

Abstract
Mutation of BLM helicase causes Blooms syndrome, a disorder associated with genome instability, high levels of sister chromatid exchanges, and cancer predisposition. To study the influence of BLM on double-strand break (DSB) repair in human chromosomes, we stably transfected a normal human cell line with a DNA substrate that contained a thymidine kinase (tk)-neo fusion gene disrupted by the recognition site for endonuclease I-SceI. The substrate also contained a closely linked functional tk gene to serve as a recombination partner for the tk-neo fusion gene. We derived two cell lines each containing a single integrated copy of the DNA substrate. In these cell lines, a DSB was introduced within the tk-neo fusion gene by expression of I-SceI. DSB repair events that occurred via homologous recombination (HR) or nonhomologous end-joining (NHEJ) were recovered by selection for G418-resistant clones. DSB repair was examined under conditions of either normal BLM expression or reduced BLM expression brought about by RNA interference. We report that BLM knockdown in both cell lines specifically increased the frequency of HR events that produced deletions by crossovers or single-strand annealing while leaving the frequency of gene conversions unchanged or reduced. We observed no change in the accuracy of individual HR events and no substantial alteration of the nature of individual NHEJ events when BLM expression was reduced. Our work provides the first direct evidence that BLM influences DSB repair pathway choice in human chromosomes and suggests that BLM deficiency can engender genomic instability by provoking an increased frequency of HR events of a potentially deleterious nature.
Keywords: double-strand break repair, homologous recombination, nonhomologous end-joining, Bloom syndrome, human cell culture
1. Introduction
The genome of a mammalian cell must contend with a variety of types of DNA damage every day [1,2]. One potentially serious threat is the DNA double-strand break (DSB). DSBs can arise either directly or indirectly from exposure to chemical or radiological agents, from metabolism of spontaneous lesions, or at stalled replication forks. It is imperative that DSBs be dealt with efficiently and accurately to avoid detrimental chromosomal rearrangements. There are two broad classes of DSB repair pathways that operate in mammalian cells: homologous recombination (HR), and nonhomologous end-joining (NHEJ) [recently reviewed in 3–6]. HR classically involves the use of a DNA template to maintain or restore genetic information, and so HR is considered to be an accurate means of DSB repair. In contrast, NHEJ is untemplated and is considered to be error-prone since it is often accompanied by deletion of sequence at the site of the healed DSB. Both HR and NHEJ are important pathways and defects in either of these pathways can lead to genomic instability and disease [3,4]. HR is active primarily during late S and G2 phases of the cell cycle, while NHEJ appears to be active throughout the cell cycle [reviewed in 7]. There remain important gaps in our understanding of how the choice is made between the HR and NHEJ pathways for DSB repair, although recent work suggests that regulation of 5' to 3' resection of DNA ends, an early step in HR, may play an important role in pathway choice [8–11].
Although HR is generally considered to be accurate, there are potential hazards involved in the execution of HR that must be avoided in order for genome integrity to be preserved. The use of inappropriate recombination partners, for example, can lead to chromosomal rearrangements such as translocations and deletions. HR events, particularly those resolving as crossovers, can also be detrimental merely by occurring at too high a rate since high levels of such events will likely increase the incidence of loss of heterozygosity (LOH). LOH has been implicated in carcinogenesis by facilitating the loss of functional copies of tumor suppressor genes [12–14]. In addition, a distinct “nonconservative” form of HR, known as single-strand annealing (SSA), does not involve the use of a DNA template and can generate deletions via the splicing together of repeated homologous sequences with the concomitant loss of sequence between the repeats [3,6]. To maintain genome stability, DSB repair in its varied forms must therefore be appropriately regulated qualitatively and quantitatively.
One protein that has gained some attention in recent years as a regulator of HR is the Bloom syndrome helicase (BLM). BLM helicase is a member of the RecQ family of DNA helicases, a highly conserved family of enzymes named for the prototypical RecQ helicase from E. coli. Five DNA helicases of the RecQ family (BLM, WRN, RecQL, RecQ4, and RecQ5), have been identified in humans. Mutations in BLM, WRN and RecQ4 lead to Bloom, Werner, and Rothmund-Thomson syndromes, respectively. Patients with Bloom syndrome (BS) display growth defects, genomic instability (including translocations and quadriradials), and a strong predisposition to a broad spectrum of cancers [15]. One hallmark of BS cells is a greatly elevated frequency of sister chromatid exchanges (SCE) [15,16]. Numerous studies of the role played by BLM helicase in maintaining genome stability have collectively revealed multiple roles for BLM in HR regulation [17–23]. Curiously, both pro-HR and anti-HR functions have been assigned to BLM.
In terms of pro-HR activities, BLM reportedly promotes 5' to 3' strand resection at DSB ends [9,10], a processing step necessary for the initiation of HR. It has also been reported recently [24] that BLM may act early in HR by stimulating the DNA strand exchange activity of Rad51. Annealing of complementary strands of DNA is another activity in BLM's repertoire [25]. BLM has been implicated in promoting regression of stalled replication forks to produce a structural configuration conducive to HR-dependent restart of replication [26,27]. BLM has additionally been shown to catalyze branch migration of Holliday junction recombination intermediates [28]. In conjunction with BLAP75 and topoisomerase IIIα, BLM has been shown to potentially play a role in late steps of HR by catalyzing what has been termed double Holliday junction (DHJ) dissolution [29–31]. In DHJ dissolution, which to date has been demonstrated only in vitro using substrates that model a DHJ recombination intermediate, two Holliday junctions are first converged by BLM and then decatenated by topoisomerase IIIα to separate the two DNA duplexes that are linked by the DHJ recombination intermediate. Dissolution leads exclusively to noncrossover recombination products. An alternative to DHJ dissolution is the process of resolution, an endonucleolytic cleavage of the DHJ that can produce both crossovers and noncrossovers [5]. BLM's role in DHJ dissolution may normally serve to suppress LOH and SCE, and may help explain the elevated levels of SCE seen in cells from BS patients [15,16] and the high levels of LOH and SCE seen in mouse and human cells experimentally depleted of BLM [32–34].
In terms of potential anti-HR activities, BLM can disrupt the D-loop structure formed following the initial strand invasion step of HR [35,36]. Although this activity may serve as a mechanism to avoid the use of inappropriate templates, it is possible that under certain circumstances displacement of the invading strand from the D-loop by BLM may actually promote HR via the synthesis-dependent stand annealing (SDSA) pathway [37–39] which proceeds without the formation of Holliday junction intermediates. In SDSA, one or both 3' ends from a DSB invades an homologous template, and DNA synthesis is primed from the invading end. The newly synthesized strand is then displaced from the template and anneals with complementary sequence from the other end of the DSB. Notably, the SDSA pathway results almost exclusively in noncrossovers [40]. BLM has also been shown to disrupt Rad51 nucleofilaments, but only when the filament is in its inactive ADP-bound form [38]. Thus, BLM may wield a type of “quality control” over HR by disrupting inappropriate events at a very early stage. Finally, the ability of BLM to anneal complementary strands as well as catalyze branch migration of Holliday junctions may implicate BLM not only in the regression of stalled replication forks but also in subsequent reversal of regressed forks. By undoing the so-called “chicken-foot” structure produced by fork regression, BLM may actually prevent recombination events at stalled replication forks [28].
Although it is clear that BLM helicase is an important regulator of HR, more work is needed to sort out BLM's varied and seemingly disparate roles. Perhaps surprisingly, most previous work on human BLM has been carried out using cell-free or plasmid-based systems while comparatively few studies have been carried out on the impact of BLM on DNA transactions as they occur within the chromosomes of living human cells. We have developed an experimental system that allows the study of DSB repair, at the nucleotide level, within the genome of mitotically dividing human cells. Our system involves the integration of a DNA substrate engineered so that DNA transactions may restore function to a selectable marker gene. Expression of endonuclease I-SceI is used to deliver a specific DSB to the genome within the integrated substrate and we can investigate repair of the DSB carried out by HR or NHEJ.
