

Abstract
The pathogenic mutation in Friedreich’s ataxia (FRDA) is a homozygous guanine-adenine-adenine (GAA) trinucleotide repeat expansion on chromosome 9q13 that causes a transcriptional defect of the frataxin gene. Deficiency of frataxin, a small mitochondrial protein, is responsible for all clinical and morphological manifestations of FRDA. This autosomal recessive disease affects central and peripheral nervous systems, heart, skeleton, and endocrine pancreas. Long expansions lead to early onset, severe clinical illness, and death in young adult life. Patients with short expansions have a later onset and a more benign course. Some are not diagnosed during life. The neurological phenotype reflects lesions in dorsal root ganglia (DRG), sensory peripheral nerves, corticospinal tracts, and dentate nuclei (DN). Most patients succumb to cardiomyopathy, and many become diabetic during the course of their disease. This review seeks to reconcile the diverse clinical features with pathological and molecular data. In the pathogenesis of the lesion in DRG, dorsal spinal roots, and sensory peripheral nerves, developmental defects and atrophy occur in combination. The progressive lesion of the DN lacks a known developmental component. Destruction of the DN, optic atrophy, and degeneration of the corticospinal tracts are intrinsic central nervous system lesions. Fiber loss in dorsal columns and spinocerebellar tracts, and atrophy of the neurons in the dorsal nuclei of Clarke are secondary to the lesion in DRG. The role of frataxin deficiency in the pathogenesis of FRDA is still unclear because the protein has multiple functions in the normal state, including biogenesis of iron–sulfur clusters; iron chaperoning; iron storage; and control of iron-mediated oxidative tissue damage.
Keywords: Cardiomyopathy, Dentate nucleus, Dorsal root ganglion, Frataxin, Friedreich’s ataxia, GAA trinucleotide repeats, Iron, Transcription
1. Introduction
Friedreich’s ataxia (FRDA) is the most common autosomal recessive ataxia, in the vast majority of patients, due to homozygous expansion of a guanine–adenine–adenine (GAA) trinucleotide repeat in intron 1 of the frataxin gene on chromosome 9q13 [1]. The mutation causes a defect of transcription [2], and lack of frataxin, a small mitochondrial protein, is the accepted cause of the entire complex clinical and pathological phenotype of FRDA. The disease affects central and peripheral nervous systems: heart, skeleton, and endocrine pancreas. The estimated carrier frequency ranges from 1:50 to 1:100 in peoples of European, North African, Middle Eastern, and Indian origin. The point prevalence may be as high as 3/100,000 [3,4]. It is higher in enclaves, in which consanguinity is more frequent. The disease is peculiarly absent from Sub-Saharan and Far Eastern populations [5]. Based on clinical studies, gene testing, and acceptance of a more variable clinical phenotype of FRDA, the number of persons with FRDA in the United States at any given time can be estimated as 9000.
2. The history of Friedreich’s ataxia
Table 1 lists the main steps in the discovery and characterization of FRDA. Friedreich [6–9] wrote 4 long articles and one postscriptum [10] about the disease that now bears his name. He recognized that the illness was hereditary [9,10] and had good insight into its pathogenesis. He did not believe that “atrophy” alone could explain the neuropathological manifestations and that a developmental defect had to be involved [10]. His erroneous interpretation of dorsal column degeneration as inflammatory [10] was presumably influenced by the prevalence of tabes dorsalis at that time. Friedreich recognized that his patients had heart disease but attributed it to the effect of abdominal “typhus” (the German term for typhoid). It is unlikely that the patients had this specific infection but rather a highly febrile state toward the end of their lives [6]. Friedreich gave a detailed description of the gross and microscopic changes in the heart. He was impressed by the thinning of the spinal cord and provided the very first microscopic description of the abnormalities in the dorsal and anterolateral columns [10]. He thought that dorsal root ganglia (DRG) were normal and did not mention the now well established lesion of the dentate nucleus (DN). Since Friedreich’s illustrations in 1877 [10], many generations of medical trainees learnt that FRDA is a primary disease of the spinal cord. This conclusion is no longer tenable. Mott [11] studied only a single case, but his detailed descriptions and drawings stand out as a classic. Betz cells have not been examined in more recent work though degeneration of the corticospinal tracts is one of the few intrinsic central nervous system lesions in FRDA. Clinical features of FRDA never left any doubt about the existence of a cerebellar component but the lesion of the DN [11] fell into oblivion until 1957 [12]. Fifteen years elapsed between the discovery of local iron excess in the hearts of patients with FRDA [13] and frataxin deficiency [1]. A large literature has accumulated about this protein but its precise function in iron–sulfur cluster (Fe–S) biogenesis [14,15], i.e. iron chaperoning, iron detoxification, and possibly iron storage in mammalian tissues, remains elusive.
Table 1.
Historical milestones in Friedreich’s ataxia.
| 1863–1877 | Friedreich [6–10] publishes extensive descriptions of FRDA. |
| 1907 | Mott [11] provides an extensive neuropathological description of a single case of FRDA, including the lesion of the dentate nucleus. |
| 1957 | Urich et al. [12] emphasize the existence of “suprasegmental” lesion in FRDA. |
| 1980 | Lamarche et al. [13] discover minute iron-positive granules in the cardiomyocytes of patients with FRDA. |
| 1996 | Campuzano et al. [1] identify the mutation in FRDA, name the lacking protein frataxin in analogy with FRDA, and recognize its role in iron metabolism. |
| 1997 | Rötig et al. [14] discover “iron–sulfur” protein deficiency (complexes I, II and III of the mitochondrial electron transport chain), and aconitase in endocardial biopsies of FRDA patients. |
| 2002 | Mühlenhoff et al. [15] recognize the importance of the yeast homolog of frataxin in the biogenesis of iron–sulfur clusters (Fe–S); Fe–S cluster deficiency is accepted as a critical factor in the pathogenesis of FRDA. |
3. Clinical features and genetics
Friedreich’s ataxia is typically a disease of young people and affects male and females alike. In a systematic study of 115 patients with FRDA from 90 families, Harding [16] determined mean ages of onset and death as 10.52±7.4 years and 37.54±14.35 years, respectively. Koeppen et al. [17] calculated the mean duration in 21 autopsy cases as 21± 12 years. De Michele et al. [18] defined “late onset” of FRDA as 24.4 years (range 21–29). After genetic testing became available in 1996, the clinical spectrum of FRDA expanded greatly, and the inclusion of older patients shifted the age of onset to 15.5±8 years (range 2–51) [19]. Table 2 lists sex, age of onset and death, disease duration, and GAA trinucleotide repeats of 30 patients from the author’s collection. Age of onset (13±10 years), age of death (40±20 years), and disease duration (26±14 years) reflect the inclusion of older patients who would not have been diagnosed prior to routine use of genetic testing. Dürr et al. [19] and Filla et al. [20] showed a distinct relationship of age of onset to the number of GAA trinucleotide repeats in the smaller allele. Since these important reports, age of death, disease severity and duration, and cardiomyopathy have also been correlated with the shorter GAA trinucleotide repeat expansion. A contribution by the longer expansion, however, cannot be ignored (see below). Onset at the age of 50 years is now occasionally recognized, and some patients are not diagnosed during life [21]. Fig. 1 displays a regression analysis of the ages of onset and death in the 30 patients shown in Table 2.
Table 2.
Sex, age of onset, age of death, disease duration, GAA repeats, and cause of death in 28 patients with Friedreich’s ataxia.
