, study of the fruit fly has been essential to much of our fundamental understanding of the development and function of the nervous system. In recent years, studies using fruit flies have provided important insights into the pathogenesis of neurodegenerative and neuromuscular diseases. Fly models of spinal muscular atrophy, spinobulbar muscular atrophy, myotonic dystrophy, dystrophinopathies and other inherited neuromuscular diseases recapitulate many of the key pathologic features of the human disease. The ability to perform genetic screens holds promise for uncovering the molecular mechanisms of disease, and indeed, for identifying novel therapeutic targets. This review will summarize recent progress in developing fly models of neuromuscular diseases and will emphasize the contribution that Drosophila has made to our understanding of these diseases.
“Everything should be made as simple as possible, but not one bit simpler.”
-- Albert Einstein
Introduction
The fruitfly, Drosophila melanogaster, has long been recognized as one of the most powerful genetic systems for studying neuromuscular development and function. Recently, Drosophila has emerged as a fruitful model system for studying neurological diseases, including neuromuscular disease. Anyone who has ever tried to swat a fly knows of its rapid response to stimuli and sophisticated motor behaviors, including an ability to walk, climb, jump, and, of course, fly. These complex behaviors are controlled by a nervous system that is simple relative to ours, and yet contains the same basic neural circuitry with sensory and motor neurons, multinucleate muscle cells, and elaborate neuromuscular junctions (NMJs). At a genetic, molecular, and physiologic level, the fundamental mechanisms of neuromuscular function are remarkably well conserved in processes ranging from action potential generation and propagation in the neuron to synaptic transmission at the NMJ to excitation-contraction coupling in the muscle. This conservation has allowed discoveries made in the fly to be directly translated into advancing our understanding of how the human nervous system functions, and indeed, how things malfunction in disease.
Drosophila began to be used to study neurological diseases about 10 years ago when, through the efforts of the Drosophila and human genome projects, it became apparent that the genetic makeup of the fruitfly was surprisingly similar to humans. About 75% of all known human disease genes have fly homologues 1. While the majority of human genes have a fly counterpart, the fly genome is much more “compact” with smaller gene families and less redundancy, fewer and smaller introns and spliced variants, and simpler noncoding regulatory regions, thus making genes easier to study and their functions easier to understand. The presence of numerous powerful genetic tools developed over the last century has allowed these genes to be manipulated rapidly to allow their in vivo function to be investigated.
The ability to use Drosophila to model neurological disease was first shown in the late 1990’s with glutamine-repeat disorders. Overexpression of polyglutamine-expanded versions of human Huntingtin or Ataxin-3 proteins in the fruitfly resulted in late-onset neurodegeneration, progressive impairment in locomotion, and ubiquitinated aggregates similar to those seen in human brains 2, 3. Furthermore, overexpression of human alpha-synuclein in fly neurons leads to specific loss of dopaminergic neurons and accumulation of protein aggregates that resemble Lewy bodies, thereby serving as a model for Parkinson’s disease 4. Thus, overexpression of mutant proteins in the fly causes neuronal degeneration with pathologic inclusions that mimic those seen in human disease 5. In the last decade, the fly has been increasingly used to model many different types of human disease.
When compared to vertebrate models, probably the greatest advantage of the fly as a model system is the speed and power of genetic screens. Flies have a rapid life cycle, taking only 10 days to complete development from embryo to fertile adult. The small size of animals, large numbers of progeny, and rapid generation time permit large-scale genetic screens to be performed that are not feasible in the mouse. These whole-genome screens for genetic modifiers of disease phenotypes can provide unbiased and often unanticipated
Insights into disease mechanisms and identify potential drug targets that can be validated in vertebrate models. For example, whole genome screens for modifiers of polyglutamine-induced toxicity identified overexpression of chaperones as potent suppressors 6, 7. This discovery provided early validation of the hypothesis that protein misfolding is a fundamental problem in polyglutamine disease, and revealed a new strategy of therapeutic intervention based on targeting chaperones with small molecules. Together, these approaches have made Drosophila a powerful, simple model system for studying the mechanisms of neuromuscular function and disease.
Neuromuscular Development in Drosophila
Before discussing how the fly has been used to study neuromuscular disease, we will first give an overview of the development and function of the fly neuromuscular system. The life cycle of the fly begins with fertilization of a female’s egg by male sperm. Embryogenesis lasts about 21 hours, and at the end of this period, a first-instar larva hatches from the embryonic shell and crawls in search of food. Thus, the development of a fully functional larval nervous system from an unfertilized egg takes less than 24 hours. Through a series of molts, the larva grows about 1000 fold in size, until emerging from the food as a third-instar larva to undergo pupariation. During metamorphosis, many of the larval tissues including muscles degenerate, and the adult structures develop from “imaginal discs”. During the four days of pupal development, motor neurons must retract their axons from degenerating larval muscles and regrow connections with newly forming adult muscle cells. At the completion of metamorphosis, an adult fly ecloses from the pupal case, and has a lifespan in culture of about two months.
The embryonic and larval nervous systems, and in particular the NMJ, have been extensively studied to understand fundamental mechanisms of nervous system development and function 8. Larvae can rapidly be filleted to display a complex array of NMJs embedded in muscles of the body wall. Shown in Figure 1A is the larval nervous system with the central nervous system comprised of two lobes of the brain (*) and the ventral nerve cord (**). Motor neurons (yellow) send axons that travel in nerves (arrow) to innervate multiple muscles, forming elaborate NMJs shown in red (arrowhead). Multiple types of sensory neurons are present in the body wall and send axons along these same nerves to the ventral nerve cord. Drosophila axons are unmyelinated, but they are surrounded by glial cytoplasmic processes, similar to unmyelinated human nerves (Figure 1A, inset). As shown in Figure 1B, a bouton of the NMJ contains multiple active zones (arrowhead), each containing multiple synaptic vesicles (arrow). The presynaptic terminal is surrounded by a synaptic cleft (arrowhead) surrounded by a subsynaptic reticulum (SSR) of the surrounding muscle cell. Growth of the larva occurs with a dramatic expansion of the NMJ, and the larval NMJ has been an important tool to understand the homeostatic mechanisms that regulate synaptic growth. As shown in figure 1C, at the ultrastructural level, the basic cell structure of fly muscle cells is conserved, including a sarcomeric structure with actin thin filaments, myosin thick filaments, Z-bands linking adjacent sarcomeres, a complex T-tubular system, and multiple peripherally-located nuclei.
Figure 1.
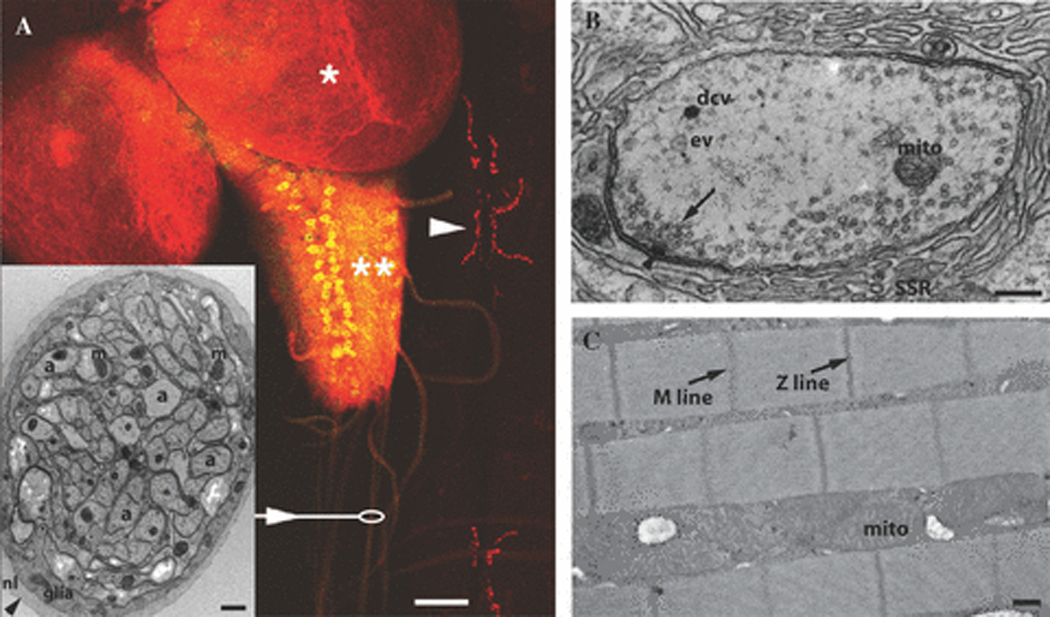
Anatomy of Drosophila Neuromuscular System: (A) Larval neuromuscular system showing lobes of the brain (*), the ventral nerve cord (**) containing motor neurons (yellow), nerves (arrow and oval), and NMJ’s (red, arrowhead). This is a third-instar larval fillet expressing green fluorescent protein (GFP) under control of a motor neuron-specific promoter and immunostained with synaptotagmin antibodies to highlight the CNS and NMJ at muscles 6 and 7. Arrowhead: Each red puncta represents a single NMJ bouton like “beads on a string”. Scale bar is 50 µm. Inset: Electron micrograph showing a cross section of a single wild type larval nerve. Nerves are surrounded by a neural lemmela and contain glial cell processes surrounding many unmyelinated axons. Axons show abundant mitochondria (mito) and microtubules. Scale bar is 2 µm. (B) Electron micrograph of a larval NMJ bouton (a small red puncta in panel A). Shown here is a synaptic bouton containing many synaptic vesicles (arrow), including many clustered at an active zone (arrowhead represents electron dense structure called the “T-Bar”). Occasional dense core vesicles (dcv), endocytic vesicles (ev), and mitochondria (mito) can be observed. Surrounding the synaptic vesicle rich presynapse is the synaptic cleft (arrowhead) and the subsynaptic reticulum (SSR), invaginations of the postsynaptic muscle membrane. Scalle bar is 200 nm. (C) Electron micrograph of adult thoracic flight muscle showing “Z lines” linking actin thin filaments of adjacent sarcomeres and “M lines” where myosin thick filaments are linked together. Abundant mitochondria are present between myofibrils. Scale bar is 500 nm.
