

Abstract
To facilitate functional analysis of neuronal connectivity in a mammalian nervous system tightly packed with billions of cells, we developed a new technique that allows inducible genetic manipulations within fluorescently labeled single neurons in mice. We term this technique SLICK for Single-neuron Labeling with Inducible Cre-mediated Knockout. SLICK is achieved by co-expressing a drug-inducible form of cre recombinase and a fluorescent protein within the same small subsets of neurons. Thus, SLICK combines the powerful cre recombinase system for conditional genetic manipulation and the fluorescent labeling of single neurons for imaging. We demonstrate efficient inducible genetic manipulation in several types of neurons using SLICK. Furthermore, we apply SLICK to eliminate synaptic transmission in a small subset of neuromuscular junctions. Our results provide evidence for the long-term stability of inactive neuromuscular synapses in adult animals. More broadly, these studies demonstrate a cre-LoxP compatible system for dissecting gene functions in single identifiable neurons.
Introduction
The mammalian nervous system contains a heterogeneous mixture of billions of neurons that are interconnected by trillions of synapses. Techniques for revealing the morphological and electrophysiological properties of single neurons have overcome this complexity and helped to lay the foundations of modern neuroscience. Today the sequencing of several mammalian genomes presents neuroscientists with unprecedented opportunities to determine the molecular mechanisms underlying the complex structure and function of the nervous system. Genetic manipulation in mice is a powerful tool for dissecting roles of individual genes in the nervous system, and large scale projects with the ultimate goal of generating conditional knockout alleles of all mouse genes are currently underway1. The difficulty however, lies in the detailed analysis of the structure and function of mutant neurons in a brain tightly packed with billions of interconnected cells. Thus, the development of tools for conditional genetic manipulation of individual neurons in mice would greatly facilitate functional genomics in the nervous system. Furthermore, a wealth of genetically encoded tools is now available to both monitor and manipulate various properties of neurons2-8. Genetic approaches that allow the application of these tools in single identifiable neurons would greatly expand their utility in probing neuronal structure and function9.
A major difficulty in performing genetic analysis on single neurons in vivo is the identification of the small numbers of genetically modified neurons among the masses of wild type cells10. We previously generated transgenic mice in which small subsets of neurons are brightly labeled with fluorescent proteins by taking advantage of a transgenic phenomenon called ‘position effect variegation’11. The fluorescent labeling of isolated single neurons in a Golgi-like fashion in living animals permits the direct in vivo visualization and imaging of neuronal dynamics in accessible regions of the nervous system12-15. Here we describe a method that combines the fluorescent labeling of single neurons with inducible genetic manipulation in these same labeled neurons. This is done by using two copies of the Thy1 promoter to simultaneously express creERT2, a drug-inducible form of the cre recombinase enzyme16 and yellow fluorescent protein (YFP) in the same cells. This method thus combines the powerful cre/loxP system for performing conditional gene knockout or transgene expression and the labeling of single neurons with a fluorescent reporter to reveal their morphology. Genetic manipulation is spatially restricted to small subsets of labeled neurons and can be controlled temporally through administration of the activating drug, tamoxifen. We therefore name this technique Single-neuron Labeling with Inducible Cre-mediated Knockout (SLICK). We validate this method by showing efficient tamoxifen-induced, cre-mediated recombination in fluorescently labeled single neurons of SLICK mice. As a proof of principle for the application of this approach, we use SLICK mice to manipulate neural activity in vivo by deleting the choline acetyl transferase (ChAT) gene in subsets of labeled motor neurons. This approach reveals remarkable long term stability of the neuromuscular synapse in the absence of synaptic transmission. More broadly, this study illustrates a powerful genetic tool that is compatible with all cre/loxP-based genetically modified mice.
Results
Transgenic co-expression of cre recombinase and YFP
The cre / loxP site specific recombinase system has been widely used to perform both conditional gene knockout and conditional gene expression in mice17. Our goal was to develop transgenic mice in which it is possible to perform inducible genetic manipulation using the cre/loxP system in small subsets of fluorescently labeled neurons throughout the nervous system. With this goal in mind we evaluated strategies to achieve transgenic co-expression of a fluorescent protein and creERT2, a tamoxifen-inducible form of cre recombinase16,18. We reasoned that this would reveal the morphology of single labeled neurons while simultaneously permitting cre-mediated gene knockout or gene expression in these same cells upon tamoxifen administration (Fig. 1a–c).
Figure 1. Strategy used for co-expression of YFP and cre in SLICK transgenic mice.
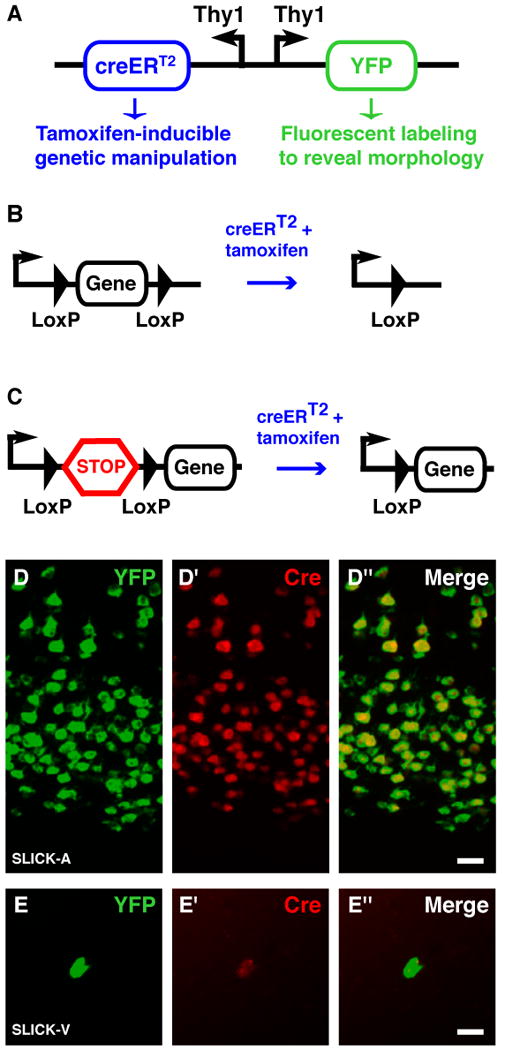
A. Schematic representation of the DNA construct used to generate SLICK transgenic mice. Two copies of the Thy1 promoter drive expression of YFP and the inducible form of cre recombinase–creERT2. CreERT2 consists of cre recombinase fused to a modified ligand binding domain from the estrogen receptor that can be activated by the synthetic ligand tamoxifen but not by endogenous estrogens16. Tamoxifen administration can thus be used to control the timing of recombination. B, C. Strategies to achieve conditional gene knockout and expression using the SLICK system. SLICK mice are first crossed to mice in which the expression of a gene of interest is control by cre-mediated recombination. For gene knockout, part or all of the coding sequence of a gene of interest is flanked by two short loxP sequences (“floxed”) and is excised upon cre activation by tamoxifen (B). For conditional transgene expression a transcriptional stop sequence (STOP cassette) is placed immediately upstream of the coding sequence of the transgene. Deletion of the stop cassette by activated cre initiates transgene expression (C). D, E. Double-fluorescent in situ hybridization for YFP (green) and cre recombinase (red) in the cortex of SLICK-A and hippocampus of SLICK-V transgenic mice respectively. Scale bar = 20μm.