Using our system in conjunction with an RNA interference approach to knock-down expression of BLM, we investigated the role of BLM in DSB repair in cultured human fibroblasts. We report here that knockdown of BLM in human cells altered genomic DSB repair by bringing about a significant shift toward repair via HR that produced deletions either by crossovers or by SSA. Our work implicates BLM as an important participant in DSB pathway choice and provides the first direct evidence that BLM deficiency may contribute to genome instability in somatic human cells by stimulating a specific type of HR event.
2. Materials and methods
2.1. Cell lines and DSB repair substrate
Cell line pLB4/11, described previously [41,42], and cell line pLB4/20 were derived from SV40-immortalized normal human fibroblast cell line GM637 (obtained from the NIGMS) and each line contains a single integrated copy of recombination substrate pLB4 (Fig. 1). pLB4 contains a tk-neo fusion gene disrupted by the insertion of a 22 bp oligonucleotide that contains the 18-bp recognition sequence for yeast endonuclease I-SceI. In addition, pLB4 contains a 2.5 kb HindIII fragment containing a complete functional herpes simplex virus type 1 tk gene. The tk gene and the tk-neo fusion gene are oriented as direct repeats.
Fig. 1.
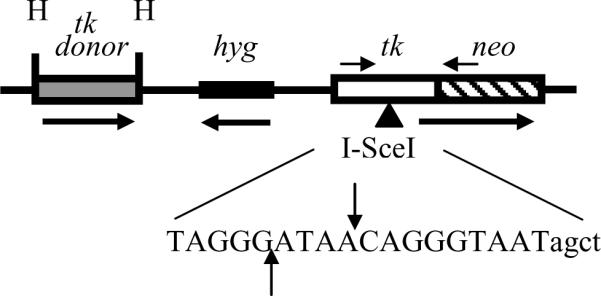
Recombination substrate pLB4. pLB4 contains a tk-neo fusion gene that is disrupted by a 22 bp oligonucleotide containing the 18 bp recognition site for endonuclease I-SceI. The sequence of the 22 bp oligonucleotide is shown, with the 18 bp I-SceI recognition site denoted in uppercase. The sites of staggered cleavage by I-SceI are indicated. pLB4 also contains a HindIII (H) fragment containing a complete tk “donor” gene to serve as a potential recombination partner for the fusion gene. The tk donor sequence is shaded to represent the small degree (<1%) of mismatch with the tk portion of the fusion gene (see Fig. 2). The direction of transcription of both the tk-neo fusion gene and the tk donor is from left to right as drawn. PCR primers AW85 and AW91 are indicated by short horizontal arrows above the fusion gene and are located 1.4 kb apart. A hygromycin resistance gene (hyg) is also contained on pLB4 to allow for stable transfection.
Human fibroblast cell line GM8505 (obtained from NIGMS) was derived from a Bloom Syndrome patient and is null for BLM.
Cells were cultured in alpha-modified minimum essential medium (Sigma) supplemented with 10% fetal bovine serum. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2.
2.2 Knockdown of BLM expression with siRNA
To knock-down BLM expression, we used two siRNAs, Hs-BLM-1-HPsiRNA936 and Hs-BLM-3-1HPsiRNA, which were purchased from Qiagen. These siRNAs are referred to as BLM-1 and BLM-3, respectively. A nonsilencing negative control siRNA was also purchased from Qiagen (catalog number 1027310). siRNAs were transfected into cells using HiPerFect Transfection Reagent (Qiagen) following the standard protocol described by the supplier. siRNA was routinely used at a concentration of 25 nM during transfection.
2.3. Western blotting analysis
Cells were harvested by trypsinization and cell lysates were prepared for Western blotting analysis by standard procedures. BLM antibody ab476 (Abcam) was used as a primary antibody, HRP-conjugated anti-rabbit IgG antibody NB730-H (Novus Biologicals) was used as a secondary antibody. HRP-conjugated tubulin antibody sc-5274 HRP (Santa Cruz Biotechnology) was used to assess protein loading. HRP signal was detected using an ECL Advance Western Blotting Detection Kit (GE Healthcare). ImageJ software [43] was used on scanned images of Western blots to estimate the degree of BLM knockdown.
2.4 Recovery of HR and NHEJ events
Plasmid pCMV3xnls-I-SceI (“pSce”) expresses endonuclease I-SceI and was generously provided by M. Jasin (Sloan Kettering). We transiently transfected pSce into pLB4/11 or pLB4/20 cells to induce a DSB at the I-SceI site within the integrated copy of pLB4. We recovered DSB repair events that occurred by HR or NHEJ under conditions of normal or reduced levels of BLM expression. To accomplish this, cells were first plated at a density of 1.5 × 105 cells per 35 mm dish in normal growth medium. Twenty-four hours later, cells were transfected with BLM-silencing siRNA, nonsilencing control RNA, or left untreated. Twenty-one hours later, cells were fed with fresh medium and after three more hours the cells were transfected a second time with BLM-silencing siRNA, nonsilencing siRNA, or left untreated. After an additional 24 hours, cells were harvested by trypsinization; 2.5 × 105 cells from each dish were removed for electroporation with pSce, while the remaining cells from each dish were plated into a 25 cm2 flask and incubated an additional 24 hours prior to harvesting for Western blotting analysis.
For each electroporation with pSce, 2.5 x 105 cells were mixed with 7.5 μg of pSce in 300 μl of phosphate buffered saline in a 0.4 cm gap cuvette and then exposed to an electric pulse of 700 V, 25 μF using a Bio-Rad Gene Pulser. Cells were allowed to recover by incubation in growth medium for two days. Cells were then plated at a density of 1 × 106 cells per 75 cm2 flask into medium containing 1000 μg/ml G418 to select for HR and NHEJ events that restored function to the tk-neo fusion gene on pLB4. Colonies were recovered after 14 days growth under selection.
2.5. Southern blotting analysis
Genomic DNA samples (8 μg each) were digested with appropriate restriction enzymes and resolved on 0.8% agarose gels. DNA was transferred to nitrocellulose membranes and hybridized with a 32P-labeled tk probe as previously described [44].
2.6. PCR amplification and DNA sequence analysis
A segment of the tk-neo fusion gene spanning the original location of I-SceI site was amplified from 500 ng of genomic DNA isolated from G418R clones using primers AW85 (5'-TAATACGACTCACTATAGGGCCAGCGTCTTGTCATTGGCG-3') and AW91 (5'-GATTTAGGTGACACTATAGCCAAGCGGCCGGAGAACCTG-3'). AW85 is composed of nucleotides 308 to 327 of the coding sequence of the HSV-1 tk gene [numbering according to 45], with a T7 forward universal primer appended to the 5' end of the primer. AW91 is composed of 20 nucleotides from the non-coding strand of the neomycin gene mapping 25 through 44 bp downstream from the neomycin start codon, with an Sp6 primer appended to the 5' end of the primer. PCR was carried out using Ready-To-Go PCR beads (GE Healthcare) and a “touchdown” PCR protocol. The annealing temperature was initially set to 72°C and was progressively decreased in steps of 2°C down to 62°C with two cycles at each temperature. An additional 20 cycles were run at an annealing temperature of 60°C. Prior to sequencing, PCR products were treated with shrimp alkaline phosphatase and exonuclease I (USB). PCR products were then sequenced from a T7 or Sp6 primer at the Environmental Genomics Core Facility at the University of South Carolina.