In addition to the genetic, cellular, and structural similarities of the fly and human neuromuscular unit, the fundamental mechanisms of synaptic transmission at the NMJ are conserved, although these synapses are glutamatergic rather than cholinergic. Sophisticated electrophysiological recordings can readily be performed at the embryonic, larval, and adult NMJ, and newer techniques have allowed electrophysiological recordings at central synapses. The third-instar larval NMJ is a particularly amenable and robust model system long used to study the mechanisms of synaptic transmission 9, even allowing one to perform genetic screens to identify mutant phenotypes 10. A recording electrode inserted in an identified muscle cell measures the muscle membrane potential, and in the presence of tetrodotoxin to inhibit action potentials, records miniature end-junction potentials (mEJPs, or “minis”), a measure of spontaneous release of single synaptic vesicles. A stimulating electrode is used to generate an action potential in larval nerves, and the evoked junction potential (EJP) is measured in the muscle cell. Complex analyses, including determining the calcium-dependence and probability of release and response to repetitive nerve stimulation are also readily performed with this preparation. Thus, the fly is an ideal model system for studying the consequences of mutations on the development, structure, and function of the peripheral nervous system.
Genetic Tools for Drosophilists
One of the major advantages of Drosophila as a model organism is the abundance of genetic tools that allow rapid investigation of gene function in vivo. One particularly valuable tool is the P element transposon which has been “hopped” throughout the genome to provide insertions in at least 40% of genes 11. Different types of transposons have been generated allowing alteration of flanked genes including overexpression and deletion via imprecise excision 12. These transposons are also used as vectors for the generation of transgenic animals, and this has been particularly useful when carried out with the UAS/GAL4 system 13. This genetic system, analogous to Cre-Lox in mice, allows tissue-specific expression of a gene of interest. When a fly containing a transgene downstream of UAS sites is crossed to a fly containing the GAL4 transcription factor expressed under the control of a tissue-specific promoter, GAL4 binding to UAS sites allows the gene of interest to be expressed selectively in the desired tissue or tissues. Recently, fly libraries containing RNAi of almost all Drosophila genes downstream of UAS sites have been generated that allow tissue-specific knockdown of all fly genes 14.
One particularly powerful use of the GAL4/UAS system has been to study the effect of gene expression in the fly eye. The Drosophila eye is a compound eye, made of about 800 ommatidia, each comprised of eight photoreceptor sensory neurons. The repetitive structure allows amplification and obvious inspection of what might otherwise be subtle phenotypes. Even subtle developmental defects can cause obvious gross phenotypes that can be used in genetic screens. Because the eye is not needed for viability and fertility, mutations that would cause death or sterility if ubiquitously expressed, can be readily studied when overexpressed in the eye.
Fly Models of Motor neuron diseases
As discussed previously, Drosophila has proven to be an excellent model system to study the molecular mechanisms of neurodegenerative diseases. Recently, Drosophila models of motor neuron diseases have been generated that replicate key features of the human disease and have led to novel insights into the mechanisms of these diseases.
Spinobulbar muscular atrophy
Spinobulbar muscular atrophy (SBMA), also known as Kennedy’s disease, is an inherited neurodegenerative disease characterized by progressive loss of motor neurons of the brainstem and spinal cord 15. SBMA is one of the polyglutamine diseases, which includes at least eight other disorders, among them Huntington’s disease, dentatorubral-pallidoluysian atrophy, and six forms of spinocerebellar ataxia. All of these diseases are caused by expanded trinucleotide (CAG) repeats in exonic regions of DNA, and all result in late-onset, progressive neurodegeneration 16. In SBMA, the CAG trinucleotide repeat is located in the androgen receptor (AR) gene, which encodes a ligand-dependent transcription factor 17. The CAG repeat length is polymorphic, with repeat lengths ranging from 9 to 36 in normal individuals, while patients with SBMA have repeat sizes greater than 40. In SBMA, there is a correlation between longer repeat lengths and more severe clinical manifestations. A remarkable feature of SBMA is gender-restricted penetrance. All males with 40 or more CAG repeats appear to be affected, whereas females with the same-sized CAG expansions are typically asymptomatic 18.
The AR belongs to the class I nuclear hormone receptor superfamily that is conserved among metazoans, including Drosophila 19. The primary ligands for AR are testosterone and its more potent derivative dihydrotestosterone (DHT). Ligand binding to the C-terminal ligand-binding domain activates AR, resulting in a conformational change, dissociation from heat shock proteins, and translocation of the active receptor/ligand complex to the nucleus where it associates with co-regulatory proteins and alters the transcription of target genes. These activities of AR are not significantly impaired by polyglutamine expansion 18, consistent with evidence that neurodegeneration results from toxic gain of function rather than loss of function.
As in the other polyglutamine diseases, it appears that the primary basis of toxicity in SBMA is a toxic gain of function in the polyglutamine-expanded protein. This concept is supported by evidence that 1) loss of AR function leads to androgen insensitivity syndrome, with no weakness or neuronal degeneration, and 2) overexpression of polyglutamine-expanded AR in mouse causes neurodegeneration in male mice. However, mild partial loss of AR function likely accounts for the mild feminization that is often found in SBMA patients. For years, the gender-restricted penetrance seen in SBMA was enigmatic, and difficult to reconcile with a mechanism that involves a toxic gain-of function. Until recently, there were two competing hypotheses to explain gender specificity in SBMA. The first proposed that females were protected from clinical manifestations by random X-inactivation resulting in only ~50% of the motor neurons expressing the mutant form of AR. This hypothesis presumed that re-sprouting from the remaining healthy motor neurons was sufficient to protect females from clinical signs and symptoms. The alternative hypothesis proposed that the toxicity of polyglutamine-expanded AR is ligand-dependent, restricting the phenotype to males because of their substantially higher androgen levels. Studies performed in Drosophila have resolved this issue with surprising clarity.
Takeyama and colleagues were the first to successfully model SBMA in Drosophila by mis-expression of full length AR protein harboring an expanded glutamine stretch in the Drosophila eye 20. In contrast to Drosophila models of other neurodegenerative diseases, including other polyglutamine diseases, no degeneration was observed. However, as shown in figure 2, exposure of these flies to dietary DHT resulted in strong, polyglutamine length-dependent eye degeneration. Ligand-dependent degeneration in SBMA has been corroborated by other investigators working with independently-generated fly and mouse models 21–23 and this is also supported by human studies 24. These data indicate that a ligand-dependent activation of AR is an essential step in pathogenesis. Consistent with this idea, flies expressing a truncated version of the AR consisting of the N-terminal transactivation domain, which includes the polyglutamine stretch, led to ligand-independent degeneration; and this phenotype was suppressed by co-expressing un-liganded C-terminal AR fragment. Furthermore, toxicity was abrogated by restricting AR to the cytoplasm, providing further evidence that translocation to the nucleus is a requisite step in pathogenesis 20.
Figure 2.

A Drosophila model of SBMA. Expression of human AR resulted in ligand-dependent, polyglutamine repeat length-dependent degeneration. Flies not treated with DHT had normal eyes (top row). Flies reared on food containing DHT showed polyQ length-dependent degeneration (bottom row). Degeneration was most severe at the posterior eye margin. There was disorganization of the ommatidial array, fusion of ommatidia, and abnormal bristles. Longer repeat length caused a more severe degenerative phenotype that extended further anteriorly. Ligand-dependent, polyQ length-dependent degeneration recapitulates two key features of human SBMA. >1000 fly eyes of each genotype have been examined. Reprinted with permission from Pandey et al.