Our initial attempts to co-express a fluorescent protein and creERT2 using the neuron-specific Thy1 promoter in conjunction with an internal ribosome entry site did not allow both proteins to be co-expressed at high levels (data not shown). We therefore switched to a strategy in which the expression of the cDNAs for each protein is driven by separate copies of the Thy1 promoter that are linked together in opposite orientations (back-to-back). One copy drives expression of the yellow fluorescent protein (YFP) and the other drives expression of creERT2 (Fig. 1a). Transgenic mice were generated using this construct and are hereafter referred to as SLICK transgenic mice. Since this strategy relies on separate copies of the Thy1 promoter to drive transcription of both mRNAs it was not clear whether both mRNAs would be expressed in the same cells, especially when transcription is influenced by position effect variegation. To test this we performed double fluorescent in situ hybridization to examine the expression of YFP and creERT2 mRNAs in SLICK mice (Fig. 1d). The expression of both mRNAs in the same cells is highly correlated, with 96.2% (+/− 0.6%) of the YFP expressing cells also expressing cre, while 99% (+/− 0.3%) of cre positive cells express YFP. This tight correlation is also observed in lines in which expression is restricted to a very small subset of neurons (Fig. 1e).
Fluorescent labeling of neurons in SLICK transgenic lines
Thirty lines of SLICK transgenic mice were generated and fluorescent labeling of neurons examined throughout the nervous system of at least 2 mice from each line. YFP expression was confined primarily to projection neurons, with variations in the extent and brightness of labeling from line to line as previously reported for the Thy1 promoter11. Similar to Thy1-XFP mice11, the expression patterns are stably inherited within each line from generation to generation (Supplementary Fig. 1). While there is some variability in the number of labeled neurons between individuals within a line, the overall pattern of labeling does not change (Supplementary Fig. 1). We are primarily interested in the lines in which small subsets of neurons are brightly labeled so that genetic manipulation and imaging can be performed in single neurons. Thus most transgenic lines with weak or very widespread YFP expression were not maintained. However, one line (SLICK-H) with both widespread and strong expression of YFP has been maintained for possible use in experiments in which imaging of neurons is not required. Table 1 summarizes the YFP labeling patterns observed in the different lines that we have maintained.
Table 1. Summary of the YFP expression patterns in 7 lines of SLICK transgenic mice.
The extent of labeling is classified into three broad categories according to the percentage of neurons in the indicated population that are labeled. Few denotes < 10% labeled; many denotes 10 – 90 % labeled and all denotes > 90% labeled. Neuronal populations with especially noteworthy labeling are highlighted in bold. RGC = retinal ganglion cell, INL = inner nuclear layer, DRG = dorsal root ganglion, SMG = submandibular ganglion, pre = presynaptic post = post synaptic, DG = Dentate granule cells, Mossy = cerebellar mossy fibers, Purk = cerebellar purkinje cells, Gran = cerebellar granule cells Olf B = Olfactory Bulb, Mitral = mitral cells, n/d = not determined
| SLICK line | Motor axons | Retina | DRG | SMG | Cortex | Hippocampus | Cerebellum | Olf B | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RGC | INL | Pre | Post | CA1 | DG | Mossy | Purk | Gran | Mitral | ||||
| SLICK-3 | All | Few | Few | Many | None | Few | Many | Many | None | Many | None | None | Few |
| SLICK-A | Few | Few | None | Few | None | None | Many | Many | Many | Many | None | None | None |
| SLICK-H | All | All | Many | All | All | Many | Many | All | Many | Many | Many | Many | Many |
| SLICK-I | All | All | n/d | All | Many | Few | Many | Many | Many | Many | Many | None | Many |
| SLICK-P | All | Many | n/d | Many | Many | None | Many | Many | Many | Many | Many | None | Many |
| SLICK-V | Few | Few | None | Few | None | None | Few | Few | Many | Few | None | Few | None |
| SLICK-X | Few | Few | None | Few | n/d | n/d | Few | Few | Many | Few | None | Few | None |
To evaluate YFP expression in the peripheral nervous system of SLICK transgenic mice, we examined motor neurons, sensory neurons (dorsal root ganglion) and parasympathetic neurons (submandibular ganglion). Several lines were obtained in which restricted subsets of cells in these neuronal populations are labeled (Table 1). In the SLICK-A line a small subset of motor axons (<5%) are labeled in most muscles examined (Fig. 2a). Exceptions to this are the extraocular muscles of the eye in which approximately 50% of axons express YFP. Similarly, a small number of dorsal root ganglion neurons are labeled in SLICK-A mice (Fig. 2b).
Figure 2. YFP labeling of distinct neuronal populations in SLICK transgenic mice.