2.7. Cell cycle analysis
Cell cycle analysis was performed by flow cytometry using a Beckman Coulter FC 500 cytometer and ModFit LT software version 3.1 (Verity Software House, Topsham, ME) essentially as described [46], except that a count of 10,000 cells was used for each analysis.
3. Results
3.1. A system for monitoring intrachromosomal DSB repair events in human cells
The centerpiece of our strategy is substrate pLB4 [41,42] shown in Fig. 1. A single copy of pLB4 was stably integrated in the genome of normal human fibroblast cell line GM637 to independently derive cell lines pLB4/11 and pLB4/20. A seminal feature of pLB4 is a tk-neo fusion gene disrupted by a 22 bp oligonucleotide inserted into the tk portion of the fusion gene. The oligonucleotide contains the 18 bp recognition site for yeast endonuclease I-SceI. Also contained on pLB4 is a complete, functional tk gene. This “donor” fragment serves as a potential recombination partner for the tk-neo fusion gene and shares about 1.7 kb of homology with the tk segment of the fusion gene. A DSB can be introduced into the fusion gene by transiently transfecting pLB4/11 or pLB4/20 cells with the I-SceI expression plasmid pSce. Repair of the DSB can restore function to the neo portion of the tk-neo fusion gene but, importantly, this restoration does not require accurate removal of the 22 bp oligonucleotide inserted into the tk portion since neo expression requires only that an appropriate reading frame be established. Selection for G418R clones following DSB induction therefore allows recovery of error-prone NHEJ as well as accurate HR between the fusion gene and the closely linked functional tk gene. Previous studies have confirmed that both NHEJ and HR events are recoverable in this system, and both noncrossover and crossover (or SSA) type events are found among recovered HR events [41,42]. We cannot always distinguish between crossovers produced via Holliday junction intermediates versus nonconservative products of SSA. For simplicity, we will refer to these latter two types of events collectively as “homology-dependent deletions” (HDD)1 because in either case the recovered product displays a loss of sequence between the tk donor and the tk-neo fusion gene along with an accurate, in-frame joining of the tk donor and fusion gene.
As shown in Fig. 2, there are 13 scattered nucleotide mismatches between the tk portion of the tk-neo fusion gene and the tk donor sequence on pLB4. These mismatches enhance sequence analysis of DSB repair products and enable an unambiguous demonstration of recombination between the tk donor sequence and the fusion gene. The mismatches are sufficiently sparse so as to preserve substantial stretches of perfect homology, including 410- and 231-bp segments of continuous homology. Critically, these stretches of continuous homology exceed the minimal amount of homology required for efficient HR [47].
Fig. 2.

Alignment of tk-neo fusion gene sequence with tk donor sequence from pLB4. Nucleotides 308 to 1622 [numbering according to reference 45] of the tk portion of the tk-neo fusion gene are aligned with the corresponding donor sequence from pLB4. The span of tk sequence shown is the tk portion of the PCR product generated by primers AW85 and AW91 and used in subsequent analyses. Mismatches between the donor and tk-neo fusion gene sequences are highlighted. The 22 bp oligonucleotide containing the 18 bp I-SceI recognition sequence inserted in the tk-neo fusion gene (absent from donor) is depicted in boldface, with the actual I-SceI recognition sequence in uppercase. AluI recognition sites (agct) are underlined and are indicated by downward arrows.
3.2 Knockdown of BLM expression does not alter the recovery of intrachromosomal DSB repair events
Cells from the pLB4/11 and pLB4/20 cell lines were electroporated with pSce to induce a genomic DSB within the integrated copy of pLB4. DSB induction and repair were allowed to occur in conjunction with treatment of cells with two different BLM-silencing siRNAs, control nonsilencing siRNA, or no siRNA. Western blotting (Fig. 3) was used to confirm that knockdown of BLM expression was achieved by transfection with BLM-1 or BLM-3 siRNA. We estimate that as much as 80% knockdown was achieved relative to expression following transfection with nonsilencing siRNA. Treatment of pLB4/11 and pLB4/20 cells with control or BLM-targeting siRNA did not affect the doubling time or plating efficiency of cells, and cell cycle analysis performed on samples collected at the time of transfection with pSce indicated that cell cycle profiles were effectively identical for cells treated with control siRNA, BLM-targeting siRNA, or no siRNA (data not shown).
Fig. 3.
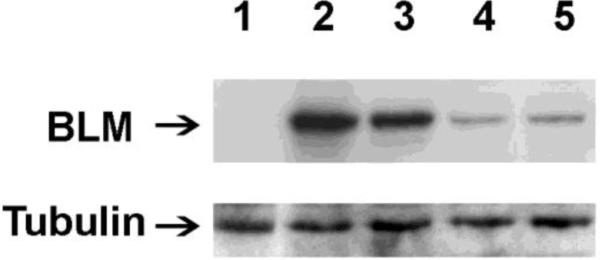
Knockdown of BLM expression. Cell line pLB4/11 was transfected with BLM-1 or BLM-3 siRNA directed against human BLM or with a control nonsilencing siRNA. 48 hours post-transfection, whole cell lysates were prepared and displayed on a Western blot (50 μg protein per lane). BLM and tubulin were visualized with appropriate antibodies. Protein extracts are from: BLM-deficient GM08505 cells (lane1), pLB4/11 cells untreated (lane 2) or treated with control nonsilencing siRNA (lane 3), BLM-1 siRNA (lane 4), or BLM-3 siRNA (lane 5).
Cells were placed under G418 selection in order to recover intrachromosomal DSB repair events that restored function to the tk-neo fusion gene. As indicated in Table 1, knockdown of BLM expression in pLB4/11 or pLB4/20 cells did not alter the recovery of G418R colonies following DSB induction suggesting that the efficiency of DSB repair overall was not sensitive to reduced BLM levels.
Table 1.
Recovery of DSB repair events from pLB4/11 and pLB4/20 cells
| siRNA Useda | Cells Plated Into G418 (105) | G418R Colonies | Colony Frequency (10−3)b |
|---|---|---|---|
| pLB4/11 cells | |||
| None | 7.55 | 3587 | 4.75 |
| Control | 6.23 | 2549 | 4.09 |
| BLM-1 | 7.72 | 3651 | 5.07 |
| BLM-3 | 5.09 | 2312 | 4.54 |
| BLM-1 + BLM-3 | 1.61 | 826 | 5.13 |
| Mockc | 4.45 | 2 | 0.004 |
| pLB4/20 cells | |||
| None | 7.50 | 2640 | 3.52 |
| Control | 7.50 | 2970 | 3.96 |
| BLM-1 | 7.50 | 3071 | 4.10 |
| BLM-3 | 7.82 | 2964 | 3.79 |
| Mockc | 3.00 | 0 | <0.003 |
For each case, with the exception of the BLM-1 + BLM-3 mixture used on pLB4/11 cells, the data presented is a pool from three independent experiments. The data presented for the BLM-1 + BLM-3 mixture is from a single experiment.
Calculated by dividing the number of G418R colonies by the number of cells plated into G418 selection.
In “mock” experiments, cells were treated with no siRNA and were electroporated with phosphate-buffered saline containing no pSce.
3.3 Knockdown of BLM expression in pLB4/11 cells increases both the percentage of DSBs repaired via HR and the percentage of HR events that produce deletions
DSB-induced G418R clones recovered following transfection of cells with pSce could have arisen via NHEJ or HR. Furthermore, both gene conversions (noncrossovers) and HDD events (crossovers or SSA) were potentially recoverable among HR events. To characterize G418R clones recovered from pLB4/11 cells, a combination of several approaches was used.