The ability to use fly models for genetic screens has been used to great advantage in the case of SBMA. Suzuki and colleagues carried out a gain-of-function screen to identify dominant modifiers of degeneration in their SBMA fly model by genetic cross with ~2000 “GS strains”, each of which over-expresses a different Drosophila gene. This study identified Rbf, the Drosophila homolog of the human protein Retinoblastoma (Rb) as a dominant modifier of neurodegeneration 25. Rb is a tumor suppressor protein and negative regulator of cell proliferation. Rb promotes terminal differentiation by inducing both cell-cycle exit and tissue-specific gene expression 26. Rb interacts with the transcription complex initiated by E2F and recruits the repressor HDAC1, inhibiting E2F-mediated transcription activation 27. Rb also interacts with AR and Suzuki et al. determined that this interaction is enhanced by polyglutamine expansion in AR. The authors suggested that AR interaction with Rb impairs recruitment of HDAC6, leading to inappropriate activation of E2F target genes and toxicity. In support of this hypothesis, the authors further demonstrated that polyQ-expanded AR toxicity was suppressed by E2F deficiency.
In a separate study, Pandey and colleagues generated a similar fly model of SBMA and carried out a loss-of-function screen to identify dominant modifiers of degeneration 21. These investigators found that deficiency in the cytoplasmic, microtubule-associated deacetylase HDAC6 strongly enhanced degeneration. They subsequently determined that over-expression of HDAC6 suppressed degeneration not only in SBMA flies, but also in flies carrying mutations in the ubiquitin-proteasome system. This study demonstrated that HDAC6 enhances autophagic degradation of ubiquitinated proteins, providing the first evidence of the compensatory relationship between the ubiquitin-proteosome system and autophagy and identifying the first molecular link between these degradation systems.
Spinal Muscular Atrophy
Spinal muscular atrophy (SMA) was first described in 1891 independently by Guido Werdnig and Johann Hoffman. Their reports presented the clinical and pathological aspects of infantile SMA, including early onset, occurrence among siblings of unaffected parents, progressive weakness, hand tremor, and death from respiratory failure in early childhood. Since the first description of this disease, a broader range of severity and age of onset has been appreciated, and a few separate disease entities have been defined. In 1991, the International Consortium on Childhood SMA defined three clinical subgroups to reflect the broad range of clinical severity 28. All types of SMA linked to chromosome 5q11 are associated with a relative deficiency in the product of the survival motor neuron (SMN) gene. SMA is one of the most common autosomal recessive diseases, affecting approximately 1 in 6000–10,000 live births, and is the leading hereditary cause of infant mortality 29. De novo mutations in the gene responsible for SMA are relatively frequent, and the carrier frequency for disease-causing mutations is estimated at approximately 1 in 50 30.
SMN exists in two copies in a duplicated region of chromosome 5q. The telomeric version (SMN1) generates a full-length SMN protein, whereas the centromeric counterpart (SMN2) gives rise mainly to an alternatively spliced form that produces little functional protein. SMN1 is deleted or mutated in >98% of SMA patients. In experimental systems, SMN function appears necessary for cell survival independent of cell type. SMN is a ubiquitously expressed gene that is highly conserved in eukaryotes 31. Therefore, it is intriguing and enigmatic that homozygous deletions of SMN1 should lead to selective motor neuron loss in patients with SMA. The basis of cell type specific neuron loss with relative depletion of SMN is a central issue that remains to be resolved. The SMN protein is a 294 amino acid polypeptide that is present in both the cytoplasm and the nucleus. In the nucleus, SMN is highly concentrated within discrete bodies called gems that overlap, or are in close proximity to, coiled bodies 31. SMN participates in the assembly of RNA-protein complexes and, in particular, is important for snRNP assembly. In neurons, SMN is also found in axons and growth cones, suggesting that it trafficks with RNA to distal sites 32.
Most metazoans have a single SMN gene, and knock out of this gene results in embryonic lethality. However, as a result of gene duplication, humans have two SMN genes. In SMA patients only SMN1 is disrupted, and the low level of SMN protein produced by the SMN2 gene permits survival, but results in specific loss of lower motor neurons and muscle wasting. The early lethality caused by complete absence of the SMN gene has made it difficult to develop animal models of SMA. To circumvent this difficulty and more closely approximate conditions that lead to the human disease, Drosophila biologists have identified partial loss-of-function (hypomorphic) mutations in smn to gain insight into mechanisms of pathogenesis.
Chan and colleagues isolated Drosophila smn mutants containing point mutations in the YxxG self-association domain that result in reduced self-association of Smn protein 33. Similar mutations have been found in SMA patients 34. Flies with these hypomorphic smn alleles showed abnormal motor behavior, disorganized synaptic boutons, and reduced excitatory post-synaptic currents at the NMJ. Interestingly, in rescue experiments they determined that smn expression in both motor neurons and muscle was required to suppress the neuromuscular defect, indicating both pre-synaptic and post-synaptic functions of SMN. Similar results were reported by Rajendra and colleagues in which hypomorphic mutations in smn caused flightlessness and muscular atrophy 35. The affected motor neurons had defects in axon path-finding and arborization. Interestingly, these investigators also reported that in wild-type muscles from both fly and mouse, SMN co-localizes with sarcomeric actin and forms a complex with alpha-actinin, the thin filament cross-linker. This latter observation suggests a muscle-specific function for SMN independent of its known role in snRNP assembly. Indeed, these investigators reported no detectable defect in snRNP assembly in flies with larval-lethal smn-null mutations, leading them to suggest that lethality is not a result of snRNP deficiency. However, Cauchi and colleagues report that gemin3-null mutations also result in larval lethality and that flies with hypomorphic gemin3 alleles develop motor behavioral defects, abnormalities in the NMJ, and muscle atrophy similar to that observed in smn hypomorphic flies 36. Since Gemin3 is an RNA helicase that interacts with the SMN complex and participates in snRNP assembly, this substantiates the hypothesis that defects in snRNP assembly contribute to SMA pathogenesis.
Chang and colleagues reported that SMN protein concentrates in the post-synaptic NMJ region and, consistent with prior studies, also found NMJ defects in flies with hypomorphic smn alleles 37. These investigators conducted a genetic screen using a large collection of transposon-induced mutations and recovered 27 dominant modifiers of the NMJ defect. Among these was wishful thinking (wit), a receptor for the growth factor BMP. Two additional components of the BMP signaling system, mothers against dpp(mad) and daughters against dpp (dad), were subsequently found to also modify NMJ defects in smn hypomorphic flies. The identification of these modifier genes implicates a role for the BMP trophic signaling pathway in SMA pathogenesis.
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease in which progressive loss of motor neurons in the cortex, brainstem and spinal cord leads to fatal paralysis with a typical course of 2 to 5 years. The age-adjusted worldwide incidence of ALS is 0.5–3 per 100,000 and the prevalence is 4–6 per 100,000 38. Most forms of ALS do not have a clear family history and are deemed sporadic, but ~5–10% of patients have an inherited form of the disease 39. As shown in table 1, more than 15 different genetic aberrations have been found to cause familial forms of ALS (fALS). When multiple genes influence a single disorder, those genes may define a pathogenic biochemical pathway. However, the 10 genetic mutations thus far identified have yet to reveal a unifying model of ALS pathogenesis, although defects in RNA metabolism and axonal trafficking are consistent themes. Nevertheless, the identification of these genes has enabled the development of animal models recapitulating features of the human disease and provided insights into the cellular and molecular basis of pathogenesis.
Table 1.
Familial ALS
| Type | Gene | Inheritance | Location |
|---|---|---|---|
| ALS 1 | SOD1 | AD | 21q22 |
| ALS 2 | ALS2 | AR | 2q33 |
| ALS 3 | AD | 18q21 | |
| ALS 4 | SETX | AD | 9q34.3 |
| ALS 5 | AR | 15q15 | |
| ALS 6 | FUS/TLS | AD | 16p11 |
| ALS 7 | AD | 20p13 | |
| ALS 8 | VAPB | AD | 20q13.33 |
| ALS 9 | Angiogenin | AD | 14q11 |
| ALS 10 | TARDBP | AD | 1p36 |
| ALS 11 | FIG4 | AD | 6q21 |
| ALS-FTD 1 | AD | 9q21-22 | |
| ALS-FTD 2 | AD | 9p13 | |
| ALS-FTD 3 | CHMP2B | AR | 2p11 |
| ALS X | X-linked | Xp11-q22 |
Source: Neuromuscular Disease Center, Washington University (neuromuscular.wustl.edu)
In 1993, mutations in the gene superoxide dismutase type 1 (SOD1) were found to be responsible for a dominantly inherited form of motor neuron disease that is clinically indistinguishable from sporadic ALS 40. SOD1 is a copper-dependent enzyme that catalyzes the conversion of toxic superoxide radicals to hydrogen peroxide and oxygen. Mutations that impair the antioxidant function of SOD1 could lead to toxic accumulation of superoxide radicals. However, a loss-of-function mechanism for familial ALS is unlikely given that no motor neuron degeneration is seen in transgenic mice in which SOD1 expression has been eliminated. Moreover, overexpression of mutant SOD1 in transgenic mice causes motor neuron disease despite elevated SOD1 activity. This supports a role for a deleterious gain of function by the mutant protein, consistent with autosomal dominant inheritance 41. A pro-oxidant role for mutant SOD1 contributing to motor neuron degeneration was a hypothesis long entertained. This hypothesis was tested by Parkes and colleagues using Drosophila, an organism whose life span is exquisitely sensitive to oxidative damage 42. They generated transgenic Drosophila expressing wild type and mutant forms of human SOD1 specifically in motor neurons and found no evidence of reduced life span or degenerative phenotype. Indeed, two groups independently determined that expression of mutant SOD1 in Drosophila actually extends lifespan, augments resistance to oxidative stress and partially rescues SOD-null mutants just as well as wild type SOD1 43, 44. More evidence against the pro-oxidant hypothesis came from ablation of the specific copper chaperone for SOD1, which deprives SOD1 of copper and eliminates enzymatic activity, and has no effect on motor neuron degeneration in mutant SOD1 transgenic mice 45.