| No | Sex | Age of onset (years) | Age of death (years) | Disease duration (years) | GAA repeats | Cause of death | |
|---|---|---|---|---|---|---|---|
| FRDA 1 | F | 7 | 38 | 31 | 750 | 594 | Cardiomyopathy; embolic stroke |
| FRDA 2 | F | 10 | 25 | 15 | 850 | 750 | Cardiomyopathy |
| FRDA 3 | F | 22 | 65 | 43 | 631 | 242 | Cachexia |
| FRDA 4 | F | 21 | 79 | 58 | 549 | 356 | Cachexia |
| FRDA 5 | F | 10 | 24 | 14 | 950 | 700 | Cardiomyopathy |
| FRDA 6 | F | 9 | 26 | 17 | 850 | 690 | Cardiomyopathy |
| FRDA 7 | M | 16 | 39 | 23 | 491 | 370 | Cardiomyopathy |
| FRDA 8 | F | 8 | 28 | 20 | 709 | 559 | Cardiomyopathy; embolic stroke |
| FRDA 9 | F | 8 | 23 | 15 | 777 | 528 | Cardiomyopathy; bronchopneumonia |
| FRDA 10 | F | 17 | 58 | 41 | 566 | 566 | Cardiomyopathy; brain hemorrhage |
| FRDA 11 | M | 10 | 38 | 28 | 934 | 249 | Cardiomyopathy |
| FRDA 12 | F | 11 | 42 | 31 | 990 | 761 | Cardiomyopathy |
| FRDA 13 | F | 7 | 55 | 48 | 793 | 644 | Cachexia |
| FRDA 14 | F | 6 | 36 | 30 | 840 | 715 | Cardiomyopathy |
| FRDA 15 | F | 8 | 15 | 7 | 840 | 840 | Cardiomyopathy |
| FRDA 16 | F | 2 | 33 | 21 | 604 | 455 | Cardiomyopathy; renal failure |
| FRDA 17 | F | 34 | 77 | 43 | 841 | 466 | Cachexia; cardiomyopathy |
| FRDA 18 | F | 20 | 61 | 41 | 694 | 593 | Cardiomyopathy |
| FRDA 19 | M | 2 | 10 | 8 | 1016 | 1016 | Cardiomyopathy |
| FRDA 20 | F | 12 | 24 | 12 | 910 | 740 | Myoglobinuria; renal failure |
| FRDA 21 | M | 3 | 23 | 20 | 1200 | 1200 | Cardiomyopathy |
| FRDA 22 | M | 14 | 50 | 36 | 750 | 400 | Cardiomyopathy; myocardial infarction |
| FRDA 23 | F | 50 | 83 | 33 | 236 | 106 | Cachexia |
| FRDA 24 | M | 10 | 24 | 14 | 1050 | 700 | Cardiomyopathy |
| FRDA 25 | M | 13 | 20 | 7 | 850 | 850 | Cardiomyopathy |
| FRDA 26 | M | 7 | 34 | 27 | 1114 | 1114 | Cardiomyopathy |
| FRDA 27 | F | 17 | 50 | 33 | 1122 | 515 | Cardiomyopathy |
| FRDA 28 | M | 15 | 19 | 4 | 1153 | 841 | Cardiomyopathy |
| FRDA 29 | F | 18 | 63 | 45 | 730 | 639 | Cardiomyopathy |
| FRDA 30 | F | 6 | 23 | 17 | 864 | 668 | Cardiomyopathy |
| Means±S.D.* | 13±10 | 40±20 | 26±14 | 822±213 | 629±247 |
S.D., standard deviation.
Fig. 1.

Ages of onset and death as a function of GAA trinucleotide repeat expansions (short alleles) in 30 patients with autopsy-confirmed FRDA (Table 2). (a) Age of onset vs. GAA trinucleotide repeats; (b) age of death vs. GAA trinucleotide repeats. The patient identified by the arrows (onset at 50, death at 83) was not diagnosed during life. An exponential trend line generated an R2=0.5087 for age of onset; a logarithmic trend line yielded R2=0.494 for age of death. Dürr et al. [19] reported an optimal fit between age of onset and GAA trinucleotide repeat expansion in the shorter allele in 140 patients (quadratic regression; R2=0.56). Filla et al. [20] obtained an R2=0.47 in 64 patients (linear regression).
Not all patients with FRDA have homozygous GAA expansions. In an estimated 2–4%, they are compound heterozygotes with one GAA expansion and point mutations or deletion on the other allele [1,22]. Patients with compound heterozygosity can have some atypical clinical features. They tend to have less dysarthria than homozygous patients but have optic pallor with greater frequency [22].
In well established cases, the clinical diagnosis is not difficult. Several authors have summarized the clinical features in large series of patients [16,19–24]. Children with beginning FRDA may raise concern in parents and teachers because they appear “clumsy” and their motor skills do not match those of unaffected sibs. Scoliosis and foot deformity (pes cavus) may also be early signs and precede ataxia. In some patients, cardiomyopathy is the first clinical manifestation whereas diabetes mellitus is invariably delayed in the course of the illness. The following neurological signs are most frequent: Gait ataxia, dysmetria of arms and legs, dysarthria, head titubation, atrophy and weakness of the distal extremities, absence of muscle stretch reflexes, Babinski signs, loss of joint and vibratory senses, and superimposed stocking-and-glove type sensory neuropathy. Muscle tone is generally normal in the arms but variable in the legs [16]. Spasticity and hyperreflexia in the legs are no longer rare. From among the 30 FRDA patients shown in Table 2, three (FRDA 13, FRDA 19, and FRDA 27) had received continuous intrathecal baclofen injections for the control of leg spasticity. Nearly all patients become paraplegic and require wheelchairs. Harding [16] estimated that over half of her patients were wheelchair-bound 16 years after onset. Many patients with FRDA have nystagmus. Advanced technology shows complex oculomotor disturbances, among which abnormal saccades and square wave jerks are characteristic [25,26]. Optic atrophy is uncommon but some patients become blind. Clinically apparent hearing loss is relatively uncommon [16,19,23] though it may be severe enough to cause deafness in approximately 1% of patients [16]. Despite normal pure-tone audiometry, patients with FRDA may have problems with speech perception [27]. A suitable term is “auditory neuropathy/dyssynchrony” [27]. There is also evidence of vestibular dysfunction [26].
Magnetic resonance imaging (MRI) in the diagnosis of FRDA was used before gene testing was available [28–31], and thinning of the cervical spinal cord was a consistent observation. Atrophy of cerebellum and brain stem was more variable, but in more recent studies [32,33], the images clearly disclosed degeneration of the superior cerebellar peduncles. This finding is not surprising because the superior cerebellar peduncles contain most of the efferent fibers of the DN. At high magnetic field strengths, iron-related hypointensity of the DN on T2-weighted images may be a potential bio-marker of FRDA [34,35].
Harding and Hewer [36] reported electrocardiographic abnormalities in 86 of 114 patients (75%), which is comparable to the frequency in a group of 32 Canadian patients [23]. Dürr et al. [19] listed cardiomyopathy in 63%, and their study included echocardiography. In the author’s autopsy series of 30 patients (Table 2), cardiomyopathy was the immediate or contributory cause of death in 25 (83.3%). Cardiomyopathy is less frequent in FRDA patients with short GAA trinucleotide expansions in one allele (Refs. [19,20] and cases FRDA 3 and FRDA 23 in Table 2). Isnard et al. [37] showed a significant positive correlation between thickness of the left ventricular wall by echocardiography and GAA trinucleotide repeat expansion. Electrocardiography, echocardiography, and, more recently, MRI and 31P spectroscopy of the heart have contributed a wealth of information on the cardiomyopathy in FRDA [36–43], highlighting the importance of heart disease in FRDA. The observations provide a rationale for antioxidant and chelation therapy, and, in selected patients, cardiac transplantation [44,45]. Cardiomyopathy of FRDA is not always concentric and hypertrophic. In some cases, the lesion is a dilated cardiomyopathy. Mural thrombi in the left cardiac ventricle are common, and some patients with FRDA have embolic strokes.
Diabetes mellitus occurs in 8–32% of FRDA patients [16,19,20,22–24], and most ultimately require insulin. Filla et al. [20] found that 8 patients from among 63 with FRDA (12.6%) had diabetes mellitus and that the mean of their GAA trinucleotide repeat lengths on the shorter allele was significantly longer (956) than those in the non-diabetic members of the group (698). This correlation was not significant in other studies that included the analysis of the frataxin gene [19,24]. Diabetes also occurs in FRDA patients who are compound heterozygotes [22].
Scoliosis is extremely common in FRDA (60–79%) [16,19,20,22–24] and is clearly progressive. Harding [16] listed scoliosis as a first manifestation of FRDA in 5.2%. Progressive deformity of the chest causes the patient great problems with sitting comfortably. Scoliosis surgery is a major procedure involving fusion over multiple levels [46,47]. Anesthetic risk is high, and the benefit is only temporary. Bracing is ineffective [46,47]. Pes cavus is almost as common in FRDA as scoliosis. Some patients may gain functional benefit from botulinum toxin injections of the gastrocnemius muscles or Achilles tendon lengthening when their equinovarus deformity impairs standing and walking [48].
4. Gross pathology
All observers agree that DRG in FRDA are smaller than normal and may be difficult to recognize during dissection. Dorsal spinal roots are thin and gray (Fig. 2). Diameters of the spinal cord are reduced at all levels, but thinning is especially evident in the thoracic region. Transverse diameters of the thoracic spinal cord are often less than 10 mm (Fig. 2). Systematic measurements of cross-sectional areas have confirmed this overall reduction: The mean cross-sectional area of 12 typical juvenile-onset FRDA cases was 20.5±5.3 mm2 (mean±standard deviation). In 6 normal spinal cords, the comparable measurement was 30.4±5.6 mm2 [49]. Transverse slices of the spinal cord also reveal smallness and gray discoloration of the dorsal columns. At the thoracic level, the lesion involves gracile and cuneate fasciculi to the same extent, but in cervical segments, degeneration is more prominent in the gracile fasciculi. The gelatinous appearance is not invariably wedge-shaped but at times appears like a cap over the dorsal columns. Fiber loss in the anterolateral fields corresponding to spinocerebellar and corticospinal tracts may also be visible to the naked eye.