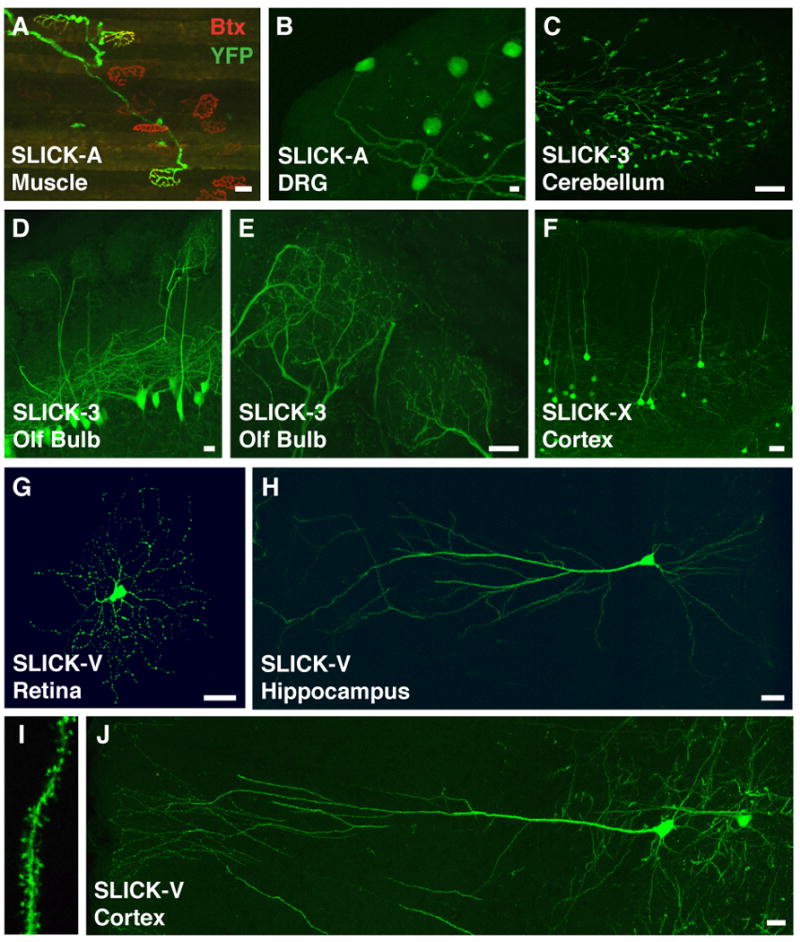
A. Labeling of motor axons in the latissimus dorsi muscle of the SLICK-A line (green). Co-staining for acetylcholine receptors with α-bungarotoxin at the neuromuscular junction (red) shows that only a subset of motor axons are labeled in this line. B. YFP expression in a small number of dorsal root ganglion (DRG) neurons in the SLICK-A line. C. Labeling of a subset of mossy fiber axons and nerve terminals in the granule cell layer of the cerebellum in the SLICK-3 line. D, E. A small subset of YFP expressing mitral cells in the olfactory bulb of SLICK-3 mice. Panel E shows a high magnification view of the mitral cell apical dendrites in two glomeruli. F. Labeling of layer five cortical neurons in the SLICK-X line G. Bright YFP labeling of a small number of retinal ganglion cells in the retina of SLICK-V line mice. H, I, J. Extremely sparse labeling of pyramidal neurons in the hippocampus (H and I) and neocortex (J) of SLICK-V mice. Fine morphological features such as dendritic spines are readily discernable at higher magnification (I). Scale bars = 20μm.
In the central nervous system, labeling of various subsets of neurons can similarly be observed in several lines. In the SLICK-3 line, a subset of mitral cells (generally 1-3 mitral cells per glomerulus) in the olfactory bulb are labeled (Fig. 2d,e). In many of the glomeruli in this line the fine structure of dendritic tufts originating from a single labeled mitral cell can be visualized, making in vivo imaging of mitral cells by 2-photon microscopy feasible (data not shown). This line also shows labeling in mossy fiber axons projecting to the granule cell layer of the cerebellum (Fig. 2c). The SLICK-V line shows much sparser labeling of neurons in the brain. For example, only 6-12 brightly labeled retinal ganglion cells are typically observed per retina (Fig. 2g). Brightly labeled single neurons are found more frequently in the hippocampus, and there is very sparse labeling of pyramidal neurons in several areas of the neocortex. (Fig. 2h,j). Labeled cells are also seen in many other regions of the brain including the amygdala, brain stem and the inferior and superior colliculi (Supplementary Fig. 2). The complete dendritic trees of pyramidal cells in the cortex and in the CA1 region of the hippocampus can be imaged in SLICK-V mice, and fine features such as dendritic spines are easily visualized (Fig. 2i). The SLICK-X line has a YFP labeling pattern quite similar to SLICK-V in many parts of the brain but with a higher number of labeled neurons in these areas compared to SLICK-V (Supplementary Fig. 3). In particular, a higher degree of labeling in the cortex of SLICK-X may be advantageous for transcranial in vivo imaging (Fig. 2f). In summary, the fluorescent labeling in SLICK transgenic mice reveals the detailed morphology of neurons in many regions of the nervous system, including several areas that are amenable to live in vivo imaging approaches. To aid in the evaluation of SLICK lines as experimental tools, a series of whole brain images showing YFP fluorescence in sagittal sections from the SLICK-V and SLICK-X lines are provided (Supplementary Figs. 2 and 3).
Validation of CreERT2 function in SLICK mice
We next wanted to test whether it was possible to perform inducible genetic manipulations within the fluorescently labeled neurons of SLICK transgenic mice. To do this we used the R26R cre reporter strain19. In this strain a neomycin gene flanked by loxP sites has been placed upstream of a LacZ sequence coding for β-galactosidase and targeted to the ROSA26 locus (Fig. 3a). Excision of the neomycin gene by cre recombinase activity allows LacZ expression from the ROSA26 promoter. Because this strain was generated using a ‘knock in’ gene targeting approach, there is only one copy of the reporter gene construct, unlike transgenic reporter lines in which multiple copies of the transgene are present. This allows us to accurately determine recombination efficiency at a single locus, which is equivalent to cre-mediated recombination for conditional gene knockout.
Figure 3. Genetic manipulation in SLICK transgenic mice.