These and related experiments led to the pro-oxidant hypothesis falling out of favor. More recently, attention has turned to the possible deleterious effects of accumulating aggregates of mutant SOD1 46. The notion that aggregation is related to pathogenesis is supported by the observation that murine models of mutant SOD1- mediated disease feature prominent intracellular inclusions in motor neurons, and in some cases within the astrocytes surrounding them as well 47.
Watson and colleagues also expressed wild type or disease-linked mutants of human SOD1 selectively in motor neurons 48. Careful analysis of flies expressing mutant SOD1 revealed mild motor deficits that were accompanied by defective neural circuit electrophysiology (Figure 3). Histopathological analysis revealed focal aggregation of human SOD1 protein in motor neurons and a stress response in surrounding glia. Thus, these studies suggest cell-autonomous injury by SOD1 to motor neurons in vivo as well as non-autonomous effects on glia that correlate with SOD1 aggregation.
Figure 3.

hSOD1 induces age-dependent electrophysiological defects in the giant fiber neural circuit. (A) schematic illustration of the giant fiber pathway responsible for jump-flight escape behavior. The giant fiber neuron (GFn) is located in the brain and descends to the thoracic ganglion, where it excites the motor neuron (TTMn) that innervates the TTM via an electrical synapse (marked with a lightning bolt). GFn also excites the peripherally synapsing interneuron (PSI) via an electrical synapse, which in turn excites five motor neurons (DLMn) innervating DLMs. Both DLM and TTM motor neurons synapse with their respective muscles via glutamatergic synapses. For illustrational purposes, the DLMn is shown outside the thoracic ganglion. (B) representative responses of DLM following 140-Hz stimulation of the GFn in control flies (top panels), flies expressing WT hSOD1 (second panels), flies expressing G85R (third panels). The muscle responded normally to each stimulus at 10 days but failed to follow each stimulus when aged (55 days) in the experimental flies. G85R mutation enhanced this effect. The arrows indicate failed responses. Reprinted with permission from Watson et al.
In early 2004, Nishimura and colleagues reported a Brazilian family with 26 members in three generations affected by late onset, autosomal dominant motor neuron disease linked to chromosome 20q13.33 49. These patients experienced an atypical form of motor neuron disease with early onset and slow progression. All patients had lower motor neuron signs affecting all four limbs, while several had bulbar signs and only one had upper motor neuron signs. Many of the patients also experienced postural tremor and disabling cramps. Electromyography in eight of these patients showed neurogenic patterns and no evidence of sensory or motor conduction abnormalities. This condition was designated ALS8. Later in 2004, these investigators reported that a missense mutation in the gene VAMP (synaptobrevin)-associated protein B (VAPB) was responsible for this condition in the original Brazilian family as well as six additional Brazilian kindreds with motor neuron disease including one family with typical ALS 50. Mutations in VAPB have not been found associated with sporadic ALS 51.
VAP proteins (human VAPB/ALS8, Drosophila VAP33, and C. elegans VPR-1) are homologous proteins with an amino-terminal major sperm protein (MSP) domain and a transmembrane domain. The MSP domain is named for its similarity to the C. elegans MSP protein, a sperm-derived hormone that binds to the Eph receptor and induces oocyte maturation 52. Studies conducted in Drosophila have contributed significantly to understanding the normal function of VAPB and the consequence of the disease-causing mutation. As early as 2002, Giusy Pennetta, working with Hugo Bellen, had demonstrated that Drosophila VAP33 encodes a protein that is localized to the NMJ 53. They reported that loss of VAP33 caused a severe decrease in the number of boutons and a corresponding increase in bouton size. Conversely, presynaptic overexpression of VAP33 induced an increase in the number of boutons and a decrease in size of the NMJ. Recent insights revealed that the conserved MSP domains of VAP proteins are cleaved and secreted ligands for Eph receptors that define a critical signaling pathway that regulates formation of the NMJ 52, 54. The P58S point mutation responsible for ALS8 leads to a failure in secretion of this protein and abnormalities in NMJ formation similar to those observed in VAP33 loss-of-function mutants 52, 54–56. Recapitulation of a loss of function was unexpected given that ALS8 has a dominant mode of inheritance. However, in these animals mutant VAP33 is found to aggregate early in the secretory pathway, initiating an unfolded protein response, and also traps wild type VAP33 in pathological aggregates, potentially leading to a dominant negative effect, both of which could contribute to a dominant mode of inheritance 52, 56. One study suggests that loss of VAP33 function results in defective signaling through the BMP pathway, suggesting potential mechanistic overlap with the defects observed in smn deficiency 37, 56.
While Drosophila and other model systems have contributed insights into the pathogenesis of SOD1- and VAPB-related motor neuron disease, these two genes together account for less than 2% of all ALS. Furthermore, the pathological features specific to these two forms of motor neuron disease are not shared by common, sporadic ALS or even other forms of FALS. The neuropathology of ALS is characterized by the accumulation of proteinaceous inclusions in the cytoplasm of degenerating motor neurons 57, 58. Until recently, little was known about the composition of these neuronal cytoplasmic inclusions, except that they were immuno-positive for ubiquitin. NCIs, which a number of studies have confirmed to be a highly sensitive and specific marker for ALS, are most common in lower motor neurons and most often appear as either filamentous skeins or compact, round bodies 57, 58. Two years ago, the TAR DNA binding protein, TDP-43, was identified as a major component of the NCI in sporadic ALS (sALS), some forms of familial ALS (fALS), as well as in frontotemporal lobar dementia with ubiquitinated inclusions (FTLD-U), the most common pathological subtype of frontotemporal dementia 59. A role for TDP-43 in the pathogenesis of ALS is further supported by the recent identification of TDP-43 mutations in familial and sporadic cases of ALS (reviewed in 60). TDP-43 is a heterogeneous ribonucleoprotein that binds single-stranded nucleic acid and has been implicated in regulation of transcription and alternate RNA splicing. Very recently, mutations in FUS/TLS, a related RNA processing protein, were identified as the cause of familial ALS type 6. The pathology of both TDP-43-related and FUS/TLS-related ALS is characterized by redistribution of a nuclear RNA-binding protein to the cytoplasm, and suggests the possibility of related mechanisms 61, 62. Considering the functions of TDP-43 and FUS/TLS together with SMN, many recent insights point to a possible defect in RNA metabolism as a frequent underlying cause of motor neuron disease. All of these genes are highly conserved in Drosophila which provides an ideal model system for investigating the role of these genes in motor neuron disease pathogenesis in coming years.
Fly Models of Neuropathies
In contrast to motor neuron diseases described above, there are very few fly models of neuropathies generated to date. This is not due to a lack of fly homologues of genes mutated in Charcot Marie Tooth (CMT) disease or other inherited neuropathies, and in fact, of the 34 genes known to be affected in CMT, 79% have fly orthologues that are on average 45% identical and 63% similar (determined using Blastp searches with help from the “Homophila” database63). Three-fourths of these fly homologues have mutants already available, and all have RNAi transgenic flies available that can be rapidly used to study the effects of gene knockdown in fly neurons. Thus, the fly holds tremendous potential for studying the function of genes that are mutated in inherited neuropathies.