Fig. 2.
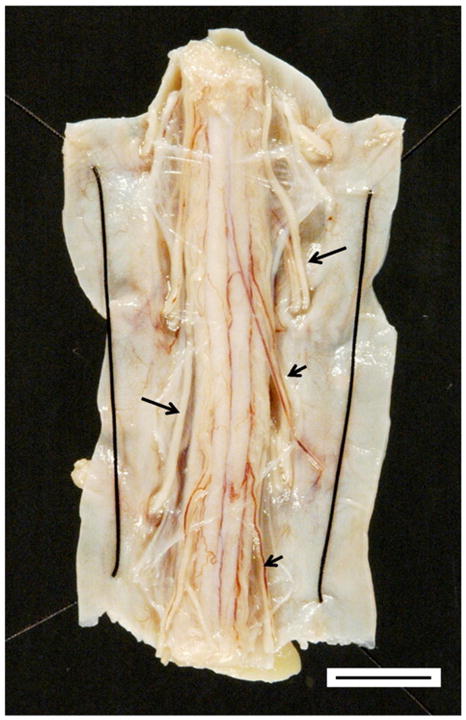
The thoracic spinal cord in FRDA. The transverse diameter of the spinal cord is less than 1 cm (bar). This view of the dorsal surface of the spinal cord shows thick white anterior roots (long arrows) that stand out in contrast to thin dorsal roots (short arrows).
Atrophy of the DN and its efferent fibers stand out against the normal cerebellar cortex and white matter. The collapse of the DN is especially apparent after a macroscopic iron stain (Fig. 3). Blue reaction product does not coincide with the gray matter ribbon of the nucleus but is distributed more diffusely in the afferent and efferent white matter (Fig. 3). Spatz [50] illustrated this peculiar localization of iron in his 1922 color illustration of the normal DN. The current author republished this figure more recently in a review of the history of brain iron [51].
Fig. 3.
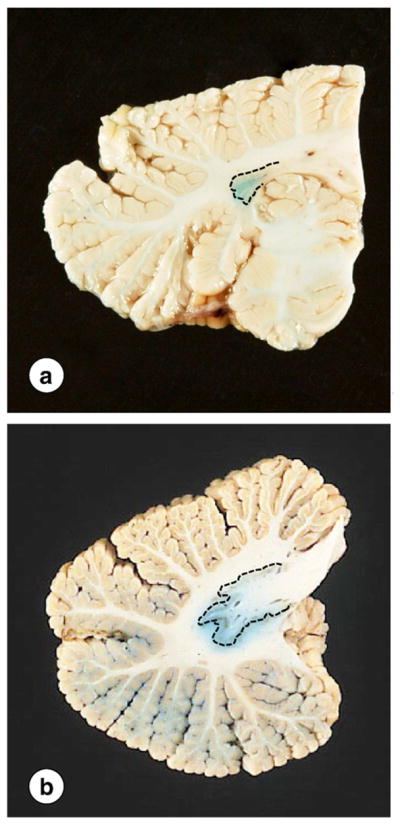
Gross appearance of the DN in FRDA. (a) FRDA; (b) normal control. The interrupted lines indicate the approximate location of the DN gray matter. The small size of the nucleus in FRDA is particularly apparent on a macrostain for iron (a). The normal DN shows the typical meandering gray matter ribbon (b). Iron reaction product does not co-localize with the gray matter but extends into the white matter of hilus and fleece.
Fig. 4 illustrates the gross pathology of the heart in two patients with hypertrophic cardiomyopathy. Heart weights are often very high (600–800 g). Normal weights for adult men and women are 365±71 g and 312±78 g, respectively (mean±standard deviation) [52]. The thickness of left ventricular wall and interventricular septum often exceeds 2 cm (normal, less than 1.5 cm). The right ventricular wall is also thickened above the average normal of 0.5 cm (Fig. 4a). The heart illustrated in Fig. 4b shows dilatation of the right ventricle. The “marble”-like discoloration of the myocardium described by Friedreich [6] is especially apparent in the heart shown in Fig. 4a.
Fig. 4.

Gross appearance of the heart in FRDA. (a) Concentric cardiac hypertrophy and discoloration of the myocardium. In this case, the ventricles are narrowed. (b) Cardiac hypertrophy affecting only the left ventricular wall and interventricular septum. The right ventricle is dilated. Bars, 1 cm.
5. Histopathology
Friedreich’s ataxia causes numerous changes in the DRG (Ref. [53] and Figs. 5 and 6). The lesion affects the entire DGR but is most prominent in subcapsular regions (Fig. 5). Cell stains reveal an overall reduction in the size of ganglion cells, as previously quantified by Inoue et al. [54]. Very large neurons are absent (Fig. 5a). Clusters of nuclei represent “residual nodules” (of Nageotte) (Fig. 5a) and indicate an invasion-like entry of satellite cells into the cytoplasm of neurons. Cytoskeletal stains, such as for class-III-β-tubulin (Fig. 5b and e), support progressive destruction of neuronal cytoplasm (Fig. 5b). In a normal DRG, satellite cells form a relatively thin layer around nerve cells that can be shown especially well by immunocytochemistry of S100α protein (Fig. 5f). This rim of cells is greatly thickened in FRDA (Fig. 5c), and the cells constituting residual nodules remain strongly reactive with anti-S100α. This observation allows the conclusion that the nodules derive from satellite cells.
Fig. 5.

Dorsal root ganglion in FRDA. (a)–(c), FRDA; (d)–(f), normal control. The hematoxylin-and-eosin stain (a; d) shows the overall size reduction of subcapsular nerve cells in the DRG and several residual nodules. The arrow (a) points to one nodule of Nageotte. Smaller size and frank destruction of neuronal cytoplasm are especially apparent after immunostaining for class-III-β-tubulin (arrows in [b]). Immunocytochemistry for S100α shows more prominent reaction product in satellite cells about smaller neurons in FRDA (c). Residual nodules are also S100α-positive (arrows in [c]). Bars: 50 μm.
Fig. 6.
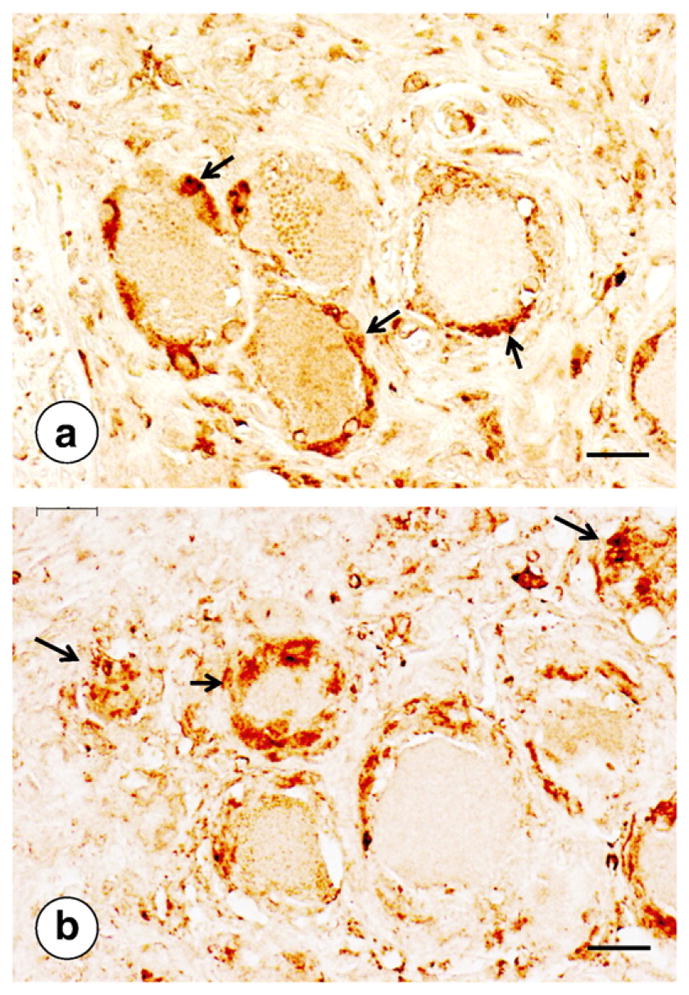
Ferritin in DRG of FRDA and a normal control. (a) Normal; (b) FRDA. In the normal state, immunocytochemistry of ferritin labels a few cells in the satellite layer around neurons (arrows in [a]). In FRDA, ferritin is present in a thicker layer of satellite cells (short arrow in [b]) and persists in residual nodules after complete neuronal atrophy (longer arrows in [b]). Bars, 20 μm.