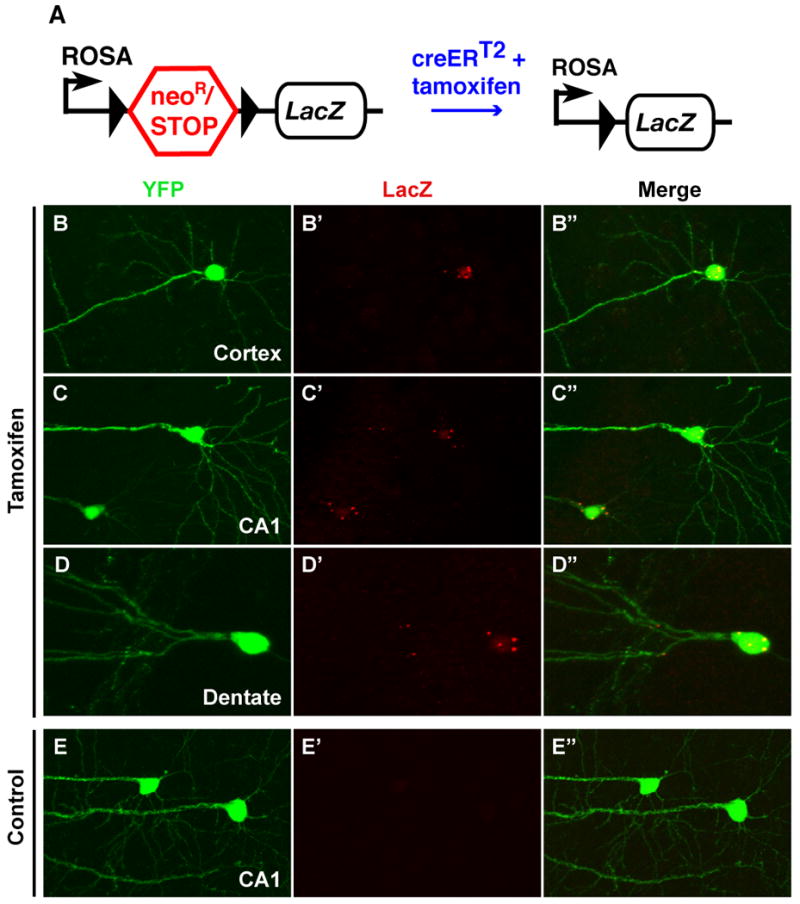
A. Schematic representation of R26R cre reporter strain19. A neomycin resistance gene and transcriptional stop sequence are removed upon cre-mediated recombination to allow expression of the LacZ reporter gene coding for β-galactosidase. The black triangles represent loxP sites. SLICK-V transgenic mice that had been crossed to the R26R reporter were treated with either tamoxifen to induce cre recombinase activity or corn oil as a control. B-E. Efficient genetic manipulation as indicated by Lac-Z expression is seen in fluorescently-labeled neurons of tamoxifen-treated mice (B,C, D) but not control animals (E). B. A layer five cortical neuron. C and E. Pyramidal neurons in the CA1 region of the hippocampus D. A granule cell in the dentate gyrus.
SLICK-V mice were crossed to the R26R reporter, and double heterozygous animals were treated with tamoxifen to induce cre activity (Fig. 3a). Tamoxifen or vehicle alone (corn oil) was administered by oral gavage once a day, for five consectutive days. Recombination was assayed by immunofluorescent staining for LacZ expression two weeks following the final treatment with tamoxifen (Fig. 3b–e). LacZ positive cells are never seen in vehicle-treated animals (Fig. 3e). In tamoxifen-treated animals we observe robust LacZ staining in YFP positive neurons (Fig. 3b–d) and only rarely observe LacZ positive cells in which YFP fluorescence is not detected (< 1% of all LacZ positive cells; not shown). Thus the SLICK-V/R26R animals show no “leaky” reporter gene expression, and cre activity is inducible and confined to fluorescently labeled neurons as expected. Recombination occurred within 24-48 hours after tamoxifen administration, while robust LacZ expression was observed by 5 days post treatment (Supplementary Fig. 4). We next quantified the efficiency of recombination in brightly fluorescent neurons from several brain regions of SLICK-V/R26R mice (Table 2, Supplementary Fig. 5). The efficiency of recombination exceeds 95% in most of the neuronal populations examined (Table 2). Similar results were obtained for the SLICK-X line (Table 2, Supplementary Fig. 6). The SLICK system therefore allows one to reliably identify and image genetically manipulated single neurons.
Table 2. Efficiency of recombination assessed in SLICK-V and SLICK-X/R26R double transgenic mice.
| Brain Stem | Thalamus | Hippocampus CA1 | Dentate Gyrus | Cortex | |
|---|---|---|---|---|---|
| SLICK-V | 98% (62/63) | 90% (17/19) | 96% (204/213) | 95% (131/138) | 95% (72/76) |
| SLICK-X | 97% (140/144) | 90% (150/166) | 93% (289/312) | 98% (643/659) | 92% (274/298) |
All brightly labeled YFP positive cells in at least seven 50μm sagittal sections were counted. The numbers in parentheses indicate LacZ positive cells / YFP positive cells. Percentages were calculated from these numbers.
Conditional gene knockout in SLICK mice
Understanding the mechanisms by which neural activity influences the formation and plasticity of neural circuits is a major focus of neuroscience research20,21. To address this issue using the SLICK system we took advantage of mice carrying a conditional knockout allele of the choline acetyl transferase (ChAT) gene22 (Fig. 4a). ChAT is required for the synthesis of the neurotransmitter acetylcholine, and genetic ablation of ChAT has been shown to eliminate neurotransmission at cholinergic synapses such as the neuromuscular junction (NMJ), resulting in neonatal lethality22,23. To examine the consequences of long-term inhibition of neurotransmission at adult neuromuscular synapses, we used the SLICK-A line to knock out ChAT in a small subset of YFP-labeled motor neurons. SLICK-A+/ChATflox/− mice were treated with either tamoxifen to induce ChAT knockout, or with corn oil as a control (Fig. 4b–e). Motor axons and neuromuscular synapses were examined eight weeks following tamoxifen treatment. ChAT expression was monitored by immunofluorescent staining and postsynaptic acetylcholine receptors were labeled with α-bungarotoxin. In untreated control animals ChAT is present in both YFP-labeled (arrows) and unlabeled axons and nerve terminals (Fig. 4b,d). By contrast, in tamoxifen-treated animals ChAT is undetectable in 99% (111/112) of YFP-labeled motor axons and 97% (73/75) of YFP-labeled nerve terminals (Fig. 4c,e; arrowheads). ChAT is present in almost all YFP-negative axons and terminals of tamoxifen-treated animals (Fig. 4b,d), although occasionally ChAT is not detectable in non-YFP-labeled axons and terminals (not shown; see Supplementary Discussion online for discussion of this observation). Similar overall results for ChAT knockout are observed four weeks following tamoxifen administration (data not shown). Thus NMJs examined at an eight week timepoint would have been completely depleted of ChAT for at least four weeks. In summary, extremely efficient recombination of the floxed ChAT allele in YFP expressing cells is observed. This demonstrates that the SLICK system can be used to achieve gene knockout at rates that are as high, or even higher than, those observed for reporter gene expression (see Table 2).