A recent study on the fly homologue of GARS (glycyl-tRNA synthetase), mutated in CMT-2D, illustrates the potential of fly models for understanding mechanisms of neuropathy 64. In a screen for mutations that affect dendritic or axonal morphogenesis, a loss-of-function allele of gars was identified that caused a severe reduction in terminal dendritic and axonal arborizations. Mutant dendritic and axon terminals developed normally in early pupal stages, but arbors were lost in late pupal stages, suggesting that GARS is necessary for maintaining these terminal branches. Interestingly, the mitochondrial isoform was essential to maintain dendritic but not axonal arbors, whereas the cytoplasmic isoform was required for both. Importantly, wild type human GARS was able to fully rescue the null phenotype, whereas mutant GARS containing mutations present in CMT2D patients (E71G and L129P) showed little or no rescue. These data strongly suggest that these CMT2D –causing mutations result from loss-of-function, and suggest that the autosomal dominant nature of the disease is likely due to haploinsufficiency, rather than a gain-of-function as had been proposed. Given that mutations in tyrosine-tRNA synthetase (YARS) also cause CMT 65, peripheral nerves may be uniquely sensitive to subtle disruptions of protein translation, and this study suggests that the mechanism may be destabilization of terminal arborizations.
While other fly models of CMT have not yet been generated, Drosophila models have been generated for Friedreich’s Ataxia (FRDA), a neurodegenerative disease with neuropathy as a major feature. FRDA is a recessively-inherited disorder caused by a GAA triplet repeat expansion in the first intron of the frataxin gene that causes reduced gene expression. Decreased frataxin expression impairs the formation of iron-sulfur clusters essential to the function of some mitochondrial enzymes, leading to impaired oxidative phosphorylation and increased production of reactive oxygen species. Fly models for FRDA have been generated using RNAi which remarkably replicate the key features of this disease including abnormal iron metabolism, sensitivity to oxidative stress, and reduced lifespan 66, 67. In contrast, overexpression of frataxin in neurons increases resistance to oxidative stress and increases lifespan 68. Interestingly, selective knockdown of Drosophila frataxin in the peripheral nervous system leads to decreased lifespan, suggesting that this is truly a fly model of neuropathy 66, 69. Importantly, overexpression of a hydrogen peroxide scavenger, catalase, but not superoxide scavengers such as SOD, markedly suppresses these RNAi-induced phenotypes 69. The authors also show that aconitase, a mitochondrial enzyme that is irreversibly damaged by reactive oxygen species in FRDA patients and in their fly model, regains enzymatic activity with catalase overexpression. This simple in vivo model suggests that therapies targeted at scavenging hydrogen peroxide may have efficacy in patients with this disease.
Fly Models of Myopathies
Drosophila muscle cells share many of the molecular, cellular, and physiologic features of vertebrate muscle, and thus it’s not surprising that this powerful genetic system has been used to study the molecular pathogenesis of inherited myopathies. Most muscular dystrophies are caused by mutations that disrupt the function of the dystrophin-dystroglycan complex, a multiprotein complex that links the actin cytoskeleton of muscle cells to the extracellular matrix. Fly models of dystrophinopathies have been generated and are yielding new insights into the function of this complex. Other muscular dystrophies such as oculopharyngeal muscular dystrophy (OPMD) and myotonic dystrophy types 1 and 2 (DM1 and DM2) are repeat expansion disorders that are thought to be caused by toxic nuclear accumulation of protein (OPMD) or RNA (DM). Recently, fly models for these disorders have been generated that replicate both the progressive muscle degeneration and nuclear inclusions seen pathologically in patients with these diseases, and hold promise for uncovering novel disease mechanisms and identifying new therapeutic agents.
Dystrophin-Dystroglycan Complex
Duchenne and Becker muscular dystrophies, the most common inherited muscle diseases, are caused by absence (Duchenne) or reduction (Becker) of functional dystrophin protein. Patients with Duchenne muscular dystrophy (DMD) develop progressive muscle degeneration beginning in early childhood, typically leading to wheelchair-dependence, dilated cardiomyopathy, and respiratory failure in their mid-teens. Studies of the vertebrate dystrophin protein suggest that it likely plays a key structural role in maintaining muscle membrane stability during contraction by linking the sarcolemmal protein dystroglycan to the actin cytoskeleton. However, dystrophin is also an important scaffold for signal transduction, and the mechanisms whereby lack of dystrophin causes muscle degeneration is unclear 70. Use of mouse models of DMD has been complicated by partial redundancy of the dystrophin homologue utrophin and a profound muscle regeneration response, leading to a subtle non-lethal phenotype in the commonly used mdx model 71.
All known components of the dystrophin-dystroglycan complex (DGC) are present in the fly, including a single, highly conserved dystrophin ortholog that is predominantly expressed in muscle and the nervous system 72, 73. Similar to the vertebrate protein, fly dystrophin is a huge protein with multiple isoforms, the largest containing an amino-terminal actin binding domain, multiple spectrin repeats, and a highly conserved cysteine-rich carboxy-terminal domain capable of interacting equally well with fly and human dystroglycan 74. Interestingly, as shown in figure 4, muscle-specific RNAi-mediated knockdown of all dystrophin isoforms or of dystroglycan leads to age-dependent, progressive climbing deficits and severe muscle degeneration in adult flies 74, 75. Similar phenotypes were observed with deletion mutants of dystrophin. Furthermore, mutations in fly dystrophin also cause heart defects with age-dependent disruption of cardiac myofibrillar architecture, chamber dilation, and diastolic dysfunction reminiscent of the dilated cardiomyopathy seen in DMD patients 76. Similar muscle and heart defects with reduced lifespan are also seen in fly mutants of δ-sarcoglycan, a member of the DGC mutated in limb-girdle muscular dystrophy 2F 77.
Figure 4.

Dg and Dys mutants manifest age-dependent muscle degeneration. Light microscopy of histological transverse sections of IFMs stained with H&E. (I–K) The mesoderm-specific RNAi-based reduction of Dg and Dys (dsDys C-term and dsDg30A/24B-Gal4) at 20 days after eclosion, but not at 3 days after eclosion (I) show obvious IFM muscle pathology: the loss of fiber density and vacuolization (asterisks). Reproduced with permission from Ruohola-Baker et al.
In addition to a role in maintaining muscle membrane stability, members of the DGC are enriched at the NMJ and may play a role in synaptic transmission 78. Dystrophin and dystroglycan are highly expressed at the postsynaptic NMJ in Drosophila, and loss of dystroglycan leads to loss of synaptic localization of dystrophin, laminin, and spectrin 79, 80, indicating a central role of dystroglycan in organizing the DGC at synapses. Interestingly, deletion or muscle-specific RNAi-mediated knockdown of a dystrophin isoform (DLP2) that localizes to the postsynaptic NMJ in Drosophila leads to a ~ 45% increase in quantal content of neurotransmitter release at the larval NMJ 79. This phenotype is rescued by postsynaptic expression of DLP2, and detailed electrophysiological analysis suggests that this defect is due to an increased probability of neurotransmitter release. A similar phenotype is observed in heterozygous deletion mutants, indicating that this phenotype is due to haploinsufficiency. This presynaptic phenotype suggests that dystrophin in muscle cells modulates a retrograde signaling pathway to motor neuron terminals. Such homeostatic mechanisms regulating synaptic transmission have been observed in vertebrate and fly NMJ’s, and in the fly are believed to be mediated via the type II TGF-β receptor Wit 81, 82. Indeed, mutations of wit block this enhancement in neurotransmitter release in dystrophin mutants, demonstrating that the dystrophin-modulated retrograde signal goes through the wit signaling pathway. In contrast to the increase in neurotransmitter release seen in dystrophin mutants, loss of dystroglycan leads to a reduction in synaptic transmission 80, 83. This phenotype is observed with both postsynaptic RNAi-mediated knockdown of dystroglycan and mutant alleles. Interestingly, rescue of the mutant allele defect is only observed with expression both pre- and post-synaptically, demonstrating a critical function for dystroglycan in neurons.
Many patients with congential muscular dystrophies (CMD) have mental retardation and brain pathology, and about 30% of DMD patients have intellectual impairment. These central nervous system manifestations are believed to be due to a disruption of DGC function in neurons 84, and may be related to disruption of synaptic transmission. In the absence of a CNS-specific isoform of dystrophin called Dp186, evoked neurotransmission is enhanced at a central synapse, and again this defect can be rescued by expressing Dp186 postsynaptically 85, similar to what is seen at the NMJ. In contrast to the NMJ, though, this enhancement is not altered in a wit mutant background, demonstrating that a different retrograde signal is involved.
Although dystroglycan mutations have not been found in patients, presumably due to an essential function of dystroglycan during development 86, mutations in genes that glycosylate dystroglycan are known causes of CMD. Some patients with Walker-Warburg syndrome and LGMD2K have mutations in the glycosylation enzyme, protein O-mannosyl transferase 1 (POMT). DiAntonio and colleagues identified mutations in a fly POMT (called dPOMT1) in a screen for mutations affecting postsynaptic glutamate receptor subunit composition at the NMJ 83. As in vertebrates, POMT1 is required in vivo for glycosylation of dystroglycan in Drosophila. dPOMT1, dPOMT2, and dystroglycan genetically interact 83, 87, and dPOMT1 mutants have the same electrophysiologic phenotype as dystroglycan mutants, namely a severe reduction in evoked neurotransmitter release. Together, these data strongly suggest that a postsynaptic DGC regulates presynaptic neurotransmitter release.