Fig. 6 illustrates increased ferritin immunoreactivity in satellite cells of a DRG in FRDA. In the normal state, ferritin reaction product occurs in some but not all satellite cells, and the layer of this protein around neurons is relatively thin (Fig. 6a). In FRDA, the rim of ferritin-reactive satellite cells is thicker, and reaction product persists in residual nodules (Fig. 6b). Koeppen et al. [53] have interpreted the increased ferritin reaction product as indirect evidence of regional iron excess in DRG of patients with FRDA. A second iron-responsive protein, ferroportin, undergoes similar up-regulation in satellite cells in FRDA [53]. In contrast to ferritin, however, ferroportin is also present in the cytoplasm of DRG neurons. The disease causes progressive loss from this location and dorsal spinal roots [53] (not illustrated).
Fig. 7 compares a dorsal root of the lumbar spinal cord in FRDA and a normal control. Axonal density is not visibly different (Fig. 7a and c), but the lack of large myelinated fibers in FRDA is readily apparent (Fig. 7b). Computer-assisted fiber counting confirmed an equal number of axons/unit area in the dorsal roots of FRDA patients when compared to normal controls [53]. Surprisingly, the percentage of myelinated fibers was significantly higher in dorsal roots of FRDA (66%) than in controls (55%) [53]. Thinning of myelinated fibers on histograms was matched by an increase of thinner axons [53]. There was no difference in fiber density or thickness in ventral roots [53] (not illustrated).
Fig. 7.

Dorsal spinal roots in FRDA. (a)–(b), FRDA; (c)–(d) normal control; (a) and (c), immunocytochemistry of phosphorylated neurofilament protein to visualize axons; (b) and (d), immunostain of myelin basic protein. Axons are present in normal abundance in FRDA (a) when compared to a normal control (c). In contrast, myelin sheaths in FRDA are much thinner (b) than in the normal control (d). Bars, 20 μm.
Fig. 8 displays the striking smallness of the spinal cord in FRDA and loss of myelin and axons in dorsal columns and spinocerebellar and corticospinal tracts. The dorsal nuclei of Clarke often show total loss of large round nerve cells (Fig. 8c) that normally contain abundant peripherally placed chromatin (Fig. 8e).
Fig. 8.
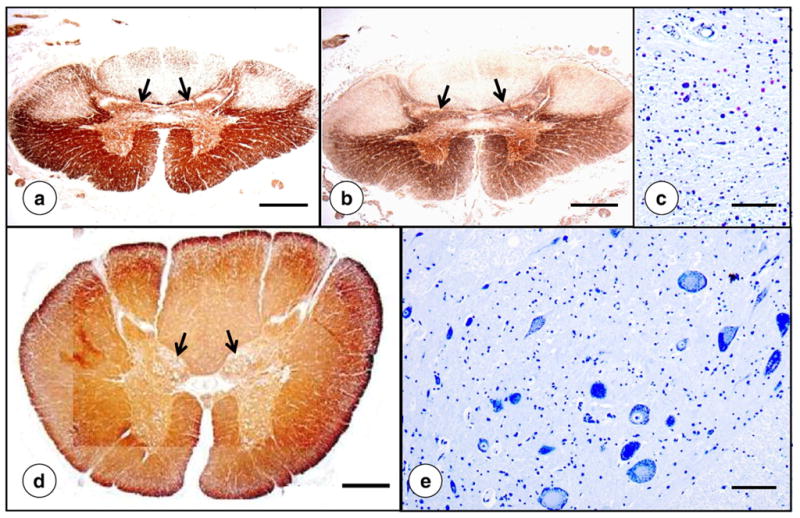
Upper lumbar spinal cord in FRDA. (a)–(c), FRDA; (d)–(e), normal control. (a) and (d), immunostain of myelin basic protein; (b) immunostain of phosphorylated neurofilament protein in axons; (c) and (e), Cresyl Violet. The overall area of the spinal cord in FRDA ([a] and [b]) is greatly reduced in comparison with a normal control (d). In FRDA, the nucleus dorsalis of Clarke (c) is devoid of large round chromatin-rich neurons. Panel (e) illustrates the normal nucleus. Atrophy of the dorsal nuclei in FRDA is also apparent on low-power magnification of the spinal cord (arrows in [a] and [b]). In the normal spinal cord, dorsal nuclei show a distinct “bulge” (arrows in [d]). Bars: (a), (b), and (d), 1 mm; (c) and (d), 100 μm.
The lesion of the DN in FRDA (Fig. 9) is remarkable because it resembles spinocerebellar ataxia type 3 (SCA-3; Machado–Joseph disease) [21]. Large neurons disappear (Fig. 9a) while many small neurons remain. In typical cases of FRDA, immunostaining with an antibody to glutamic acid decarboxylase (GAD) shows severe loss of γ-aminobutyric acid (GABA)-containing terminals (Fig. 9c), implying impaired corticonuclear connections. Immunostaining with anti-GAD also reveals grumose degeneration (inset in Fig. 9b). It is likely that all small neurons that remain in the DN of FRDA patients are GABA-ergic because they display GAD-reaction product in their cytoplasm (arrows in Fig. 9b). They are the parent nerve cells of the dentato-olivary tracts that assure the integrity of the inferior olivary nuclei. Similar to SCA-3, the severe lesion in the DN of FRDA does not cause transsynaptic degeneration in olivary neurons.
Fig. 9.

The dentate nucleus in FRDA. (a) and (b), FRDA; (c) and (d), normal control. (a) and (c), immunocytochemistry of neuron-specific enolase (NSE); (b), inset in (b), and (d), glutamic acid decarboxylase (GAD). NSE-reaction product shows severe loss of large neurons while small neurons remain (arrows in [a]). GAD reaction product shows loss of corticonuclear terminals in the dentate nucleus of an FRDA patient (b) and grumose degeneration (inset in [b]). Small neurons display GAD-reaction product in their cytoplasm (arrows in [b]). In the normal dentate nucleus, the great abundance of GAD-positive terminals obscures the reactive cytoplasm in small neurons though one small GABA-ergic is visible (arrow in [d]). Bars: (a) and (c), 100 μm; (cb) and (d), 50 μm; inset in (b), 25 μm.
Fig. 10 illustrates the status of frataxin in the DN of patients with FRDA. The protein is susceptible to autolysis, but rapidly fixed specimens of the cerebellum are suitable for immunocytochemistry with anti-frataxin. Frataxin immunofluorescence shows punctate reaction product in areas known to be rich in mitochondria, namely, neuronal cytoplasm and synaptic terminals (Fig. 10d–f). In FRDA, double visualization of synaptophysin and frataxin shows severe cluster-like modification of corticonuclear terminals and lack of frataxin reaction product (Fig. 10a–c). Immunostaining of mitochondria (not illustrated) reveals abundant reaction product in terminals about the nerve cells of the dentate nucleus and in grumose degeneration. This observation suggests that mitochondria remain while they become frataxin-deficient.
Fig. 10.

The dentate nucleus in FRDA. (a)-(c), FRDA; (d)-(f), normal control. Double-label confocal immunofluorescence of synaptophysin ([a] and [d]; yellow-green; fluorescein isothiocyanate) and frataxin ([b] and [e]; red; Quantum dot 655); (c) and (f), merged images. In FRDA ([a]-[c]), synaptic terminals are modified to clusters of excessively large endings, representing grumose degeneration. Frataxin reaction product (b) is very sparse, and the merged images (c) show no co-localization of synaptophysin and frataxin. In the normal state ([d]–[f]), frataxin fluorescence is abundant in the cytoplasm of a nerve cell and in the rim of axosomatic terminals (e). The interrupted line in the lower panel ([d]–[f]) indicates the neuronal nucleus. Bars: 25 μm.
Fig. 11 shows the lesion of sural nerves in FRDA. The main abnormality is the paucity of myelinated fibers and absence of large fibers (Fig. 11b). Systematic counts of axons revealed normal fiber density though only 11% of axons were myelinated (controls: 36%) [55]. Histograms of myelin sheaths confirmed the lack of larger myelin sheaths and a shift to thinner axons [55]. Lack of myelin and tightly spaced groups of unmyelinated fibers are visible on electron micrographs of the sural nerve affected by FRDA (Fig. 11c; and Ref. [55]).
Fig. 11.