Figure 4. Inhibition of neurotransmission in subsets of motor neurons through ChAT knockout in SLICK-A mice.
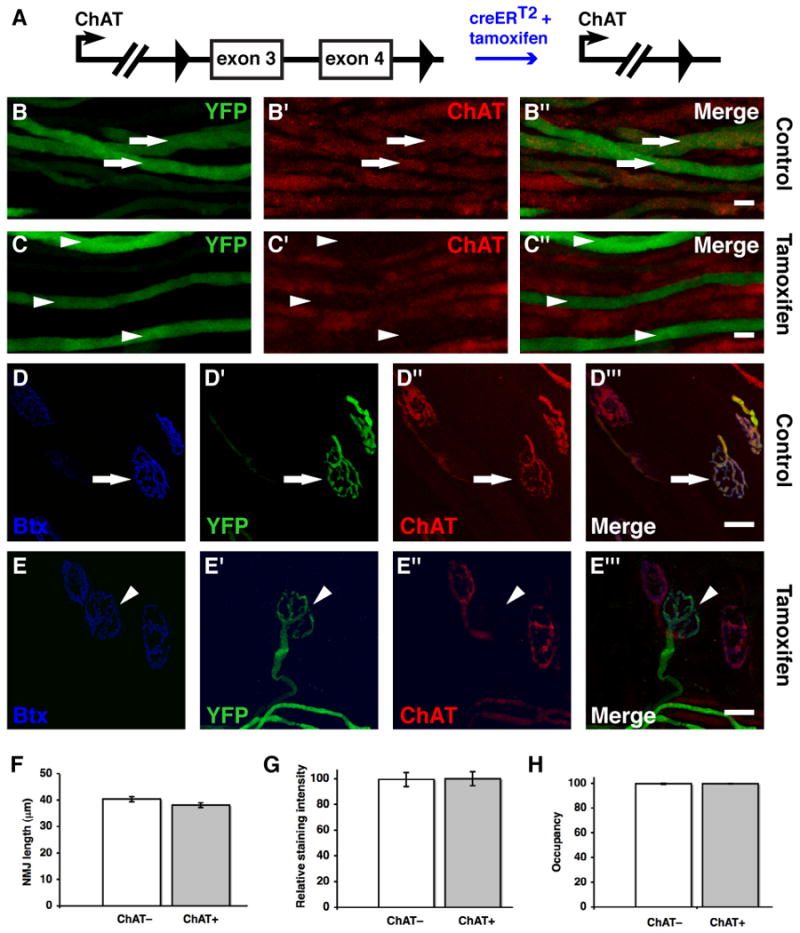
A. Schematic representation of the conditional ChAT allele. Exons 3 and 4 are deleted upon cre-mediated recombination. B, C. Immunofluorescence staining for ChAT in segments of nerve entering the extraocular muscles. Efficient ChAT knockout is observed in YFP-labeled motor axons of tamoxifen-treated (C) but not control animals (B). Arrows and arrowheads show ChAT+ and ChAT− axons respectively. D, E. Staining of NMJs for ChAT as well as α-bungarotoxin (to label acetylcholine receptors). All nerve terminals show strong ChAT staining in control animals but ChAT is specifically deleted from YFP-labeled terminals in tamoxifen-treated animals. The arrow and arrowhead show ChAT+ and ChAT− NMJs respectively. F, G, H. Quantification of aspects of NMJ morphology for ChAT+ and ChAT− NMJs. The length of NMJs was determined as a measure of overall junction size in F (n = 75 junctions). Staining intensity for α-bungarotoxin was quantified as a measure of junctional acetylcholine receptor density in G (n = 40 junctions). The area of α-bungarotoxin staining at NMJs that was occupied by YFP-labeled ChAT− and ChAT+ presynaptic nerve terminals and was measured in H (n = 97 junctions). Data from at least three muscles were used in all quantifications. Scale bars = 20μm. Error bars in F,G,H represent the standard error of the mean.
To determine whether NMJ stability is altered by long-term inhibition of neurotransmission in a subset of motorneurons we compared YFP-labeled ChAT− NMJs to unlabeled ChAT+ NMJs. As a measure of overall NMJ size we quantified the maximal length of NMJs as revealed by α-bungarotoxin labeling. We also examined the expression levels of postsynaptic acteylcholine receptors by quantifying the intensity of α-bungarotoxin staining. We find no significant difference between ChAT+ and ChAT− junctions for either of these parameters (Fig. 4f and 4g). We next examined the the morphology of YFP expressing ChAT− nerve terminals. We find that the YFP labeling of nerve terminals precisely matchs postsynaptic α-bungarotoxin staining at ChAT− junctions. The occupancy of α-bungarotoxin labeled post synaptic sites by ChAT− nerve terminals is 99.7% (n= 97 junctions). This is identical to the occupancy of postsynaptic sites by YFP-labeled ChAT+ terminals in untreated control animals (Fig. 4h). Thus there is no evidence of retraction of inactive terminals from NMJs. Moreover, we do not observe any aberrant growth or sprouting of inactive axons or any gross abnormalities in the complexity or structure of inactive terminals. In summary, eight weeks following ChAT knockout, inactive ChAT− NMJs are morphologicaly indistinguishable from neighbouring active junctions and there is no evidence of replacement of inactive terminals by active ones.