In addition to functioning in synaptic transmission, the DGC complex has also been implicated in other neuronal functions. Loss-of-function mutations or neuron-specific knockdown of fly dystrophin and dystroglycan cause defects in central axon pathfinding 74. The DGC is required in both neurons and glia, highlighting a cell non-autonomous function of this complex in axon guidance. The authors showed strong dominant genetic interactions of dystroglycan mutations with alleles of the Insulin receptor and an SH2/SH3 adaptor molecule named Nck/Dock, suggesting a potential signaling mechanism for dystroglycan-mediated pathfinding. A large-scale genetic screen for modifiers of loss-of-function dystroglycan and dystrophin phenotypes identified many genes known to function in axon pathfinding and cytoskeletal remodeling, including the semaphorin-plexin, netrin-frazzled, and slit-robo pathways 88. Additional modifier genes identified in these screens include members of the Notch, TGF-β, and EGFR signaling pathways, further highlighting the role of the DGC as a platform for intracellular signaling. Together, these data suggest that the fly is an excellent model for understanding central nervous system phenotypes of muscular dystrophies, as well as for identifying the complex array of regulatory and downstream signaling molecules of the DGC.
Myotonic Dystrophy and OPMD
Myotonic dystrophy (DM) is an autosomal dominant disease caused by CUG or CCUG repeat expansions in noncoding regions of messenger RNA. Patients have a wide range of clinical features including myotonia, muscle atrophy, cardiac conduction defects, testicular atrophy, cataracts, hypersomnolence, and cognitive impairment 89. Considerable recent evidence, including that provided by Drosophila models, suggests that DM is an RNA-mediated disease caused by a gain-of-toxicity of expanded CUG or CCUG repeats, and has little to do with the gene product in which the repeats are present. A current model for DM pathogenesis is that these toxic RNA species sequester RNA-binding proteins, including muscleblind-like proteins (MBNL), leading to a disruption of mRNA splicing. As discussed below, fly models of DM support this disease mechanism, and have led to new insight into the pathogenesis of this disease.
De Haro and Colleagues generated the first fly model of DM by expressing a noncoding RNA containing an expanded, interrupted CUG repeat (iCUG)480 in Drosophila 90. When overexpressed in muscle, (iCUG)480 colocalizes with muscleblind (mbl) protein in nuclear foci (figure 5) and causes progressive muscle degeneration, similar to what has been observed in muscle from patients with this disease 91. Similar degenerative phenotypes were observed with overexpression of (iCUG)480 in the fly eye. Expression of fewer than 200 CUG repeats led to accumulation of nuclear foci containing Mbl, but not muscle degeneration, demonstrating that the presence of these ribonuclear foci were not sufficient to cause toxicity 92. Interestingly, overexpression of human MBNL1 suppresses these degenerative phenotypes whereas reduction of endogenous muscleblind enhances them, supporting the model that sequestration of muscleblind protein is a key step in the pathogenesis of the disease.
Figure 5.

Co-localization of MBNL1 protein with the CUG-containing nuclear foci. (A–C) Images of nuclei from larval somatic muscle co-expressing MBNL1 and (iCUG)480 after immunofluorescence and in situ hybridization. MBNL1 protein is detected in muscle nuclei when stained with an antibody against MBNL1 (A, white arrows, green). The (iCUG)480 transcripts are also visualized in the form of nuclear foci after in situ hybridization (B, white arrows, red). Both MBNL1 and (iCUG)480 RNA co-localize when the images are merged (C, white arrows, yellow). Muscles are also stained with anti-CBP antibody to visualize the nuclei (white). (D–E) Images of nuclei from larval somatic muscle expressing MBNL1 but no (iCUG)480 after immunofluorescence. Anti-CBP antibody revealing nuclei outline (D, white), MBNL1 signal shown in green (E). (F) Comparative quantification of nuclei with single versus multiple foci in flies expressing (iCUG)480 alone, or together with MBNL1. Images below the chart illustrate the types of nuclear foci found; no nucleus of any genotype was found without at least one foci. Notice that co-expression of MBNL1 together with (iCUG)480 causes an overall decrease in the number of nuclei with multiple foci when compared with the repeats alone. Error bars represent standard deviation. Data was analyzed using Student’s t (P 1/4 0.006 in both groups). Total muscle nuclei (200 per larva) from four to five larvae per genotype were analyzed. Scale bar: (A–F) 5 mm. Genotypes are A–C, F: w; UAS-MBNL1[M10M]/ +; Mhc-GAL4/UAS-(iCUG)480 [M5T]; D–E: w; UAS-MBNL1[M10M]/ +; Mhc-GAL4/ + . Reprinted with permission from de Haro et al.
A second RNA-binding protein implicated in the pathogenesis of DM is CUG binding protein 1 (CUG-BP1). This protein is upregulated in muscle from myotonic dystrophy patients, and overexpression of CUG-BP1 in mammalian cells mimics some of the protein splicing abnormalities seen in DM patients. Interestingly, overexpression of CUG-BP1 in flies enhances the toxicity of (iCUG)480, supporting a role for this protein in the pathogenesis of disease 93. Furthermore, several transcripts of muscle proteins known to be misspliced in DM muscle are mis-spliced in this model, including troponin T and α-actinin 94, 95.
To use this fly model of DM to identify novel therapeutic targets, Artero and colleagues performed genetic and chemical modifier screens of CUG-mediated toxicity 95. A screen of 695 lethal P-element alleles and additional candidate genes for dominant genetic interactions with flies overexpressing (iCUG)480 in the fly eye identified 15 genetic modifiers. These modifier genes are thought to function in several diverse cellular processes including transcription, cell adhesion, and apoptosis. Interestingly, a loss-of-function allele of Aly, a mRNA export factor, enhanced CUG-mediated toxicity, supporting the hypothesis that a failure to export CUG-binding transcripts is important to disease pathogenesis. Furthermore, a screen of 400 compounds with known biological activity found 10 chemical suppressors of CUG-mediated neuronal toxicity. Intriguingly, most of these compounds are inhibitors of neuronal excitation, including dopamine, acetylcholine, and histamine receptor blockers, inhibitors of monoamine reuptake, and compounds that affect sodium and calcium metabolism. These data suggest that CUG repeats may induce excitotoxicity, a finding that requires additional study in other model systems. Most importantly, this study demonstrates the utility of Drosophila in identifying novel genetic and chemical suppressors of phenotypes associated with neuromuscular diseases.
Like DM, Oculopharyngeal Muscular Dystrophy (OPMD) is also caused by a trinucleotide repeat expansion, has characteristic nuclear inclusions, and has been modeled in the fly. In contrast to Myotonic Dystrophy, though, OPMD results from a polyalanine expansion of poly(A)-binding protein 1 (PABPN1), and the RNA species itself has not been directly implicated in the pathogenesis of the disease. Flies overexpressing a mutant human protein containing 17 alanine repeats (normal is 10 and greater than 12 is pathogenic) develop progressive, age dependent muscle degeneration with rimmed vacuoles and nuclear inclusions that are very similar to those seen in OPMD patients 96. While the polyalanine expansion enhances toxicity, even PABPN1 protein deleted of the polyalanine domain is toxic to flies, despite the absence of nuclear inclusions, demonstrating that neither the polyalanine expansion nor the presence of nuclear inclusions is sufficient for disease. Interestingly, deletion of the RRM (RNA recognition motif) RNA-binding domain or point mutations in the RRM domain that disrupt RNA binding completely abolish PABPN1-mediated muscle degeneration. These data suggest that the expanded polyalanine domain of PABPN1 itself is not deleterious, but rather alters the level or structure of the protein such that the RRM domain gains toxicity. This elegant study suggests that this polyalanine disease has a very different mode of toxicity than has been proposed for polyglutamine diseases, whereby the polyglutamine repeat itself is toxic.
Other neurologic diseases studied at the NMJ
Because of it’s usefulness as a model system, the Drosophila larval peripheral nervous system has been useful in studying a large number of neurological diseases from neurodegenerative diseases such as Huntington’s and Alzheimer’s disease to mental retardation and autism-spectrum disorders such as Fragile X disease. The larval NMJ is much easier to study than central synapses, and has some similarities with vertebrate central synapses including using glutamate as neurotransmitter. Discoveries and principles learned at this peripheral synapse can later be tested on central synapses in the fly and vertebrate models.
For example, analysis of the fly homologue of Fragile X mental retardation 1 (dfmr1) has led to novel insights into the mechanism of this disease, and many of these discoveries were made through analysis of its function at the NMJ. Loss of function dfmr1 mutations in Drosophila cause overgrowth of the NMJ and excessive branching of sensory dendrites, whereas overexpression of dfmr1 causes the opposite phenotype, suggesting that dfrm1 normally inhibits synaptic growth 97. dfmr1 was found to be a translational repressor of the microtubule-associated protein, MAP1B, and loss of MAP1B suppresses the synaptic overgrowth and increased neurotransmission of dfmr1 mutant animals. These data suggest that misregulation of cytoskeletal components are a component of Fragile X disease pathogenesis.