The sural nerve in FRDA (autopsy specimens). (a)–(c), FRDA; (d)–(f), normal control. (a) and (e), immunostain of phosphorylated neurofilament protein; (b) and (e), immunostain of myelin basic protein; (c) and (f) electron microscopy. The main abnormality in FRDA is lack of myelin sheaths, especially those of larger diameter (b). Axons remain numerous in FRDA (a) though their overall size appears smaller in comparison with the normal state (e). Paucity of thicker myelin sheaths and greater abundance of unmyelinated fibers (c) are also apparent at the electron microscope level in FRDA (c) when compared to the normal state (f). umf, clusters of unmyelinated fibers. Bars: (a)–(b) and (d)–(e), 20 μm; (c) and (f), 5 μm.
Fig. 12 illustrates the histopathology of the heart in FRDA. The transverse section of the interventricular septum (Fig. 12a) shows abnormal fiber size variation, fiber splitting, abnormal nuclei, and an excess of endomysial connective tissue. Iron stains reveal collections of tiny reactive inclusions in a small percentage of cardiomyocytes that are arranged in parallel with myofibrils (Fig. 12b). It is likely that these collections contain cytosolic (Fig. 12c) and mitochondrial ferritin (Fig. 12d), as reported before [56]. Ultrastructural examination of FRDA heart muscle after enhancement of ferritin by bismuth subnitrate [57] shows electron-dense inclusions in mitochondria (Fig. 12e and f). Due to their location, they may represent accumulation of mitochondrial rather than cytosolic ferritin [56]. They also differ from lipofuscin. In more severe cases of cardiomyopathy, sections of heart also show fiber necrosis and an inflammatory reaction [56,58,59]. Inflammation may be quite severe (Fig. 4 in Ref. [59]).
Fig. 12.

The histopathology of the heart in FRDA. (a) Hematoxylin and eosin; (b) iron histochemistry; (c) immunocytochemistry of cytosolic ferritin; (d) immunocytochemistry of mitochondrial ferritin; (e) and (f) electron microscopy after enhancement of ferritin by bismuth subnitrate [55]. For ultrastructural study, tissue samples were fixed by paraformaldehyde–glutaraldehyde mixtures and osmium tetroxide but other contrasting agents, such as lead citrate and uranyl acetate, were omitted. A cross-section of heart muscle (a) displays highly variable fiber sizes, excessive endomysium, and bizarre sarcoplasmic nuclei. The iron stain in (b) shows a single fiber with a collection of reactive granules. They are distributed in row-like manner, seemingly in parallel with muscle fibrils. Frequency and distribution of cytosolic (c) and mitochondrial ferritin (d) resemble the iron-containing granules in (b). The ultrastructural images were obtained from the heart of an FRDA patient with numerous iron deposits (as in [b]). The electron dense inclusions are thought to represent iron-laden mitochondria rather than lipofuscin granules. The arrow in (e) indicates partial involvement of a mitochondrion. Bars: (a)–(b), 50 μm; (c)–(d), 20 μm; (e)–(f), 1 μm.
Fig. 13 is a display of pancreatic β-cells in a diabetic (Fig. 13a–b) and a non-diabetic patient with FRDA (Fig. 13c–d). Synaptophysin immunostaining is suitable to show lack and progressive destruction of the islets of Langerhans in the diabetic patient [60] (Fig. 13a–b). The section of pancreas of the non-diabetic FRDA patient displays many more and larger islets of β-cells (Fig. 13c–d). In FRDA, synaptophysin-positive islets lose their sharp demarcation, and β-cells appear to “fade” into the surrounding exocrine pancreas (Fig. 13b).
Fig. 13.

The endocrine pancreas in FRDA. (a)–(b) Diabetic patient; (c)–(d) non-diabetic patient; synaptophysin immunocytochemistry. In the diabetic patient, only few small islets remain (arrows in [a]). The interrupted line in (a) shows an islet that has lost most of its synaptophysin immunoreactivity. Normal islets are over 200 μm in diameter (c). Panel (b) suggests that in diabetic FRDA patients, β-cells lose their synaptophysin immunoreactivity as the disease progresses. The arrows in (c) and (d) show invaginations of normal exocrine pancreatic tissue into islets. They are synaptophysin-negative and must be distinguished from non-reactive cells in the diabetic patient (a). Bars, (a) and (c), 100 μm; (b) and (d), 20 μm.
6. Pathogenesis of FRDA
Classification of FRDA as a “neurodegenerative disease” implies that affected patients are normal at birth and for a period thereafter until the “age of onset” is established by the clinical phenotype. All patients with FRDA have at least some frataxin, but tissues are not equally vulnerable. The concept of frataxin deficiency in FRDA, rather than the total absence of the protein, received a boost from a murine model [61]. Total failure of frataxin biosynthesis causes embryonic lethality, and there is no accumulation of iron in the products of conception [61]. Some neuropathological lesions in the human condition are likely due to hypoplasia and atrophy. The author and his collaborators previously summarized the evidence for a combined process in DRG [53] and sensory peripheral nerves [55]. In DRG, the reduction in average neuronal size [49,54] is probably not the result of selective atrophy of larger neurons. Satellite cells invade nerve cells of large and small size (Fig. 5b). Residual nodules may be considered an active process that is superimposed on DRG hypoplasia. Morral et al. [55] presented evidence for a similar combination in sural nerves of patients with FRDA. It is likely that the spinal cord is thinner than normal from birth [21]. The occurrence of scoliosis as a first clinical manifestation of FRDA [16] and narrowing of the cervical spinal canal on plain X-ray films [62] support the interpretation that the spinal cord never grows to normal size. Little information exists about the progression of the disease in the DN. In cases coming to autopsy after an average disease duration of 26 years (Table 2 and Ref. [49]), loss of neurons, synaptic modification, and grumose degeneration are very similar. Rarely, cardiomyopathy causes death before the neurological phenotype is fully developed [49]. In a patient with disease duration of only 4 years and fatal cardiomyopathy, the DN was almost normal, and it may be suggested that the lesion in more typical cases of FRDA is purely degenerative.
Iron accumulates in a small percentage of cardiomyocytes in FRDA and in yeast lacking the frataxin homologue Yfh1p [63]. The relationship to iron–sulfur cluster deficiency is unknown, and cytoplasmic iron depletion in vivo remains speculative. Iron-rich granules in the heart occur early and do not correlate well with other histopathological features of FRDA cardiomyopathy [56]. The deposits are not progressive as shown in a comparison between biopsy and autopsy tissues from the heart of the same patient (17-year-interval), implying turnover of the affected cardiomyocytes [56]. In support of this interpretation, iron granules and ferritin also occur in endomysial phagocytes of FRDA heart near damaged fibers [56]. Total iron and holoferritin in FRDA hearts do not increase, which differs greatly from cardiac hemochromatosis. Though lack of high-energy phosphates in the heart is beyond dispute, the role of iron in the in vivo synthesis of tissue-damaging oxygen species is still unproved.
Mitochondrial ferritin is a distinct species of an iron-carrying protein that is not widely expressed in mammalian tissues [64]. Its presence in FRDA heart [56] may be important because it may be protective against oxidative damage. Mitochondrial ferritin shows a distinct benefit in cultured fibroblasts of patients with FRDA [65].
In contrast to the cardiac and nervous system lesions, the pathogenesis of diabetes mellitus in FRDA appears rather straightforward. Selective frataxin-depletion of β-cells in a murine model of FRDA causes diabetes mellitus [66]. Double-label immunofluorescence revealed persistence of glucagon in the rims of damaged islets where frataxin expression remained [66]. Ristow et al. [66] also showed strong evidence that the destruction of β-cells is due to an excess of reactive oxygen species and apoptosis.
7. Functions of frataxin and lessons learnt from mouse models of FRDA
Mature frataxin is a 14.2 kDa mitochondrial protein (see review in Ref. [67]) that derives from a cytosolic 23 kDa precursor (210 amino acids) through enzymatic cleavage [68,69]. The protein retains amino acids 81–210 (130 residues) though longer isoforms have been described by several authors. Beyond participation in the biogenesis of iron–sulfur clusters [14,15], frataxin may have several other functions [70,71]. “Iron-trafficking” in mitochondria [71] is of special interest in FRDA research. Routine iron stains show reactive granules only in the heart while the evidence for iron dysmetabolism in DN and DRG is indirect: Light chain ferritin biosynthesis increases in the DN [72], and immunocytochemistry of the protein shows a shift from juxtaneuronal oligodendroglia in the DN to microglia [72]. A recent technology of metal-mapping in tissues, X-ray fluorescence, reveals a net accumulation of iron in the DRG of some cases of FRDA [53], matching the more prominent ferritin reaction product in satellite cells ([51] and Fig. 6).