Discussion
Genetic manipulation in mice has proven a powerful tool in dissecting mechanisms underlying the development, maintenance and plasticity of neuronal connectivity24. Here we describe the generation of SLICK transgenic mice that allow genetic manipulations in single neurons brightly labeled with YFP. Recombination efficiencies of greater than 95% are observed for both inducible transgene expression and gene knockout. The ability to genetically manipulate labeled neurons in such a precise manner has several advantages. First, the Golgi-like labeling with YFP permits the analysis of detailed morphology and synaptic connections of individual mutant neurons. Second, the restriction of genetic manipulation to a labeled, small subset of neurons allows analysis to be performed in a predominantly wild-type background, preventing large scale disruption of neural circuits and allowing cell-autonomous gene function to be distinguished from indirect effects of widespread gene knockout. This should facilitate the examination of competition between mutant and wild-type neurons in developmental processes such as synapse formation and elimination. Third, the inducible nature of genetic manipulation using the SLICK system permits gene function to be analyzed at late developmental or adult stages, thereby avoiding the potential complications of embryonic lethality, early developmental defects or secondary compensatory mechanisms. Another important feature of the SLICK system is that the YFP labeling of neurons is constitutive. This allows labeled neurons from untreated animals to be used as controls for genetically manipulated cells. Furthermore, in vivo imaging of YFP-labeled neurons should make it possible to image the same cells before and after a genetic manipulation, allowing the examination of neuronal properties in the presence and absence of a given gene in the same neuron in vivo. Such time-lapse imaging of genetically manipulated neurons will also allow dynamic aspects of cellular morphology and synaptic plasticity to be studied.
The goal of our method is similar to a recently described technique termed mosaic analysis with double marker (MADM)10. Mosaic analysis makes use of mitotic recombination to generate clones of cells that are homozygous null for a gene of interest and has been widely used in Drosophila25. MADM is an adaptation of mosaic analysis for use in mice. The goal of both MADM and SLICK is to perform genetic manipulations within fluorescently labeled populations of cells. However there are many intrinsic differences between the two methods. For example, MADM operates in mitotic cells and is mostly applicable in neurons at early developmental stages when neuronal precursors are still dividing. SLICK by contrast would be more applicable in late embryonic, postnatal and adult mice since it relies on the Thy1 promoter that is most active at these stages. For gene knockout experiments the sets of genes that can be used by each technique are different. MADM can be used for all types of mutations (targeted, spontaneous or chemically induced) when markers are eventually placed on each arm of all mouse chromosomes. By comparison, SLICK is compatible with all floxed conditional knockout alleles but not other types of mutations. MADM has the advantage that is not restricted to neurons and can in principle be used in any cells or tissue. SLICK is neuron specific, but has the advantage of being able to temporally control genetic manipulations in defined types of neurons. Thus, although the goals of both methods are broadly similar, in practice they are likely to find different but very complementary applications in neuroscience research.
To demonstrate the utility of SLICK for gene knockout, we use the system to inhibit cholinergic neurotransmission by knocking out essential exons in the ChAT gene22. This prevents acetylcholine synthesis, effectively silencing cholinergic neurons. Global disruption of cholinergic transmission is lethal due to respiratory failure22,23. We avoided lethality by using the SLICK-A line of mice to restrict ChAT knockout to a very small subset of YFP-labeled motor neurons in most muscles. This provided us with a unique opportunity to inhibit neurotransmission in vivo for long periods of time in subsets of nerve terminals while leaving the activity of their neighbors unperturbed. We have examined the consequences of this manipulation on the stability of the NMJ. We find that the gross morphology of the NMJ is unchanged under such conditions. There is no alteration in the level of receptor expression at silenced junctions, in agreement with observations following short term pharmacological inhibition of action potentials26,27. We also considered the possibility that active axons might invade or take over inactive NMJs, similar to the manner in which denervated NMJs can be innervated by sprouting axons from intact neighboring junctions28. However, we find no evidence of ChAT− nerve terminals vacating inactive junctions, nor do we find active axons replacing inactive ones (See Supplementary Discussion online for further discussion of these findings). In conclusion, the adult NMJ remains extremely stable for long periods in the absence of neurotransmission, despite potential competition from neighboring active nerve terminals.
In addition to its use for targeted gene knockout, SLICK can be used to target expression of any gene of interest to single, fluorescently labeled neurons in vivo. Many genetically encoded tools to both monitor and manipulate various physiological properties of neurons have been developed in recent years2-8,29-32. These include fluorescent reporters of synaptic transmission and ion concentrations, as well as reagents to modulate neuronal excitability and neurotransmitter release. SLICK will allow expression of such probes in single neurons in vivo, while simultaneously revealing the morphology of the neurons for imaging or identifying them for electrophysiological analysis. Used in this manner, SLICK will permit many genetically-encoded probes to be applied with single-cell resolution in mice for the first time. Although some fluorescent reporters are based on YFP and would not be compatible with the SLICK mice described here, this problem could be circumvented by generating SLICK mice that express spectrally distinct fluorescent proteins33. SLICK therefore has the potential to increase the scope and utility of a wide variety of recently developed tools for monitoring and manipulating neurons and neural circuits.
SLICK is based on the popular cre/loxP system that has been used extensively to achieve conditional gene knockout in mice17,34,35. The availability of mice harboring conditional knockout alleles is continually increasing and a large scale project to generate “floxed” conditional knockout alleles is underway1. The SLICK system can be used to tap into this resource and should permit cellular phenotypes to be studied in these mice at the single-neuron level. Thus, SLICK has considerable potential as a functional genomics tool in studying function and dysfunction of the nervous system.
Methods
Generation of transgenic DNA constructs
The Thy1 vector has been described previously and was provided to us by Pico Caroni11,36. The vector pCreERT2 containing the creERT2 coding sequence was obtained from Daniel Metzger and Pierre Chambon16. To generate the Thy1-YFP: Thy1-creERT2 double promoter construct, creERT2 was cut from pCre-ERT2 using EcoRI and cloned into the XhoI site of the Thy1 vector by blunt end ligation. Thy1-YFP was generated as previously described11. Thy1-creERT2 and Thy1-YFP were then joined via an EcoRI site at the 5′ end of the Thy1 promoter. The final construct was linearized using NotI to remove the vector backbone prior to injection into oocytes.