For each G418R clone analyzed, a fragment of DNA surrounding the original location of the I-SceI site in pLB4 was PCR-amplified from genomic DNA using primers AW85 and AW91 (Fig. 1) and the products were examined by agarose gel electrophoresis (Fig. 4A). The parental PCR product from cell line pLB4/11 was 1.4 kb in length, so G418R clones that arose by HR or by NHEJ events with short sequence deletions (or insertions) would produce products about 1.4 kb in length (Fig. 4A, lanes 1,2,5,6 and 7). Clones producing PCR products that were visibly shorter or longer than 1.4 kb (Fig. 4A, lanes 3 and 4) likely arose from NHEJ events with associated deletions or insertions greater than about 40 bp (based on the resolution achievable on the agarose gels). In addition to sizing PCR products by gel electrophoresis, the expedient use of three AluI sites located within the tk portion of the tk-neo fusion gene allowed for a screen for HR events among the PCR products that appeared to be full-length (Fig. 4B). The two AluI sites closely flanking the I-SceI site (Fig. 2) were present in the tk-neo fusion gene but were absent from the tk donor gene. Following HR, these latter two AluI sites were expected to be lost from the tk-neo fusion gene. The absence of these two AluI sites from the fusion gene produced a diagnostic 990 bp AluI fragment (Fig. 4B, lanes 1 and 2), and provided a reliable screen for HR. The retention of the AluI sites near the DSB produced 653 bp and 337 bp fragments upon AluI digestion (Fig. 4B, lane 3) and suggested NHEJ.
Fig. 4.
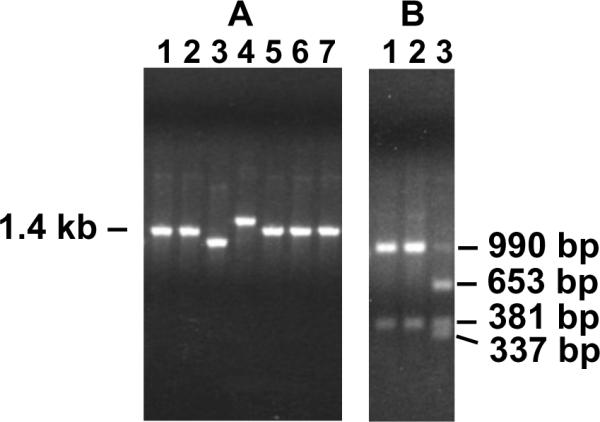
Representative PCR analysis. Genomic DNA samples isolated from G418R clones were amplified using PCR primers AW85 and AW91 shown in Fig. 1. (A) Lengths of PCR products were estimated by agarose gel electrophoresis. The parental PCR product was 1.4 kb, and clones generating similarly sized PCR products likely arose from HR or from NHEJ with small deletions or insertions. PCR products visibly shorter or longer than 1.4 kb were indicative of relatively large deletions or insertions at the DSB site. (B) PCR products that appeared to be 1.4 kb in length were further analyzed by digestion with AluI. Clones that arose from HR would have lost the two AluI sites closest to the DSB (see Fig. 2) and would therefore produce a 990 bp fragment (lanes 1 and 2). Clones producing 653 bp and 337 bp fragments (lane 3) retained the AluI sites near the DSB. Retention of one or both of the AluI sites near the DSB was consistent with NHEJ. See text for details.
More precise characterization of G418R clones was accomplished by nucleotide sequencing of representative PCR products. Thirteen scattered nucleotide mismatches between the tk-neo fusion gene and the donor gene, as indicated in Fig. 2, enabled an unequivocal demonstration of the occurrence of HR in clones that displayed a transfer of one or more of these nucleotide markers from the donor sequence to the tk-neo fusion gene. Clones displaying a sequence deletion (and/or insertion) at the I-SceI site with no transfer of markers from donor to fusion gene were categorized as having arisen via NHEJ. For clones arising via HR, the pattern of nucleotide markers present in the sequenced PCR product aided in distinguishing between HDD and gene conversions. Nucleotide sequence analysis of clones is discussed in greater detail below.
Southern blotting was also used as an important tool in the characterization of HR events. As illustrated at the top of Fig. 5, gene conversions and HDD events produced distinct restriction fragments upon digestion of genomic DNA with HindIII and XbaI. Southern blotting using a tk-specific probe therefore provided an effective way to distinguish gene conversions from HDD. A representative Southern blotting analysis of clones arising from HR events is shown at the bottom of Fig. 5. The G418R clones displayed in lanes 1–6 were recovered from cells treated with nonsilencing siRNA, while the clones in lanes 7–12 were recovered from cells treated with BLM-silencing siRNA. The clones in lanes 1, 6–10, and 12 each exhibited the 3.6 kb fragment diagnostic for HDD, while the balance of the clones displayed the 5.9 kb and 2.5 kb bands expected for gene conversions. Southern blotting also allowed an assessment of whether any gross chromosomal rearrangements were recovered. No gross rearrangements were found among 275 HR events examined by Southern blotting.
Fig. 5.

Southern blotting analysis of HR events. At the top of the figure are schematic illustrations of a gene conversion and an HDD event that each produces a functional tk-neo fusion gene. In the illustration of gene conversion, the conversion tract is denoted by the shaded segment of the tk portion of the fusion gene. The conversion tract eliminates the I-SceI site by transfer of sequence from the tk donor. For HDD, the joining together of donor and fusion gene sequences is denoted by the abutment of shaded and unshaded tk sequence. HDD, which can be produced via Holliday junction intermediates or via SSA, eliminates the I-SceI site as well as vector sequences (including the hyg gene) between the donor and the fusion gene. Expected fragments produced by digestion of genomic DNA with HindIII (H) and XbaI (X) are shown. At the bottom of the figure are Southern blots displaying genomic DNA samples (8 μg) from G418R colonies recovered from pLB4/11 cells treated with control siRNA (lanes 1–6) or BLM-3 siRNA (lanes 7–12). DNA fragments were visualized using a probe specific for tk to distinguish gene conversions from HDD. The samples in lanes 1, 6–10, and 12 displayed a single 3.9-kb band diagnostic for HDD, while the samples in lanes 2–5 and 11 displayed the 2.5 and 5.9 kb bands diagnostic for gene conversions.
In total, 338 G418R clones arising from pLB4/11 were characterized unambiguously by a combination of PCR size and restriction analysis, nucleotide sequencing, and Southern blotting as described above. The results of the analyses are summarized in the upper portion of Table 2.
Table 2.
Characterization of DSB repair events
| HR Events |
|||||
|---|---|---|---|---|---|
| siRNA Used | Clones Analyzed | Gene Conversions | HDD | Total HR | NHEJ |
| pLB4/11 cells | |||||
| None | 77 | 32 (41±6)a | 22 (29±3) | 54 (70±2) | 23 (30±2) |
| Control | 95 | 50 (53±4) | 22 (23±4) | 72 (76±6) | 23 (24±6) |
| BLM-1 | 72 | 21 (29±3) | 42 (58±5) | 63 (87±2) | 9 (13±2) |
| BLM-3 | 71 | 15 (21±8) | 49 (69±10) | 64 (90±4) | 7 (10±4) |
| BLM-1 + BLM-3 | 23 | 6 (26) | 16 (70) | 22 (96) | 1 (4) |
| pLB4/20 cells | |||||
| None | 56 | 40 (71±10) | 4 (7±1) | 44 (78±10) | 12 (22±10) |
| Control | 57 | 42 (74±2) | 5 (8±2) | 47 (82±3) | 10 (18±3) |
| BLM-1 | 21 | 14 (66±10) | 5 (24±7) | 19 (90±8) | 2 (10±8) |
| BLM-3 | 60 | 40 (67±2) | 12 (20±3) | 52 (87±3) | 8 (13±3) |
The total number of each particular type of event recovered is listed. In parenthesis, following each entry, is the percentage of clones that displayed the particular type of event. For each of these latter percentages, the standard deviation calculated from three independent trials is indicated. (For the BLM-1 + BLM-3 mixture for cell line pLB4/11, only one experiment was conducted and so no standard deviations are indicated.)