Gakh et al. [73] recently presented evidence that one isoform of FRDA (amino acids 42–210) was preferentially reduced in cerebellar tissues of two patients with FRDA when compared to mature frataxin (amino acids 81–210). Additional insight into the function of frataxin comes from heme biosynthesis by ferrochelatase [74]. Frataxin is needed to deliver iron to the enzyme for introduction into the porphyrin ring [74]. In this process, the protein is a classic iron chaperone but patients with FRDA do not have anemia. It is uncertain whether ferrochelatase is involved in the damaging effect of frataxin deficiency on nervous and cardiac tissues, or the β-cells of the pancreas.
After the discovery that total knockout of the frataxin gene in a mouse model of FRDA is incompatible with postnatal life [61], several genetically engineered murine models with postnatal survival have become available [75–78]. In all, the purpose was to reduce frataxin, either globally [76,78] or selectively by targeting tissues that are known to bear the brunt of human FRDA [75,77].
The introduction of homozygous human GAA expansions (230 repeats) into mice did not cause a sufficient reduction of frataxin (to 75%) to produce a functional deficit or tissue changes that resembled human FRDA. Deletion of one allele for the purpose of generating frda−/230GAA animals reduced tissue frataxin levels further (to 25–35%), which matches the human condition. Nevertheless, the animals remained well. In a different approach involving GAA trinucleotide repeat expansions, Al-Mahdawi et al. [78] generated mice without endogenous frataxin that were “rescued” by the introduction of expanded human GAA trinucleotide repeats in the correct genomic context. The human frataxin levels in the animals were sufficiently lowered to cause neurobehavioral and morphological changes that resembled FRDA. The cardiac lesion included the accumulation of iron but no fibrosis. Dorsal root ganglia displayed neuronal vacuolation that were similar to the lesion in a model with frataxin gene disruption targeted to the nervous system [75,77], but lesions of the central nervous system were absent. Nodules of Nageotte in DRG do not seem to occur in any murine model. Simon et al. [77] observed defects in Purkinje cell dendrites, neuronal loss in the granular layer, degeneration of neurons in the nerve cells of the dorsal and ventral horns of the spinal cord, and in the nerve cells of the dorsal nuclei of Clarke. These lesions were present in addition to vacuolation of neurons in DRG.
Mouse models of FRDA are of obvious importance in the systematic analysis of the multiple roles of frataxin in iron metabolism. Whitnall et al. [79] and Huang et al. [80] reached the conclusion that in the murine model with frataxin deficiency in cardiac muscle [75], mitochondrial iron excess is associated with depletion of cytosolic iron. Some of the observations are at variance with the analysis of iron-handling proteins in the hearts of patients with FRDA [56]. Nevertheless, FRDA models convey the advantage of being free of post mortem autolysis. They are also excellent research avenues for the study of emerging FRDA therapy, such as the promising inhibition of histone deacetylase [81].
The observation that all patients with FRDA have at least some frataxin has generated much interest in the possible reversal of the transcriptional block that is brought about by the overly long GAA trinucleotide repeats [2]. Frataxin gene transcription in FRDA is subject to epigenetic effects such as gene silencing by the formation of triplex deoxyribonucleic acid, deoxyribonucleic acid-ribonucleic acid hybrids, and heterochromatin (review in Ref. [82]). Histone deacetylation is a critical step in the formation of heterochromatin, and inhibitors of the enzyme may improve frataxin deficiency in patients with FRDA [83]. Koeppen et al. [49] recently reported that patients with late onset and long survival have significantly shorter GAA trinucleotide repeat expansions on both alleles. In 5 patients with a mean age of onset of 28±13 years and mean disease duration of 47±11 years, the expansions were 456±238 and 283±201. In 14 patients with a mean age of onset of 10±5 years and mean disease duration of 33±11 years, the triplet repeats were 808±201 and 626±253. A plausible explanation is a lower degree of gene silencing by more limited heterochromatin formation [82]. It has become apparent that the clinical FRDA phenotype is not solely controlled by the shorter allele.
Acknowledgments
The author receives financial support from Friedreich’s Ataxia Research Alliance, National Ataxia Foundation, National Institutes of Health, and Neurochemical Research, Inc. The illustrations derive from work in the laboratories of the Veterans Affairs Medical Center in Albany, N.Y., USA. Dr. Benjamin B. Gelman, Galveston, TX, USA provided the electron microscopic illustrations (Fig. 11c and f).
References
- 1.Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–7. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 2.Grabczyk E, Usdin K. The GAA•TTC triplet repeat expanded in Friedreich’s ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucl Acids Res. 2000;28:2815–22. doi: 10.1093/nar/28.14.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cossée M, Schmitt M, Campuzano V, Reutenauer L, Moutou C, Mandel JL, Koenig M. Evolution of the Friedreich’s ataxia trinucleotide repeat expansion: founder effect and premutations. Proc Natl Acad Sci USA. 1997;94:7452–7. doi: 10.1073/pnas.94.14.7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delatycki M, Williamson R, Forrest SM. Friedreich ataxia: an overview. J Med Genet. 2000;3:1–8. doi: 10.1136/jmg.37.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Labuda M, Labuda D, Miranda C, Poirier J, Soong BW, Barucha NE, et al. Unique origin and specific ethnic distribution of the Friedreich ataxia GAA expansion. Neurology. 2000;54:2322–4. doi: 10.1212/wnl.54.12.2322. [DOI] [PubMed] [Google Scholar]
- 6.Friedreich N. Ueber degenerative Atrophie der spinalen Hinterstränge. Virchows Arch Pathol Anat Physiol Klin Med. 1863;26:391–419. [Google Scholar]
- 7.Friedreich N. Ueber degenerative Atrophie der spinalen Hinterstränge. Virchows Arch Pathol Anat Physiol Klin Med. 1863;26:433–59. [Google Scholar]
- 8.Friedreich N. Ueber degenerative Atrophie der spinalen Hinterstränge. Virchows Arch Pathol Anat Physiol Klin Med. 1863;27:1–26. [Google Scholar]
- 9.Friedreich N. Ueber Ataxie mit besonderer Berücksichtigung der hereditären Formen. Virchows Arch Pathol Anat Physiol Klin Med. 1876;68:145–245. [Google Scholar]
- 10.Friedreich N. Ueber Ataxie mit besonderer Berücksichtigung der hereditären Formen. Nachtrag Virchows Arch Pathol Anat Physiol Klin Med. 1877;70:140–52. [Google Scholar]
- 11.Mott FW. Case of Friedreich’s disease, with autopsy and systematic microscopical examination of the nervous system. Arch Neurol Psychiat Lond. 1907;3:180–200. [Google Scholar]
- 12.Urich H, Norman RM, Lloyd OC. Suprasegmental lesions in Friedreich’s ataxia. Confin Neurol. 1957;17:360–71. doi: 10.1159/000105212. [DOI] [PubMed] [Google Scholar]
- 13.Lamarche JB, Côté M, Lemieux B. The cardiomyopathy of Friedreich’s ataxia. Morphological observations in 3 cases. Can J Neurol Sci. 1980;7:389–96. doi: 10.1017/s0317167100022927. [DOI] [PubMed] [Google Scholar]
- 14.Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, et al. Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–7. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 15.Mühlenhoff U, Richhardt N, Ristow M, Kispal G, Lill R. The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum Mol Genet. 2002;11:2025–36. doi: 10.1093/hmg/11.17.2025. [DOI] [PubMed] [Google Scholar]
- 16.Harding AE. Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981;104:589–620. doi: 10.1093/brain/104.3.589. [DOI] [PubMed] [Google Scholar]