Furthermore, the accessibility of larval nerves for imaging has made this an excellent model for investigating the mechanisms of axonal transport 98, 99. Defects in axonal transport occur early in ALS and have been implicated in the pathogenesis of many neurodegenerative diseases 100. Loss of Drosophila Huntingtin or Amyloid Precursor Protein (APP) and overexpression of mutant forms of these proteins present in Huntington’s and Alzheimer’s disease, respectively, cause accumulations of vesicular structures in “axon jams” present in larval nerves, phenotypes indicative of axonal transport defects 101–103. A combination of live imaging of axonal transport of organelles with genetic manipulations in Drosophila holds great promise for uncovering the precise mechanisms whereby disruption of axonal transport contributes to neurodegeneration.
Closing Thoughts
The recent proliferation of Drosophila models of neuromuscular diseases attests to the potential of this model organism for illuminating the molecular bases of these diseases. In some cases, Drosophila may be used to understand the function of a newly-discovered gene that has been implicated in a disease. In other cases, Drosophila may be used to generate a tractable animal model that recapitulates the fundamental molecular and cellular phenotypes of the human disease. As we have described, such models have been used to great success in genetic screens to reveal unanticipated insights into disease mechanism and suggest novel strategies of therapeutic intervention. Furthermore, there is tremendous untapped potential for using Drosophila in large scale, high-throughput small molecule screens in vivo that would not be feasible in mice. Just as Drosophila has been the indispensable organism for biologists past, it promises to be a vital player in the future of neurology research.
Contributor Information
Thomas E. Lloyd, Department of Neurology, Johns Hopkins University School of Medicine, 600 N. Wolfe Street/ Meyer 6-181C, Baltimore, MD 21287, tlloyd4@jhmi.edu, Phone: (410) 955-1223, Fax: (410) 502-6737.
J. Paul Taylor, Department of Developmental Neurobiology, St. Jude Children’s Research Hospital, MS 343, D-4026, 262 Danny Thomas Place, Memphis, TN 38105-3678, jpaul.taylor@stjude.org, Phone: (901) 595-6047, Fax: (901) 595-2032.
References
- 1.Bier E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat Rev Genet. 2005;6:9–23. doi: 10.1038/nrg1503. [DOI] [PubMed] [Google Scholar]
- 2.Warrick JM, et al. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–949. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 3.Jackson GR, et al. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998;21:633–642. doi: 10.1016/s0896-6273(00)80573-5. [DOI] [PubMed] [Google Scholar]
- 4.Feany MB, Bender WW. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 5.Bonini NM, Fortini ME. Human neurodegenerative disease modeling using Drosophila. Annu Rev Neurosci. 2003;26:627–656. doi: 10.1146/annurev.neuro.26.041002.131425. [DOI] [PubMed] [Google Scholar]
- 6.Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science. 2000;287:1837–1840. doi: 10.1126/science.287.5459.1837. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez-Funez P, et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- 8.Budnik V, Ruiz-Cäanada C. The fly neuromuscular junction : structure and function. San Diego, Calif.: Elsevier/Academic Press; 2006. [Google Scholar]
- 9.Jan LY, Jan YN. Properties of the larval neuromuscular junction in Drosophila melanogaster. J Physiol. 1976;262:189–214. doi: 10.1113/jphysiol.1976.sp011592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frank CA, Pielage J, Davis GW. A presynaptic homeostatic signaling system composed of the Eph receptor, ephexin, Cdc42, and CaV2.1 calcium channels. Neuron. 2009;61:556–569. doi: 10.1016/j.neuron.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellen HJ, et al. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004;167:761–781. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venken KJ, Bellen HJ. Transgenesis upgrades for Drosophila melanogaster. Development. 2007;134:3571–3584. doi: 10.1242/dev.005686. [DOI] [PubMed] [Google Scholar]
- 13.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 14.Dietzl G, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 15.Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset A sex-linked recessive trait. Neurology. 1968;18:671–680. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- 16.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 17.La Spada AR, et al. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 18.Jiang YM, et al. Gene expressions specifically detected in motor neurons (dynactin 1, early growth response 3, acetyl-CoA transporter, death receptor 5, and cyclin C) differentially correlate to pathologic markers in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2007;66:617–627. doi: 10.1097/nen.0b013e318093ece3. [DOI] [PubMed] [Google Scholar]
- 19.Truss M, Beato M. Steroid hormone receptors: interaction with deoxyribonucleic acid and transcription factors. Endocr Rev. 1993;14:459–479. doi: 10.1210/edrv-14-4-459. [DOI] [PubMed] [Google Scholar]
- 20.Takeyama K, et al. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron. 2002;35:855–864. doi: 10.1016/s0896-6273(02)00875-9. [DOI] [PubMed] [Google Scholar]
- 21.Pandey UB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 22.Katsuno M, et al. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–854. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- 23.Chevalier-Larsen ES, et al. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci. 2004;24:4778–4786. doi: 10.1523/JNEUROSCI.0808-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt BJ, et al. Expression of X-linked bulbospinal muscular atrophy (Kennedy disease) in two homozygous women. Neurology. 2002;59:770–772. doi: 10.1212/wnl.59.5.770. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki E, et al. Aberrant E2F activation by polyglutamine expansion of androgen receptor in SBMA neurotoxicity. Proc Natl Acad Sci U S A. 2009;106:3818–3822. doi: 10.1073/pnas.0809819106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harbour JW, Dean DC. Rb function in cell-cycle regulation and apoptosis. Nat Cell Biol. 2000;2:E65–E67. doi: 10.1038/35008695. [DOI] [PubMed] [Google Scholar]
- 27.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 28.Munsat TL, Davies KE. International SMA consortium meeting. (26–28 June 1992, Bonn, Germany); Neuromuscul Disord; 1992. pp. 423–428. [DOI] [PubMed] [Google Scholar]
- 29.Emery AE. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 30.Ogino S, et al. Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet. 2002;110:301–307. doi: 10.1002/ajmg.10425. [DOI] [PubMed] [Google Scholar]
- 31.Liu Q, Dreyfuss G. A novel nuclear structure containing the survival of motor neurons protein. Embo J. 1996;15:3555–3565. [PMC free article] [PubMed] [Google Scholar]
- 32.Rossoll W, et al. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan YB, et al. Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Genet. 2003;12:1367–1376. doi: 10.1093/hmg/ddg157. [DOI] [PubMed] [Google Scholar]
- 34.Lorson CL, et al. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 35.Rajendra TK, et al. A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J Cell Biol. 2007;176:831–841. doi: 10.1083/jcb.200610053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cauchi RJ, Davies KE, Liu JL. A motor function for the DEAD-box RNA helicase, Gemin3, in Drosophila. PLoS Genet. 2008;4:e1000265. doi: 10.1371/journal.pgen.1000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang HC, et al. Modeling spinal muscular atrophy in Drosophila. PLoS ONE. 2008;3:e3209. doi: 10.1371/journal.pone.0003209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurtzke JF. Epidemiology of amyotrophic lateral sclerosis. Adv Neurol. 1982;36:281–302. [PubMed] [Google Scholar]
- 39.Hand CK, Rouleau GA. Familial amyotrophic lateral sclerosis. Muscle Nerve. 2002;25:135–159. doi: 10.1002/mus.10001. [DOI] [PubMed] [Google Scholar]
- 40.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 41.Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65 Suppl 1:S3–S9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- 42.Parkes TL, et al. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat Genet. 1998;19:171–174. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- 43.Elia AJ, et al. Expression of human FALS SOD in motorneurons of Drosophila. Free Radic Biol Med. 1999;26:1332–1338. doi: 10.1016/s0891-5849(98)00333-5. [DOI] [PubMed] [Google Scholar]
- 44.Mockett RJ, et al. Phenotypic effects of familial amyotrophic lateral sclerosis mutant Sod alleles in transgenic Drosophila. Proc Natl Acad Sci U S A. 2003;100:301–306. doi: 10.1073/pnas.0136976100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramaniam JR, et al. Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat Neurosci. 2002;5:301–307. doi: 10.1038/nn823. [DOI] [PubMed] [Google Scholar]
- 46.Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- 47.Cleveland DW, Liu J. Oxidation versus aggregation - how do SOD1 mutants cause ALS? Nat Med. 2000;6:1320–1321. doi: 10.1038/82122. [DOI] [PubMed] [Google Scholar]