Several aspects of the data presented in Table 2 were striking. First, the fraction of DSB events accomplished by HR, rather than by NHEJ, was higher for pLB4/11 cells treated with BLM-silencing siRNA than for cells treated with control siRNA or with no siRNA. This difference in HR recovery was highly statistically significant (p = 1.3 × 10−4 by a Fisher exact test) when pooled data for cells treated with BLM-1 and/or BLM-3 siRNA was compared with pooled data for cells treated with control or no siRNA. Second, the percentage of HDD among recovered HR events was higher for cells treated with BLM-silencing siRNA than for cells treated with control siRNA or with no siRNA. This difference in HDD recovery was highly statistically significant (p = 8.7 × 10−10) when pooled data for cells treated with BLM-1 and/or BLM-3 siRNA was compared with pooled data for cells treated with control or no siRNA. For cells treated specifically with BLM-1 siRNA, HDD recovery among HR events was greater than for cells treated with either control siRNA or no siRNA (p = 3.3 × 10−5 or p = 5.7 × 10−3, respectively). For cells treated specifically with BLM-3 siRNA, HDD recovery was greater than for cells treated with either control siRNA or no siRNA (p= 7.5 × 10−8 or p= 1.3 × 10−4, respectively). Notably, while BLM depletion increased the overall frequency of HR events, this increase in HR was due specifically to an increase in HDD since the absolute frequency of gene conversions actually decreased. In summary, our results demonstrated that the level of BLM expression can influence both the balance of HR versus NHEJ, as well as the nature of HR events. The combined effects of reduction in BLM expression acted to quite substantially increase the fraction of all DSB repair events in pLB4/11 cells that occurred via HDD. The increase in HDD events was decidedly statistically significant (p = 5.9 × 10−13 ) when pooled data for cells treated with BLM siRNA was compared with pooled data for cells treated with control siRNA or no siRNA.
3.4. Nucleotide sequence analysis of G418R clones
Representative G418R clones recovered from pLB4/11 were examined at the nucleotide level to gain further insight into the possible influence of BLM expression level on the nature of DSB repair events. For each clone sequenced, at least 600bp of DNA sequence were determined from PCR products amplified from the region surrounding the healed DSB.
Twenty-one NHEJ clones recovered from cells treated with control siRNA, and all 16 NHEJ events recovered from BLM-1 or BLM-3 treated pLB4/11 cells were analyzed by DNA sequencing, and the results are summarized in Tables 3 and 4. Deletions associated with NHEJ ranged from 1 bp to 1212 bp, with no evident differences in deletion sizes for events recovered from control cells versus BLM-knockdown cells. Several NHEJ events from control and BLM-knockdown cells were associated with an insertion of DNA sequence at the site of the DSB. Of particular note were clones 17 and 20 from control siRNA-treated cells (Table 3) which each displayed an insertion of more than 100 bp of sequence derived from the integrated substrate pLB4. Other than deletions and insertions at the DSB site, no other sequence alterations were found associated with NHEJ events recovered from either control or BLM-knockdown cells.
Table 3.
NHEJ events recovered from pLB4/11 cells treated with control siRNA
| Clonea | Deletion size (bp)b | Microhomologyc | Insertiond |
|---|---|---|---|
| 1 | 1 (972) | A | -- |
| 2 | 1 (972) | A | -- |
| 3 | 22 (952–973) | AGG | -- |
| 4 | 28 (947–974) | GGG | -- |
| 5 | 43 (932–974) | GG | -- |
| 6 | 46 (935–980) | T | -- |
| 7 | 58 (933–990) | GC | -- |
| 8 | 79 (910–988) | ATGCC | -- |
| 9 | 205 (770–974) | G | -- |
| 10 | 208 (925–1132) | CCG | -- |
| 11 | 229 (864–1092) | GC | -- |
| 12 | 244 (731–974) | GGGT | -- |
| 13 | 472 (934–1405) | C | -- |
| 14 | 604 (758–1361) | GAGC | -- |
| 15 | 885 (450–1334) | CGTGCCGC | -- |
| 16 | 889 (605–1493) | CC | -- |
| 17 | 12 (966–977) | GGG, 0 | 176 bp, HSV upstream from tk |
| 18 | 279 (966–1244) | AAATT | |
| 19 | 363 (804–1166) | CAGTG | |
| 20 | 1193 (383–1575) | G,0 | 436 bp, inverted tk sequence |
| 21 | 1212 (375–1586) | GAGGCTGGGAG |
G418R clones are listed in order of deletion size with the exception that the five clones containing sequence insertions are grouped together at the bottom of the list.
Number of nucleotides deleted at the induced DSB. The nucleotide positions of the deletion endpoints are indicated in parentheses, using the nucleotide numbering system shown in Fig. 2.
Shown is the sequence homology shared by the DNA termini that were joined. In the case of clones with sequence insertions at the DSB, microhomologies associated with end-joining events at both ends of the inserted sequence are indicated when such a determination was possible. A “0” indicates that no homology was present.
For each of the five clones with sequence insertions, a description of the sequence inserted or the actual sequence itself is presented. For clones 17 and 20, the source of the inserted sequence appeared to be the integrated copy of pLB4. In clone 17, herpes virus sequence on pLB4 mapping just upstream from the tk gene was inserted, while in clone 20 a sequence from the tk gene was inserted with an inverted orientation.
Table 4.
NHEJ events recovered from pLB4/11 cells treated with BLM-silencing siRNA
| Clonea | Deletion size (bp)b | Microhomologyc | Insertiond |
|---|---|---|---|
| B1-1 | 4 (971–974) | 0 | -- |
| B1-2 | 4 (969–972) | 0 | -- |
| B3-1 | 7 (966–972) | A | -- |
| B3-2 | 7 (966–972) | A | -- |
| B1 3 | 22 (956–977) | AGCT | -- |
| B1-4 | 22 (960–981) | T | -- |
| B1-5 | 193 (780–972) | CAGG | -- |
| B1-6 | 274 (902–1175) | CAT | -- |
| B3-3 | 289 (970–1258) | TA | -- |
| B3-4 | 367 (624–990) | GCC | -- |
| B3-5 | 445 (939–1383) | CATATCGGGG | -- |
| B1-7 | 949 (587–1535) | C | -- |
| B1-8 | 1030 (557–1586) | CCA | -- |
| B3-6 | 1161 (486–1646) | 0 | -- |
| B3-7 | 0 | GTGTA | |
| B1-9 | 14 (961–974) | T |
G418R clones are listed in order of deletion size with the exception that the two clones containing sequence insertions are grouped together at the bottom of the list. The clones with names beginning with “B1” were recovered from cells treated with BLM-1 siRNA, while “B3” clones were recovered from cells treated with BLM-3 siRNA.
Number of nucleotides deleted at the induced DSB. The nucleotide positions of the deletion endpoints are indicated in parentheses, using the nucleotide numbering system shown in Fig. 2.
Shown is the sequence homology shared by the DNA termini that were joined. A “0” indicates that no homology was present.
For the two clones with sequence insertions, the actual sequence inserted is shown.
Microhomologies at the sites of DNA end-joining were also examined for NHEJ events (Table 3 and 4). Curiously, a significantly greater (p = 0.014) number of GC basepairs were present within the stretches of microhomology associated with NHEJ events recovered from control cells compared to microhomology used in BLM-knockdown cells.