- 17.Koeppen AH, Dickson AC, Lamarche JB, Robitaille Y. Synapses in the hereditary ataxias. J Neuropathol Exp Neurol. 1999;58:748–64. doi: 10.1097/00005072-199907000-00009. [DOI] [PubMed] [Google Scholar]
- 18.De Michele G, Filla A, Barbieri F, Perretti A, Santoro L, Trombetta L, et al. Late onset recessive ataxia with Friedreich’s disease phenotype. J Neurol Neurosurg Psychiatry. 1989;52:1398–401. doi: 10.1136/jnnp.52.12.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dürr A, Cossée M, Agid Y, Campuzano V, Mignard C, Penet C, et al. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med. 1996;335:1169–75. doi: 10.1056/NEJM199610173351601. [DOI] [PubMed] [Google Scholar]
- 20.Filla A, De Michele G, Cavalcanti F, Pianese L, Monticelli A, Campanella G, et al. The relationship between trinucleotide repeat length and clinical features in Friedreich ataxia. Am J Hum Genet. 1996;59:554–60. [PMC free article] [PubMed] [Google Scholar]
- 21.Koeppen AH. Neuropathology of the inherited ataxias. In: Manto MU, Pandolfo M, editors. The cerebellum and its disorders. Cambridge: Cambridge University Press; 2002. pp. 387–405. [Google Scholar]
- 22.Cossée M, Dürr A, Schmitt M, Dahl N, Trouillas P, Allinson P, et al. Friedreich’s ataxia: point mutations and clinical presentation of compound heterozygotes. Ann Neurol. 1999;45:200–6. doi: 10.1002/1531-8249(199902)45:2<200::aid-ana10>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 23.Andermann E, Remillard GM, Goyer C, Blitzer L, Andermann F, Barbeau A. Genetic and family studies in Friedreich’s ataxia. Can J Neurol Sci. 1976;3:287–301. doi: 10.1017/s0317167100025476. [DOI] [PubMed] [Google Scholar]
- 24.Delatycki MB, Paris DB, Gardner RJ, Nicholson GA, Nassif N, Storey E, et al. Clinical and genetic study of Friedreich ataxia in an Australian population. Am J Med Genet. 1999;87:168–74. doi: 10.1002/(sici)1096-8628(19991119)87:2<168::aid-ajmg8>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 25.Spieker S, Schulz JB, Petersen D, Fetter M, Klockgether T, Dichgans J. Fixation instability and oculomotor abnormalities in Friedreich’s ataxia. J Neurol. 1995;242:517–22. doi: 10.1007/BF00867423. [DOI] [PubMed] [Google Scholar]
- 26.Fahey MC, Cremer PD, Aw ST, Millist L, Todd MJ, White OB, Halmagyi M, Corben LA, Collins V, Churchyard AJ, Tan K, Kowal L, Delatycki MB. Vestibular, saccadic and fixation abnormalities in genetically confirmed Friedreich ataxia. Brain. 2008;131:1035–45. doi: 10.1093/brain/awm323. [DOI] [PubMed] [Google Scholar]
- 27.Rance G, Fava R, Baldock H, Chong A, Barker E, Corben L, et al. Speech perception ability in individuals with Friedreich’s ataxia. Brain. 2008;131:2002–12. doi: 10.1093/brain/awn104. [DOI] [PubMed] [Google Scholar]
- 28.Wessel K, Schroth G, Diener HC, Müller-Forell W, Dichgans J. Significance of MRI-confirmed atrophy of the cranial spinal cord in Friedreich’s ataxia. Eur Arch Psychiatry Neurol Sci. 1989;238:225–30. doi: 10.1007/BF00381470. [DOI] [PubMed] [Google Scholar]
- 29.Wüllner U, Klockgether T, Petersen D, Naegele T, Dichgans J. Magnetic resonance imaging in hereditary and idiopathic ataxia. Neurology. 1993;43:318–25. doi: 10.1212/wnl.43.2.318. [DOI] [PubMed] [Google Scholar]
- 30.Ormerod IE, Harding AE, Miller DH, Johnson G, MacManus D, du Boulay EP, et al. Magnetic resonance imaging in degenerative ataxic disorders. J Neurol Neurosurg Psychiatry. 1994;57:51–7. doi: 10.1136/jnnp.57.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mascalchi M, Salvi F, Piacentrini S, Bartolozzi C. Friedreich’s ataxia: MR findings involving the cervical portion of the spinal cord. Am J Roentgenol. 1994;163:187–91. doi: 10.2214/ajr.163.1.8010211. [DOI] [PubMed] [Google Scholar]
- 32.Pagani E, Ginestroni A, Della Nave R, Agosta F, Salvi F, De Michele G, et al. Assessment of brain white matter fiber bundle atrophy in patients with Friedreich ataxia. Radiology. 2010;255:882–9. doi: 10.1148/radiol.10091742. [DOI] [PubMed] [Google Scholar]
- 33.Akhlaghi H, Corben L, Georgiou-Karistianis N, Bradshaw J, Storey E, Delatycki MB, et al. Superior cerebellar peduncle atrophy in Friedreich’s ataxia correlates with disease symptoms. Cerebellum. 2010 doi: 10.1007/s12311-010-0232-3. [DOI] [PubMed] [Google Scholar]
- 34.Waldvogel D, van Gelderen P, Hallett M. Increased iron in the dentate nucleus of patients with Friedreich’s ataxia. Ann Neurol. 1999;46:123–5. doi: 10.1002/1531-8249(199907)46:1<123::aid-ana19>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 35.Boddaert N, Le Quan Sang KH, Rötig A, Leroy-Willig A, Gallet S, Brunelle F, et al. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 2007;110:401–8. doi: 10.1182/blood-2006-12-065433. [DOI] [PubMed] [Google Scholar]
- 36.Harding AE, Hewer L. The heart in Friedreich’s ataxia: a clinical and electrocardiographic study of 115 patients, with an analysis of serial electrocardiographic changes in 30 cases. Q J Med NS. 1983;52:489–502. [PubMed] [Google Scholar]
- 37.Isnard R, Kalotka H, Dürr A, Cossée M, Schmitt M, Pousset F, et al. Correlation between left ventricular hypertrophy and GAA trinucleotide repeat length in Friedreich’s ataxia. Circulation. 1997;95:2247–9. doi: 10.1161/01.cir.95.9.2247. [DOI] [PubMed] [Google Scholar]
- 38.Alboliras ET, Shub C, Gomez MR, Edwards WD, Hagler DJ, Reeder GS, Seward JB, Tajik AJ. Spectrum of cardiac involvement in Friedreich’s ataxia: clinical, electrocardiographic and echocardiographic observations. Am J Cardiol. 1986;58:518–24. doi: 10.1016/0002-9149(86)90026-3. [DOI] [PubMed] [Google Scholar]
- 39.Giunta A, Maione S, Biagini R, Filla A, De Michele G, Campanella G. Noninvasive assessment of systolic and diastolic function in 50 patients with Friedreich’s ataxia. Cardiology. 1988;75:321–7. doi: 10.1159/000174394. [DOI] [PubMed] [Google Scholar]
- 40.Dutka DP, Donnelly JE, Nihoyannopoulos P, Oakley CM, Nunez DJ. Marked variation in the cardiomyopathy associated with Friedreich’s ataxia. Heart. 1999;81:141–7. doi: 10.1136/hrt.81.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer C, Schmid G, Görlitz S, Ernst M, Wilkens C, Wilhelms I, et al. Cardiomyopathy in Friedreich’s ataxia-assessment by cardiac MRI. Mov Dis. 2007;22:1615–22. doi: 10.1002/mds.21590. [DOI] [PubMed] [Google Scholar]
- 42.Lodi R, Rajagopalan B, Blamire AM, Cooper JM, Davies CH, Bradley JL, et al. Cardiac energenetics are abnormal in Friedreich ataxia patients in the absence of cardiac dysfunction and hypertrophy: an in-vivo 31P magnetic resonance spectroscopy study. Cardiovasc Res. 2001;52:111–9. doi: 10.1016/s0008-6363(01)00357-1. [DOI] [PubMed] [Google Scholar]
- 43.Bunse M, Bit-Avragim N, Rieffiin A, Perrot A, Schmidt O, Kreuz FR, et al. Cardiac energetics correlates to myocardial hypertrophy in Friedreich’s ataxia. Ann Neurol. 2003;53:121–3. doi: 10.1002/ana.10419. [DOI] [PubMed] [Google Scholar]
- 44.Segovia J, Alonso-Pulpon L, Burgos R, Silva L, Serrano S, Castedo E, et al. Heart transplantation in Friedreich’s ataxia and other neuromuscular diseases. J Heart Lung Transplant. 2001;20:169. doi: 10.1016/s1053-2498(00)00313-2. [DOI] [PubMed] [Google Scholar]
- 45.Sedlak L, Chandavimol M, Straatman L. Cardiac transplantation: a temporary solution for Friedreich’s ataxia-induced dilated cardiomyopathy. J Heart Lung Transplant. 2004;23:1304–6. doi: 10.1016/j.healun.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 46.Milbrandt TA, Kunes JR, Karol LA. Friedreich’s ataxia and scoliosis: the experience at two institutions. J Pediatr Orthop. 2008;28:234–8. doi: 10.1097/BPO.0b013e318164fa79. [DOI] [PubMed] [Google Scholar]
- 47.Cady RB, Bobechko WP. Incidence, natural history, and treatment of scoliosis in Friedreich’s ataxia. J Pediatr Orthop. 1984;4:673–6. doi: 10.1097/01241398-198411000-00003. [DOI] [PubMed] [Google Scholar]