Mice
SLICK transgenic mice were generated by injection of gel-purified DNA into fertilized oocytes, using standard techniques37. Embryos for injection were obtained from matings between (C57BL6/J and CBA) F1 hybrids. Transgenic founders were backcrossed to C57BL6/J mice for analysis of expression patterns. Primers and protocols used for genotyping are described in the Supplementary Methods online. The R26R line of cre reporter mice19 was from The Jackson Laboratory (Stock No: 003474; Bar Harbor, ME, USA). ChAT conditional knockout mice were obtained from Joshua Sanes22. Experiments involving animals were conducted in accordance with the institutional protocols of Duke University. Animal experiments at University College Cork were approved by the University Ethics Committee and conducted under a license from the Department of Health and Children.
In Situ Hybridization
Probe sequences comprised the complete coding sequences of YFP and cre recombinase were cloned into pBluescript SKII vector (Stratagene, La Jolla, CA, USA). Digoxygenin or fluorescein-labeled RNA probes were generated by in vitro transcription using the MaxiScript kit (Ambion, Austin, TX,USA) according to the manufacturers instructions. For in situ hybridization fresh tissues were dissected and quickly embedded and frozen in OCT compound (TissueTek, Torrance, CA, USA). 20 μm sections were cut on a Leica CM 1850 cryostat, fixed for 5 minutes with 4% paraformaldehyde and rinsed 3 times in PBS. Double-fluorescent in situ hybridization was carried out using the tyramide signal amplification (TSA) system (Perkin Elmer, Waltham, MA, USA). YFP mRNA was detected using a fluorescein-labeled probe and fluorescein-tyramide. Cre mRNA was detected using a digoxygenin-labeled probe and Cy3-tyramide. Images were acquired on a Zeiss Axioskop 2 fluorescent microscope using a 20× objective.
Fluorescence imaging of SLICK mice
For imaging of YFP fluorescence, mice were anesthetized and perfused through the heart, first with lactated Ringer's solution, then with 4% para-formaldehyde. Images of muscles dorsal root ganglia and retinae were taken from whole-mount tissues. For brains, 100 μm sections were cut using a vibrotome. Confocal images of both whole mount tissue and sections were acquired on a Nikon Eclipse confocal microscope using 20× and 63× objectives. For two-color imaging of neuromuscular junctions acetylcholine receptors were labeled with rhodamine-conjugated α-bungarotoxin (1:10,000) for 2-3 hours and washed 3 times in PBS. Three-color imaging of muscles stained for ChAT was performed on a Leica DM IRE2 confocal microscope using a HCX PLAPO 63× objective. YFP was excited using the 488 nm laser line and fluorescence emission collected between 508 and 550 nm. Cy3 was excited at 543 nm and fluorescence collected between 555 and 627 nm. Alexa647 was excited at 633 nm and fluorescence collected between 650 and 750 nm.
Antibodies and Immunohistochemistry
Details of antibodies and protocols for immunohistochemistry are described in the Supplementary Methods online.
Tamoxifen administration
Tamoxifen (Sigma, St Louis, MO, USA; catalog number T5648) was dissolved in corn oil (Kroger brand) at a concentration of 20 mg/ml by rocking overnight at room temperature. This was stored at room temperature for immediate use. Adult mice were administered 0.25 mg/g body weight of tamoxifen per day by oral gavage for five days. For induction of recombination in SLICK-V/R26R mice a second 5 day treatment was performed two weeks after the first. SLICK-V/R26R mice were 12–16 weeks old on the date of first treatment. To induce ChAT gene knockout, mice were treated with tamoxifen for one period of 5 days at 10 weeks of age. These mice were then killed for analysis 5 or 9 weeks later.
Image Analysis and Quantification
Analysis of in situ hybridization data and β-galactosidase immunohistochemistry was performed by manually counting cells in merged two color images made using Adobe Photoshop software. Selection of cells to be counted was performed in a blinded manner prior to merging of the images. In determining the recombination efficiency in SLICK-V/R26R mice only bright cells that would be suitable for imaging of detailed cellular morphology were considered.
Analysis of ChAT knockout efficiency and NMJ structure was performed entirely on whole mount stained extraocular muscles. This was because the higher degree of labeling in these muscles provides ChAT+ and ChAT− NMJ in approximately equal numbers for statistical analysis. To examine neuromuscular junctions, Z-series of confocal images were collected and maximum intensity projections of each series made. All image analysis was performed using ImageJ software. The size of ChAT− and ChAT+ NMJ from the same muscle were quantified by measuring the maximal dimension of the α-bungarotoxin staining. Statistical comparison of ChAT− and ChAT+ junction size was performed using an unpaired Student's T-test. Quantification of acetylcholine receptor density was performed by measuring the fluorescence intensity from pairs of ChAT− and ChAT+ NMJs in the same image. The same threshold was applied to the entire image and the area of NMJs was selected using the wand tool in ImageJ. The average pixel intensity over an entire NMJ was then measured. Only NMJs laying flat on the muscle fiber and only pairs of nearby ChAT− and ChAT+ NMJ at a similar focal depth were used for analysis. Statistical analysis was performed using a paired T-test. Occupancy of NMJs by nerve terminals was calculated by making a line tracing of the pretzel-like pattern of bungarotoxin staining including all branches using ImageJ. The length of this trace was measured and compared to a line tracing of the nerve terminal based on the YFP fluorescence.
Online Resources and Availability of SLICK mice
Data describing neuronal labeling and recombination patterns in various lines of SLICK mice is available on the Feng Laboratory website (http://guopingfenglab.net/resources.html). The SLICK-A and SLICK-V lines are currently available from The Jackson Laboratory, Bar Harbor, Maine, USA (http://jaxmice.jax.org/). For information about the availability of other lines and how to request them, please see the Feng Laboratory website.
Supplementary Material
Acknowledgments
We thank Joshua Sanes for providing the ChAT conditional knockout mice and anti-β-galactosidase antibody. We thank Adi Mizrahi for performing 2-photon imaging of SLICK-3 mice and Robert Kotloski for advice regarding tamoxifen administration. We also thank the Duke Neurotransgenic Core Facility for generating SLICK mice. We thank James McNamara, Michael D. Ehlers and Mary McCaffrey for access to confocal microscopes. We are grateful to Benjamin Arenkiel, Michael D. Ehlers, Anne West, Kellie Dean and members of the Feng laboratory for critically reading and discussing the manuscript. The support of Ruth K. Broad Biomedical Research Foundation, Science Foundation Ireland and the National Institute of Health is also acknowledged.