Among HR events recovered, 21 HDD events from pLB4/11 cells treated with control siRNA and 13 HDD events from cells treated with BLM-3 siRNA were sequenced (Fig. 6). With the exception of two HDD events (discussed below), each event represented an accurate in-frame joining between the tk donor sequence and the tk-neo fusion gene with no mutations found. All HDD events resulted in a functional “wild-type” tk-neo gene with no remnant of the 22 bp oligonucleotide insertion that originally contained the I-SceI recognition site. In HDD clones 1, 7, 8, and 18–20 from control cells, and clone 4 from BLM knockdown cells, there was a stretch of sequence in which the origin of mismatched nucleotide markers (Fig. 2) alternated between tk donor and the tk-neo fusion gene. This alternation was likely due to patchy or discontinuous repair of heteroduplex DNA that formed during HR. No notable differences between HDD events recovered from control versus BLM-knockdown cells were observed.
Fig. 6.

HDD events. At the top of the figure is a schematic illustration of the 13 mismatches between the tk-neo fusion gene and the tk donor sequence on pLB4. These mismatches served as markers that aided in the analysis of recombination events. The location of the I-SceI site in the fusion gene is denoted by ▽. Listed below the schematic are representations of 20 HDD clones recovered from pLB4/11 cells treated with control nonsilencing siRNA, 12 HDD clones from cells treated with BLM-3 siRNA, and two exceptional HDD clones. For each HDD clone, the identity of each of the nucleotide markers is shown. Also, for each clone an “X” is placed between two markers to designate an approximate location of joining of the tk donor to the fusion gene; all markers downstream from the location of the “X” in each clone originated from the fusion gene. Exceptional HDD clone 1E, recovered from control cells, had an NHEJ-associated deletion spanning nucleotide positions 970 through 1358 of the tk-neo fusion gene (numbering as in Fig. 2) and an insertion of 19 bp as shown. Exceptional HDD clone 2E, recovered from BLM-3 siRNA-treated cells, had an NHEJ-associated deletion spanning nucleotide positions 960–981 of the fusion gene.
Curiously, two exceptional HDD events displayed a distinct NHEJ-associated deletion encompassing the I-SceI site in addition to a crossover or SSA event (Fig. 6, bottom). One clone (1E), recovered from control cells, had a 389 bp deletion and a 19 bp insertion in addition to a crossover or SSA event. The second clone (2E), recovered from BLM-3 siRNA-treated cells, had a 118 bp deletion in addition to a crossover or SSA event. A possible mechanism for the generation of these clones is presented in the Discussion below.
Twelve gene conversions from control cells, 19 gene conversions from BLM-1 siRNA-treated, and 15 gene conversions from BLM-3 siRNA-treated cells were analyzed at the nucleotide level (data not shown). Each of the 46 clones sequenced revealed an accurate exchange of sequence information from the tk donor sequence to the tk-neo fusion gene that resulted in the reconstruction of a wild-type fusion gene in which the 22 bp oligonucleotide insertion that originally contained the I-SceI site had been eliminated. Based on the nucleotide mismatches between the donor and fusion gene sequences, we determined that the minimal lengths of conversion tracts ranged from 3 bp to 343 bp for control cells and from 3 bp to 924 bp for BLM knockdown cells. For 38 out of the 46 gene conversions sequenced, only the two mismatched nucleotide markers closest to the DSB were converted, giving a minimal conversion tract length of 21 bp (the distance spanning the markers). No notable differences between gene conversion products from control versus BLM-knockdown cells were observed.
3.5 Knockdown of BLM expression in a second cell line promotes DSB repair via HR events that produce deletions
As described above, depletion of BLM in cell line pLB4/11 resulted in a substantial increase in DSB repair events occurring via HDD. We wanted to learn if a similar impact of BLM depletion could be replicated in another cell line. To this end, we used cell line, pLB4/20, which was independently derived from GM637 cells and also contained a single integrated copy of substrate pLB4. DSB repair was monitored in pLB4/20 cells under conditions of normal and reduced BLM expression, and the results are summarized in the lower portion of Table 2. We observed that the overall percentage of DSB repair events accomplished by HDD was considerably lower for cell line pLB4/20 compared with cell line pLB4/11 under conditions of either normal or reduced BLM expression. Nonetheless, as was the case for cell line pLB4/11, the percentage of HDD events among recovered HR events was higher for pLB4/20 cells treated with BLM-silencing siRNA than for cells treated with control siRNA or with no siRNA. This difference in HDD recovery was statistically significant (p = 0.02) when pooled data for cells treated with BLM-1 or BLM-3 siRNA was compared with pooled data for cells treated with control or no siRNA. Reduction in BLM expression in cell line pLB4/20 also increased the fraction of all DSB repair events that occurred via HDD. The increase in HDD events among all events was statistically significant (p = 0.01) when pooled data for pLB4/20 cells treated with BLM siRNA was compared with pooled data for cells treated with control siRNA or no siRNA. We thus concluded that an increase in DSB repair by HR, and by HDD specifically, is a general consequence of BLM depletion.
4. Discussion
In this paper we report the first direct evidence that the level of expression of BLM helicase influences the manner in which a DSB is repaired in the genome of human cells. More specifically, BLM depletion significantly increased the frequency of homology-dependent DSB repair events that engendered chromosomal deletion.
The loss of BLM activity in the rare genetic disorder BS leads to genomic instability and a strong predisposition to a wide spectrum of cancers. BLM defects have also been reported in cancer cells from patients that do not have BS [48,49], suggesting that the type of genomic instability associated with loss of BLM has broad relevance to carcinogenesis. A hallmark characteristic of cells from BS patients is a highly elevated frequency of SCE [15,16]. SCE are assayed cytologically and so the question of whether SCE are produced by HR events that resolve as crossovers is an issue that is difficult to address rigorously. There is some evidence that SCE are produced by HR in chicken DT40 cells [50]. The increase in SCE seen in BS has been interpreted as evidence for a hyperrecombination phenotype. Additional evidence for hyperrecombination in the absence of BLM comes from the observation that genetic knockout of BLM in normal human fibroblasts results in high levels of SCE, increased rates of LOH, and increased levels of targeted HR between endogenous and transfected DNA sequence [34]. Knockdown of BLM in the latter study did not lead to an increase in gross chromosomal rearrangements such as gain or loss of complete chromosomes. It has also been shown that knockout of BLM in mice results in elevated levels of SCE, an increase in gene-targeting frequency in ES cells derived from the mice, an elevated rate of LOH, and an increase in a wide spectrum of cancer [32]. Such studies have led to the inference that loss of BLM increases the rate of somatic HR and that elevated HR is an underlying mechanism for cancer predisposition in BS, most likely due to enhanced LOH. Our current work demonstrates that reduction in BLM expression in human cells can provoke a shift in DSB repair toward HR, with an increase specifically in HR events that produce deletions by crossovers or SSA. Our findings thus reinforce the view that an increase in, or dysregulation of, HR may be a key contributor to the cancer predisposition associated with loss of BLM.
Our nucleotide-level analysis of DSB repair events recovered from cell line pLB4/11 indicates that BLM deficiency promotes an increased frequency of HR events without an increased incidence of mutation in the products of such events. Of 80 gene conversions and HDD events recovered from control or BLM-knockdown cells that were analyzed by DNA sequencing, 78 events revealed no evidence of mutation in the HR products. We found two exceptional HDD events, one recovered from control cells and one recovered from BLM knockdown cells, that each displayed an NHEJ-associated deletion in addition to a crossover or SSA event between the tk donor and the fusion gene (Fig. 6, clones 1E and 2E). One of these clones, 1E, also contained a 19 bp sequence inserted at the NHEJ deletion site. The actual site of homology-dependent joining of the tk donor to the tk-neo fusion gene in each clone was distinctly removed from the NHEJ deletion endpoint. It is difficult to know precisely how these clones arose. Perhaps the most plausible explanation we can envision is that NHEJ producing a deletion (or a deletion plus insertion) occurred initially, followed by an HR event in the form of a crossover or SSA In this unusual scenario, a DSB provoked an HR event that occurred after the DSB was actually healed.