- 48.Delatycki MB, Holian A, Corben L, Rawicki HB, Blackburn C, Hoare B, et al. Surgery for equinovarus deformity in Friedreich’s ataxia improves mobility and independence. Clin Orthop Relat Res. 2005;430:138–41. doi: 10.1097/01.blo.0000150339.74041.0e. [DOI] [PubMed] [Google Scholar]
- 49.Koeppen AH, Morral JA, McComb RD, Feustel PJ. The neuropathology of late-onset Friedreich’s ataxia. Cerebellum. 2010 doi: 10.1007/s12311-010-0235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spatz H. Über den Eisennachweis im Gehirn, besonders in Zentren des extrapyramidal-motorischen Systems. Zeitschr Ges Neurol Psychiatry. 1922;77:261–390. [Google Scholar]
- 51.Koeppen AH. The history of iron in the brain. J Neurol Sci. 1995;134(suppl):1–9. doi: 10.1016/0022-510x(95)00202-d. [DOI] [PubMed] [Google Scholar]
- 52.De la Grandmaison GL, Clairand I, Durigon M. Organ weights in 684 adult autopsies: new tables for a Caucasoid population. Forensic Sci Int. 2001;119:149–54. doi: 10.1016/s0379-0738(00)00401-1. [DOI] [PubMed] [Google Scholar]
- 53.Koeppen AH, Morral JA, Davis AN, Qian J, Petrocine SV, Knutson MD, et al. Li D. The dorsal root ganglion in Friedreich’s ataxia. Acta Neuropathol. 2009;118:763–76. doi: 10.1007/s00401-009-0589-x. [DOI] [PubMed] [Google Scholar]
- 54.Inoue K, Hirano A, Hasson J. Friedreich’s ataxia selectively involves the large neurons of the dorsal root ganglia. Trans Am Neurol Assoc. 1997;104:75–6. [PubMed] [Google Scholar]
- 55.Morral JA, Davis AN, Qian J, Gelman BB, Koeppen AH. Pathology and pathogenesis of sensory neuropathy in Friedreich’s ataxia. Acta Neuropathol. 2010;120:97–108. doi: 10.1007/s00401-010-0675-0. [DOI] [PubMed] [Google Scholar]
- 56.Michael S, Petrocine SV, Qian J, Lamarche JB, Knutson MD, Garrick MD, et al. Iron and iron-responsive proteins in the cardiomyopathy of Friedreich’s ataxia. Cerebellum. 2006;5:257–67. doi: 10.1080/14734220600913246. [DOI] [PubMed] [Google Scholar]
- 57.Ainsworth SK, Karnovsky MJ. An ultrastructural staining method for enhancing the size and electron opacity of ferritin in thin sections. J Histochem Cytochem. 1972;20:225–9. doi: 10.1177/20.3.225. [DOI] [PubMed] [Google Scholar]
- 58.Russell DS. Myocarditis in Friedreich’s ataxia. J Path Bact. 1946;63:739–48. doi: 10.1002/path.1700580414. [DOI] [PubMed] [Google Scholar]
- 59.Hewer Rl. The heart in Friedreich’s ataxia. Br Heart J. 1969;31:5–14. doi: 10.1136/hrt.31.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Titlbach M, Chejfec G, Grimelius L, Falkner S. Neuroendocrine background of the pathology of the islets of Langerhans. Exp Clin Endocrinol. 1987;89:242–50. doi: 10.1055/s-0029-1210646. [DOI] [PubMed] [Google Scholar]
- 61.Cossée M, Puccio H, Gansmuller A, Koutnikova H, Dierich A, LeMeur M, et al. Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum Mol Genet. 2000;9:1219–26. doi: 10.1093/hmg/9.8.1219. [DOI] [PubMed] [Google Scholar]
- 62.Vassilopoulos D, Spengos M, Scarpalezos S. Étude radiologique de la colonne vertébrale cervicale dans certaines maladies dégéneratives neurologiques. J Radiol Électrol Méd Nucl. 1977;58:183–6. [PubMed] [Google Scholar]
- 63.Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, et al. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276:1709–12. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 64.Drysdale J, Arosio P, Invernizzi R, Cazzola M, Volz A, Corsi B, et al. Mitochondrial ferritin: a new player in iron metabolism. Blood Cells Mol Dis. 2002;29:376–83. doi: 10.1006/bcmd.2002.0577. [DOI] [PubMed] [Google Scholar]
- 65.Campanella A, Rovelli E, Santambrogio P, Cozzi A, Taroni F, Levi S. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: hypothesis for a protective role in Friedreich ataxia. Hum Mol Gen. 2009;18:1–11. doi: 10.1093/hmg/ddn308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ristow M, Mulder H, Pomplun D, Schulz TJ, Müller-Schmehl K, Krause A, et al. Frataxin deficiency in pancreatic islets causes diabetes due to loss of β cell mass. J Clin Invest. 2003;112:527–34. doi: 10.1172/JCI18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmucker S, Argentini M, Carelle-Calmels N, Martelli A, Puccio H. The in vivo mitochondrial two-step maturation of human frataxin. Hum Mol Gen. 2008;17:3521–31. doi: 10.1093/hmg/ddn244. [DOI] [PubMed] [Google Scholar]
- 68.Koutnikova H, Campuzano V, Koenig M. Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase. Hum Mol Gen. 1998;7:1485–9. doi: 10.1093/hmg/7.9.1485. [DOI] [PubMed] [Google Scholar]
- 69.Condò I, Ventura N, Malisan F, Rufini A, Tomassini B, Testi R. In vivo maturation of human frataxin. Hum Mol Gen. 2007;16:1534–40. doi: 10.1093/hmg/ddm102. [DOI] [PubMed] [Google Scholar]
- 70.Bencze KZ, Kondapalli KC, Cook JD, McMahon S, Millán-Pacheco C, Pastor N, et al. The structure and function of frataxin. Crit Rev Biochem Mol Biol. 2006;41:269–91. doi: 10.1080/10409230600846058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Rahmanto YS, et al. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci USA. 2010;107:10775–82. doi: 10.1073/pnas.0912925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koeppen AH, Michael SC, Knutson MD, Haile DJ, Qian J, Levi S, et al. The dentate nucleus in Friedreich’s ataxia: the role of iron-responsive proteins. Acta Neuropathol. 2007;114:163–73. doi: 10.1007/s00401-007-0220-y. [DOI] [PubMed] [Google Scholar]
- 73.Gakh O, Bedekovics T, Duncan SF, Smith DY, IV, Berkholz DS, Isaya G. Normal and Friedreich ataxia cells express different isoforms of frataxin with complementary roles in iron–sulfur cluster assembly. J Biol Chem. 2010;285:38486–501. doi: 10.1074/jbc.M110.145144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yoon T, Cowan JA. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J Biol Chem. 2004;279:25943–6. doi: 10.1074/jbc.C400107200. [DOI] [PubMed] [Google Scholar]
- 75.Puccio H, Simon D, Cossée M, Criqui-Filipe P, Tiziano F, Melki J, et al. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe–S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181–6. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 76.Miranda CJ, Santos MM, Ohshima K, Smith J, Li L, Bunting M, et al. Frataxin knockin mouse. FEBS Lett. 2002;512:291–7. doi: 10.1016/s0014-5793(02)02251-2. [DOI] [PubMed] [Google Scholar]
- 77.Simon D, Seznec H, Gansmuller A, Carelle N, Weber P, Metzger D, et al. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J Neurosci. 2004;24:1987–95. doi: 10.1523/JNEUROSCI.4549-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Al-Mahdawi S, Pinto RM, Varshney D, Lawrence L, Lowrie MB, Hughes S, Webster Z, Blake J, Cooper JM, King R, Pook MA. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics. 2006;88:580–90. doi: 10.1016/j.ygeno.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Whitnall M, Rahmanto YS, Sutak R, Xu X, Becker EM, Mikhael MR, et al. The MCK mouse heart model of Friedreich’s ataxia: alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation. Proc Natl Acad Sci USA. 2008;105:9757–62. doi: 10.1073/pnas.0804261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang ML, Becker EM, Whitnall M, Rahmanto YS, Ponka P, Richardson DR. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc Natl Acad Sci USA. 2009;106:16381–6. doi: 10.1073/pnas.0906784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rai M, Soragni E, Jenssen K, Burnett R, Herman D, Coppola G, et al. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS ONE. 2008;3:e-1958. doi: 10.1371/journal.pone.0001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wells RD. DNA triplexes and Friedreich’s ataxia. FASEB J. 2008;22:1625–34. doi: 10.1096/fj.07-097857. [DOI] [PubMed] [Google Scholar]
- 83.Gottesfeld JM. Small molecules affecting transcription in Friedreich’s ataxia. Pharmacol Ther. 2007;116:236–48. doi: 10.1016/j.pharmthera.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]