Footnotes
Author Contributions: P.Y. and G.F. conceived the SLICK method, designed the experiments and wrote the manuscript. P.Y. and J.G. generated SLICK transgenic mice. P.Y., L.Q., S.Z. and D.W. characterized SLICK mice. P.Y. and S.Z. performed the quantification and data analysis.
Bibliography
- 1.Collins FS, Rossant J. A mouse for all reasons. Cell. 2007;128:9–13. doi: 10.1016/j.cell.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 2.Brecht M, et al. Novel approaches to monitor and manipulate single neurons in vivo. J Neurosci. 2004;24:9223–7. doi: 10.1523/JNEUROSCI.3344-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Callaway EM. A molecular and genetic arsenal for systems neuroscience. Trends Neurosci. 2005;28:196–201. doi: 10.1016/j.tins.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Deisseroth K, et al. Next-generation optical technologies for illuminating genetically targeted brain circuits. J Neurosci. 2006;26:10380–6. doi: 10.1523/JNEUROSCI.3863-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marek KW, Davis GW. Controlling the active properties of excitable cells. Curr Opin Neurobiol. 2003;13:607–11. doi: 10.1016/j.conb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Miesenbock G. Genetic methods for illuminating the function of neural circuits. Curr Opin Neurobiol. 2004;14:395–402. doi: 10.1016/j.conb.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Miyawaki A. Fluorescence imaging of physiological activity in complex systems using GFP-based probes. Curr Opin Neurobiol. 2003;13:591–6. doi: 10.1016/j.conb.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 8.Luo L, Callaway EM, Svoboda K. Genetic dissection of neural circuits. Neuron. 2008;57:634–60. doi: 10.1016/j.neuron.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young P, Feng G. Labeling neurons in vivo for morphological and functional studies. Curr Opin Neurobiol. 2004;14:642–6. doi: 10.1016/j.conb.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L. Mosaic analysis with double markers in mice. Cell. 2005;121:479–92. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 11.Feng G, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 12.Grutzendler J, Kasthuri N, Gan WB. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420:812–6. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- 13.Kasthuri N, Lichtman JW. Structural dynamics of synapses in living animals. Curr Opin Neurobiol. 2004;14:105–11. doi: 10.1016/j.conb.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 14.Trachtenberg JT, et al. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–94. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- 15.Mizrahi A, Katz LC. Dendritic stability in the adult olfactory bulb. Nat Neurosci. 2003;6:1201–7. doi: 10.1038/nn1133. [DOI] [PubMed] [Google Scholar]
- 16.Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun. 1997;237:752–7. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- 17.Branda CS, Dymecki SM. Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Dev Cell. 2004;6:7–28. doi: 10.1016/s1534-5807(03)00399-x. [DOI] [PubMed] [Google Scholar]
- 18.Metzger D, Chambon P. Site- and time-specific gene targeting in the mouse. Methods. 2001;24:71–80. doi: 10.1006/meth.2001.1159. [DOI] [PubMed] [Google Scholar]
- 19.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 20.Burrone J, Murthy VN. Synaptic gain control and homeostasis. Curr Opin Neurobiol. 2003;13:560–7. doi: 10.1016/j.conb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 21.Hua JY, Smith SJ. Neural activity and the dynamics of central nervous system development. Nat Neurosci. 2004;7:327–32. doi: 10.1038/nn1218. [DOI] [PubMed] [Google Scholar]
- 22.Misgeld T, et al. Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron. 2002;36:635–48. doi: 10.1016/s0896-6273(02)01020-6. [DOI] [PubMed] [Google Scholar]
- 23.Brandon EP, et al. Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci. 2003;23:539–49. doi: 10.1523/JNEUROSCI.23-02-00539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen C, Tonegawa S. Molecular genetic analysis of synaptic plasticity, activity-dependent neural development, learning, and memory in the mammalian brain. Annu Rev Neurosci. 1997;20:157–84. doi: 10.1146/annurev.neuro.20.1.157. [DOI] [PubMed] [Google Scholar]
- 25.Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–61. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, et al. Prolongation of evoked and spontaneous synaptic currents at the neuromuscular junction after activity blockade is caused by the upregulation of fetal acetylcholine receptors. J Neurosci. 2006;26:8983–7. doi: 10.1523/JNEUROSCI.2493-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, et al. Activity-dependent presynaptic regulation of quantal size at the mammalian neuromuscular junction in vivo. J Neurosci. 2005;25:343–51. doi: 10.1523/JNEUROSCI.3252-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Son YJ, Thompson WJ. Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells. Neuron. 1995;14:133–41. doi: 10.1016/0896-6273(95)90247-3. [DOI] [PubMed] [Google Scholar]
- 29.Johns DC, Marx R, Mains RE, O'Rourke B, Marban E. Inducible genetic suppression of neuronal excitability. J Neurosci. 1999;19:1691–7. doi: 10.1523/JNEUROSCI.19-05-01691.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sweeney ST, Broadie K, Keane J, Niemann H, O'Kane CJ. Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron. 1995;14:341–51. doi: 10.1016/0896-6273(95)90290-2. [DOI] [PubMed] [Google Scholar]
- 31.Arenkiel BR, et al. In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron. 2007;54:205–18. doi: 10.1016/j.neuron.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, et al. High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0700384104. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaner NC, et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–72. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 34.Lewandoski M. Conditional control of gene expression in the mouse. Nat Rev Genet. 2001;2:743–55. doi: 10.1038/35093537. [DOI] [PubMed] [Google Scholar]
- 35.Sauer B. Inducible gene targeting in mice using the Cre/lox system. Methods. 1998;14:381–92. doi: 10.1006/meth.1998.0593. [DOI] [PubMed] [Google Scholar]
- 36.Caroni P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods. 1997;71:3–9. doi: 10.1016/s0165-0270(96)00121-5. [DOI] [PubMed] [Google Scholar]
- 37.Hogan B, Constantini F, Lacey E. Production of transgenic mice. In: Hogan B, Constantini F, Lacey E, editors. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1994. pp. 217–252. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.