In addition to sequencing HR events, we also examined 37 NHEJ events at the nucleotide level and found no obvious impact on deletion size or increased mutagenesis associated with BLM depletion (Tables 3 and 4). Our results differ from two studies in which cell-free extracts were used to study the impact of BLM deficiency on NHEJ. Gamyes et al. [51] reported that BLM deficiency increased NHEJ activity and increased the size of deletions associated with NHEJ, whereas Langlund et al [52] reported that BLM deficiency lessened the use of microhomologies and actually decreased deletion size associated with NHEJ. In contrast, Onclercq-Delic et al. [53], using a system to study in vivo DSB repair occurring in plasmids transfected into cells, reported no impact of BLM deficiency on the efficiency and fidelity of NHEJ. The latter investigation also reported an increase in DSB repair by HR in the absence of BLM, consistent with our current findings. Using an intrachromosomal system conceptually related to ours to study an I-SceI-induced DSB, So et al. [54] observed no apparent impact on intrachromosomal NHEJ due to loss of BLM and concluded that repair of a two-ended DSB by NHEJ makes almost no contribution to the genomic instability seen in BS cells. We concur that depletion of BLM doesn't appear to overtly corrupt the NHEJ process in human chromosomes. Rather, we would argue that BLM depletion allows HR to co-opt DSB repair, and the shift toward HR specifically in the form of crossovers or SSA is key to the genesis of instability.
We observed that five out of 21 NHEJ clones recovered from control cells had sequence insertions (Table 3). Two such clones (clones 17 and 20) displayed insertions of 176 bp and 436 bp of sequence copied from the integrated pLB4 substrate. An additional clone (clone 21) displayed an insert size of 11 bp, while clones 18 and 19 each displayed an insert of 5 bp. In contrast, only 2 out of 16 NHEJ clones recovered from BLM knockdown cells (clones B3-7 and B1-9, Table 4), displayed insertions, and the insert sizes were 5 bp and 1 bp. We also noticed another possible subtle influence of BLM on the nature of NHEJ events in that a significantly greater (p = 0.014) number of GC basepairs were present in the collective microhomologies used in NHEJ in control cells compared to microhomologies used in BLM-knockdown cells. This suggests the possibility that interactions between DNA termini are somewhat less dependent on strong base-pairing when BLM is depleted. This finding may be related to the finding by Langlund et al [52]) that NHEJ in a cell-free extract has a decreased dependency on microhomology in the absence of BLM. At this time, however, the true significance of base composition in the vicinity of joined DNA ends is not known.
A shift away from NHEJ and toward HR provoked by BLM reduction would, by itself, be expected to possibly lead to an increase in HDD events. However, the increase in HDD engendered by BLM depletion in our studies was not due solely to an increase in HR since BLM reduction specifically increased HDD without increasing the frequency of gene conversions in either cell line pLB4/11 or pLB/20 (Table 2). The increase in HDD brought about by BLM knockdown was actually accompanied by a reduction of both NHEJ and gene conversions in cell line pLB4/11. The question of whether BLM deficiency merely increases HR overall, or whether BLM deficiency enhances only a particular type of HR event, such as crossovers, has been raised previously [55]. We believe that our current work is the first time that this issue has been addressed experimentally, with our results indicating that BLM deficiency indeed does not increase all forms of HR equally.
Relevant to this latter point, data presented in a recent study by others [56] was interpreted as evidence that HR is actually reduced in human cells in the absence of BLM. This observation was referred to as a “paradox” [56] since one might expect to see an increase in HR in the absence of BLM based on the elevated levels of SCE seen in BS cells. However, only gene conversions were recoverable in the experimental system used in the previous investigation. Our current work, in which gene conversions, crossovers, SSA, and NHEJ were all potentially recoverable, may resolve the “paradox” by revealing that although depletion of BLM may reduce the frequency of gene conversions (as seen in cell line pLB4/11), the overall level of HR actually increases due to an increase in HDD events (crossovers or SSA). Moreover, a surge in crossover events would be consistent with the elevated levels of SCE seen in BS cells. It was suggested [56] that increased SCE may result from increased lesion formation in BS cells rather than a hyperrecombination phenotype per se. However, our current results clearly show that the nature of repair of a single DSB is altered by depletion of BLM, and this alteration provokes an increase in a type of HR event that may potentially produce SCEs.
In our studies, we used an RNA interference approach rather that studying DSB repair in BLM null cells. The RNA interference approach is better-defined genetically and affords better controls. BLM null cells, for example, may accumulate multiple mutations upon propagation over multiple generations. Significant impacts of transient BLM reduction were in fact measured in our work without completely knocking-out BLM expression. We estimated that our most effective knockdown experiments reduced BLM expression by about 80%. This suggests the possibility of a phenotype for BLM haploinsufficiency. Indeed, evidence has been reported that heterozygosity for a loss-of-function mutation in BLM increases cancer risk in humans [57] as well as in mice [58]. Further, McDaniel et al. [33] showed that genomic instability and cancer predisposition is related inversely to the level of BLM expression in mice. From such studies, it appears that perhaps a small change in BLM activity can have a big impact on genomic stability.
On a molecular level, we can envision several mechanisms by which reduction in BLM expression may increase the likelihood that a DSB will be channeled into a repair pathway that produces HDD by crossovers or SSA. BLM's reported ability to disrupt D-loops [35,36] or disrupt Rad51 filaments [38], at least under certain conditions, may explain how knockdown of BLM can lead to an elevated frequency of DSB repair via HR. Once HR is initiated there is the potential for the event to proceed via the SDSA pathway, which produces noncrossovers almost exclusively [40], or via a pathway using a DHJ junction intermediate which can produce crossovers and noncrossovers. Based on BLM's reported stimulation of SDSA [37–39], it seems plausible to suggest that depletion of BLM increases the fraction of HR events proceeding via DHJ intermediates and, hence, further increases the opportunities for crossovers. BLM, in conjunction with topoisomerase IIIα and BLAP75, has also been shown to act in vitro to catalyze DHJ dissolution which produces only noncrossovers [29–31]. BLM depletion would therefore be expected to decrease the frequency of DHJ dissolution and bring about a concomitant increase in the frequency of DHJ resolution which can produce both crossovers and noncrossovers. The predicted shift in the mode of DHJ processing would thus provide an additional means for increasing the frequency of crossovers. Knockdown of topoisomerase IIIα and/or BLAP75 expression may help reveal just how much of a contribution DHJ dissolution makes to the impact of BLM knockdown. Our attempts to knock-down topoisomerase IIIα and BLAP75 have not yielded informative phenotypes (data not shown).