- 48.Watson MR, et al. A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J Biol Chem. 2008;283:24972–24981. doi: 10.1074/jbc.M804817200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nishimura AL, et al. A novel locus for late onset amyotrophic lateral sclerosis/motor neurone disease variant at 20q13. J Med Genet. 2004;41:315–320. doi: 10.1136/jmg.2003.013029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nishimura AL, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–831. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kirby J, et al. Mutations in VAPB are not associated with sporadic ALS. Neurology. 2007;68:1951–1953. doi: 10.1212/01.wnl.0000263195.50981.a6. [DOI] [PubMed] [Google Scholar]
- 52.Tsuda H, et al. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell. 2008;133:963–977. doi: 10.1016/j.cell.2008.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pennetta G, et al. Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron. 2002;35:291–306. doi: 10.1016/s0896-6273(02)00769-9. [DOI] [PubMed] [Google Scholar]
- 54.Chai A, et al. hVAPB, the causative gene of a heterogeneous group of motor neuron diseases in humans, is functionally interchangeable with its Drosophila homologue DVAP-33A at the neuromuscular junction. Hum Mol Genet. 2008;17:266–280. doi: 10.1093/hmg/ddm303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Teuling E, et al. Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J Neurosci. 2007;27:9801–9815. doi: 10.1523/JNEUROSCI.2661-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ratnaparkhi A, et al. A Drosophila model of ALS: human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS ONE. 2008;3:e2334. doi: 10.1371/journal.pone.0002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leigh PN, et al. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain. 1991;114(Pt 2):775–788. doi: 10.1093/brain/114.2.775. [DOI] [PubMed] [Google Scholar]
- 58.Lowe J. New pathological findings in amyotrophic lateral sclerosis. J Neurol Sci. 124 Suppl:38–51. doi: 10.1016/0022-510x(94)90175-9. [DOI] [PubMed] [Google Scholar]
- 59.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 60.Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kwiatkowski TJ, Jr, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 63.Chien S, et al. Homophila: human disease gene cognates in Drosophila. Nucleic Acids Res. 2002;30:149–151. doi: 10.1093/nar/30.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chihara T, Luginbuhl D, Luo L. Cytoplasmic and mitochondrial protein translation in axonal and dendritic terminal arborization. Nat Neurosci. 2007;10:828–837. doi: 10.1038/nn1910. [DOI] [PubMed] [Google Scholar]
- 65.Jordanova A, et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- 66.Anderson PR, et al. RNAi-mediated suppression of the mitochondrial iron chaperone, frataxin, in Drosophila. Hum Mol Genet. 2005;14:3397–3405. doi: 10.1093/hmg/ddi367. [DOI] [PubMed] [Google Scholar]
- 67.Llorens JV, et al. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. Faseb J. 2007;21:333–344. doi: 10.1096/fj.05-5709com. [DOI] [PubMed] [Google Scholar]
- 68.Runko AP, Griswold AJ, Min KT. Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila. FEBS Lett. 2008;582:715–719. doi: 10.1016/j.febslet.2008.01.046. [DOI] [PubMed] [Google Scholar]
- 69.Anderson PR, et al. Hydrogen peroxide scavenging rescues frataxin deficiency in a Drosophila model of Friedreich's ataxia. Proc Natl Acad Sci U S A. 2008;105:611–616. doi: 10.1073/pnas.0709691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456–1471. doi: 10.1002/1097-4598(200010)23:10<1456::aid-mus2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 71.Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev. 2002;12:349–361. doi: 10.1016/s0959-437x(02)00309-x. [DOI] [PubMed] [Google Scholar]
- 72.Greener MJ, Roberts RG. Conservation of components of the dystrophin complex in Drosophila. FEBS Lett. 2000;482:13–18. doi: 10.1016/s0014-5793(00)02018-4. [DOI] [PubMed] [Google Scholar]
- 73.Dekkers LC, et al. Embryonic expression patterns of the Drosophila dystrophin-associated glycoprotein complex orthologs. Gene Expr Patterns. 2004;4:153–159. doi: 10.1016/j.modgep.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 74.Shcherbata HR, et al. Dissecting muscle and neuronal disorders in a Drosophila model of muscular dystrophy. Embo J. 2007;26:481–493. doi: 10.1038/sj.emboj.7601503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van der Plas MC, et al. Drosophila Dystrophin is required for integrity of the musculature. Mech Dev. 2007;124:617–630. doi: 10.1016/j.mod.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 76.Taghli-Lamallem O, et al. Dystrophin deficiency in Drosophila reduces lifespan and causes a dilated cardiomyopathy phenotype. Aging Cell. 2008;7:237–249. doi: 10.1111/j.1474-9726.2008.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Allikian MJ, et al. Reduced life span with heart and muscle dysfunction in Drosophila sarcoglycan mutants. Hum Mol Genet. 2007;16:2933–2943. doi: 10.1093/hmg/ddm254. [DOI] [PubMed] [Google Scholar]
- 78.Blake DJ, et al. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 79.van der Plas MC, et al. Dystrophin is required for appropriate retrograde control of neurotransmitter release at the Drosophila neuromuscular junction. J Neurosci. 2006;26:333–344. doi: 10.1523/JNEUROSCI.4069-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bogdanik L, et al. Muscle dystroglycan organizes the postsynapse and regulates presynaptic neurotransmitter release at the Drosophila neuromuscular junction. PLoS ONE. 2008;3:e2084. doi: 10.1371/journal.pone.0002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marques G, et al. The Drosophila BMP type II receptor Wishful Thinking regulates neuromuscular synapse morphology and function. Neuron. 2002;33:529–543. doi: 10.1016/s0896-6273(02)00595-0. [DOI] [PubMed] [Google Scholar]
- 82.McCabe BD, et al. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- 83.Wairkar YP, et al. Synaptic defects in a Drosophila model of congenital muscular dystrophy. J Neurosci. 2008;28:3781–3789. doi: 10.1523/JNEUROSCI.0478-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Montanaro F, Carbonetto S. Targeting dystroglycan in the brain. Neuron. 2003;37:193–196. doi: 10.1016/s0896-6273(03)00032-1. [DOI] [PubMed] [Google Scholar]
- 85.Fradkin LG, et al. The dystrophin Dp186 isoform regulates neurotransmitter release at a central synapse in Drosophila. J Neurosci. 2008;28:5105–5114. doi: 10.1523/JNEUROSCI.4950-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cohn RD, et al. Disruption of DAG1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell. 2002;110:639–648. doi: 10.1016/s0092-8674(02)00907-8. [DOI] [PubMed] [Google Scholar]
- 87.Haines N, Seabrooke S, Stewart BA. Dystroglycan and protein O-mannosyltransferases 1 and 2 are required to maintain integrity of Drosophila larval muscles. Mol Biol Cell. 2007;18:4721–4730. doi: 10.1091/mbc.E07-01-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kucherenko MM, et al. Genetic modifier screens reveal new components that interact with the Drosophila dystroglycan-dystrophin complex. PLoS ONE. 2008;3:e2418. doi: 10.1371/journal.pone.0002418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wheeler TM, Thornton CA. Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol. 2007;20:572–576. doi: 10.1097/WCO.0b013e3282ef6064. [DOI] [PubMed] [Google Scholar]
- 90.de Haro M, et al. MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1. Hum Mol Genet. 2006;15:2138–2145. doi: 10.1093/hmg/ddl137. [DOI] [PubMed] [Google Scholar]
- 91.Miller JW, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. Embo J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Houseley JM, et al. Myotonic dystrophy associated expanded CUG repeat muscleblind positive ribonuclear foci are not toxic to Drosophila. Hum Mol Genet. 2005;14:873–883. doi: 10.1093/hmg/ddi080. [DOI] [PubMed] [Google Scholar]
- 93.Danzeisen R, et al. Superoxide dismutase 1 modulates expression of transferrin receptor. J Biol Inorg Chem. 2006;11:489–498. doi: 10.1007/s00775-006-0099-4. [DOI] [PubMed] [Google Scholar]
- 94.Machuca-Tzili L, et al. Flies deficient in Muscleblind protein model features of myotonic dystrophy with altered splice forms of Z-band associated transcripts. Hum Genet. 2006;120:487–499. doi: 10.1007/s00439-006-0228-8. [DOI] [PubMed] [Google Scholar]
- 95.Garcia-Lopez A, et al. Genetic and chemical modifiers of a CUG toxicity model in Drosophila. PLoS ONE. 2008;3:e1595. doi: 10.1371/journal.pone.0001595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chartier A, Benoit B, Simonelig M. A Drosophila model of oculopharyngeal muscular dystrophy reveals intrinsic toxicity of PABPN1. Embo J. 2006;25:2253–2262. doi: 10.1038/sj.emboj.7601117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang YQ, et al. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- 98.Pilling AD, et al. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Russo GJ, et al. Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J Neurosci. 2009;29:5443–5455. doi: 10.1523/JNEUROSCI.5417-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.De Vos KJ, et al. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- 101.Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32:389–401. doi: 10.1016/s0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- 102.Gunawardena S, et al. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- 103.Lee WC, Yoshihara M, Littleton JT. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington's disease. Proc Natl Acad Sci U S A. 2004;101:3224–3229. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]