As mentioned above, HDD may be produced by the nonconservative HR pathway referred to as SSA [3,6] rather than through crossovers produced via Holliday junction intermediates. In SSA, 5' DNA end-resection uncovers complementary sequences of direct repeats. The complementary strands anneal, DNA single-strand tails are clipped, and gaps are filled. Although we cannot rigorously determine whether most recovered HDD events arose via crossovers or SSA, several considerations are pertinent to this matter. In general, the reported functions of BLM do not lend themselves to a ready explanation of how BLM depletion would lead to a specific increase in SSA with concomitant decreases in both NHEJ and gene conversions as observed for cell line pLB4/11 (Table 2). We also note that the I-SceI DSB in pLB4 is positioned within the tk portion of the fusion gene rather than between the homologous repeated tk sequences on pLB4. This placement of the DSB is not well-suited for promotion of SSA. Further, in the absence of DSB induction, cell line pLB4/11 gives rise spontaneously to G418R segregants generated both by gene conversions and HDD, with DSB induction by I-SceI inducing both types of events to the same extent, about 100- fold (data not shown). It is difficult to reconcile this latter observation with the notion that most HDD are produced via SSA. Finally, it appears that HDD clone number 7 from control cells, as well as HDD clones 1E and 2E (Fig. 6) cannot be explained by SSA since a nucleotide marker from the tk-neo fusion gene is present upstream from the DSB site in each of these clones. We therefore posit that most HDD events recovered in our experiments represent crossovers rather than SSA. We do note a recent report in which an inference is made that BLM depletion in Drosophila leads to an increase in SSA [59]. A salient point is that regardless of the particular mechanism for the genesis of the HDD events in our study, BLM depletion shifted DSB repair toward repair by a type of HR event that places a human cell at greater risk for sequence loss or rearrangement. We also emphasize that the events that we classify as HDD are indeed driven by homology and are distinct from the events we classify as NHEJ. It is therefore clear that BLM depletion stimulates a type of homology-dependent repair at DSBs.
Further dissection of the molecular mechanism(s) by which BLM deficiency destabilizes the genome are warranted, but the present work takes an important step by establishing that BLM deficiency corrupts DSB repair in human cells in a way that increases the frequency of HR events that are likely to be detrimental. Our current work adds to an emerging picture of BLM as a key and limiting factor in the regulation of DNA transactions in human cells. Future studies will undoubtedly further elucidate the diverse roles that BLM and other RecQ family members play in the maintenance of genome integrity.
Acknowledgements
This work was supported by Public Health Service grant GM081472 from the National Institute for General Medical Sciences to A.S.W. We thank members of the Waldman lab for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviation: HDD, homology-dependent deletion
Conflict of Interest Statement The authors declare that there are no conflicts of interest.
REFERENCES
- [1].Bernstein C, Bernstein H. Aging, Sex, and DNA Repair. Academic Press, Inc.; San Diego: 1991. [Google Scholar]
- [2].Waldman AS. Ensuring the fidelity of recombination in mammalian chromosomes. BioEssays. 2008;30:1163–1171. doi: 10.1002/bies.20845. [DOI] [PubMed] [Google Scholar]
- [3].Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].O'Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat. Rev. Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- [5].Pardo B, Gómez-González B, Aguilera A. DNA double-strand break repair: how to fix a broken relationship. Cell Mol. Life Sci. 2009;66:1039–1056. doi: 10.1007/s00018-009-8740-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sonoda E, Hochegger H, Saberi A, Taniguchi Y, Takeda S. Differential usage of nonhomologous end-joining and homologous recombination in double strand break repair. DNA Repair. 2006;5:1021–1029. doi: 10.1016/j.dnarep.2006.05.022. [DOI] [PubMed] [Google Scholar]
- [7].Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2009;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- [8].Bunting SF, Callén E, Wong N, Chen H-T, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng C-X, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. USA. 2008;105:6906–16911. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].You Z, Bailis JM. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010;20:402–409. doi: 10.1016/j.tcb.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bielas JH, Venkatesan RN, Loeb LA. LOH-proficient embryonic stem cells: a model of cancer progenitor cells? Trends Genet. 2007;23:154–157. doi: 10.1016/j.tig.2007.02.009. [DOI] [PubMed] [Google Scholar]
- [13].Donahue SL, Lin Q, Cao S, Ruley HE. Carcinogens induce genome-wide loss of heterozygosity in normal stem cells without persistent chromosomal instability. Proc. Natl. Acad. Sci. USA. 2006;103:11642–11646. doi: 10.1073/pnas.0510741103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Knudson AG., Jr. Retinoblastoma: a prototypic hereditary neoplasm. Semin. Oncol. 1978;5:57–60. [PubMed] [Google Scholar]
- [15].German J. Bloom syndrome: a Mendelian prototype of somatic mutational disease. Medicine (Baltimore) 1993;72:393–406. [PubMed] [Google Scholar]
- [16].Chaganti RS, Schonberg S, German J. A manyfold increase in sister chromatid exchanges in Bloom's syndrome lymphocytes. Proc. Natl. Acad. Sci. USA. 1974;1974;71:4508–4512. doi: 10.1073/pnas.71.11.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bennett RJ, Keck JL. Structure and function of RecQ DNA helicases. Crit. Rev. Biochem. Mol. Biol. 2004;39:79–97. doi: 10.1080/10409230490460756. [DOI] [PubMed] [Google Scholar]
- [18].Cheok CF, Bachrati CZ, Chan KL, Ralf C, Wu L, Hickson ID. Roles of the Bloom's syndrome helicase in the maintenance of genome stability. Biochem. Soc. Trans. 2005;33:1456–1459. doi: 10.1042/BST0331456. [DOI] [PubMed] [Google Scholar]
- [19].Mimitou EP, Symington LS. Nucleases and helicases take center stage in homologous recombination. Trends Biochem. Sci. 2009;34:264–272. doi: 10.1016/j.tibs.2009.01.010. [DOI] [PubMed] [Google Scholar]
- [20].Ouyang KJ, Woo LL, Ellis NA. Homologous recombination and maintenance of genome integrity: cancer and aging through the prism of human RecQ helicases. Mech. Ageing Dev. 2008;129:425–440. doi: 10.1016/j.mad.2008.03.003. [DOI] [PubMed] [Google Scholar]
- [21].Payne M, M., Hickson ID. Genomic instability and cancer: lessons from analysis of Bloom's syndrome. Biochem. Soc. Trans. 2009;37:553–559. doi: 10.1042/BST0370553. [DOI] [PubMed] [Google Scholar]
- [22].Singh DK, Ahn B, Bohr VA. Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology. 2009;10:235–252. doi: 10.1007/s10522-008-9205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wu L. Role of the BLM helicase in replication fork management. DNA Repair. 2007;6:936–944. doi: 10.1016/j.dnarep.2007.02.007. [DOI] [PubMed] [Google Scholar]
- [24].Bugreev DV, Mazina OM, Mazin AV. Bloom syndrome helicase stimulates RAD51 DNA strand exchange activity through a novel mechanism. J. Biol. Chem. 2009;284:26349–26359. doi: 10.1074/jbc.M109.029371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cheok CF, Wu L, Garcia PL, Janscak P, Hickson ID. The Bloom's syndrome helicase promotes the annealing of complementary single-stranded DNA. Nucleic Acids Res. 2005;33:3932–3941. doi: 10.1093/nar/gki712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Machwe A, Xiao L, Groden J, Orren DK. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry. 2006;45:13939–13946. doi: 10.1021/bi0615487. [DOI] [PubMed] [Google Scholar]
- [27].Ralf C, Hickson ID, Wu L. The Bloom's syndrome helicase can promote the regression of a model replication fork. J. Biol. Chem. 2006;281:22839–22846. doi: 10.1074/jbc.M604268200. [DOI] [PubMed] [Google Scholar]
- [28].Karow JK, Constantinou A, Li JL, West SC, Hickson ID. The Bloom's syndrome gene product promotes branch migration of Holliday junctions. Proc. Natl. Acad. Sci. USA. 2000;97:6504–6508. doi: 10.1073/pnas.100448097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Raynard S, Bussen W, Sung P. A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIα, and BLAP75. J. Biol. Chem. 2006;281:13861–13864. doi: 10.1074/jbc.C600051200. [DOI] [PubMed] [Google Scholar]
- [30].Wu L, Bachrati CZ, Ou J, Xu C, Yin J, Chang M, Wang W, Li L, Brown GW, Hickson ID. BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proc. Natl. Acad. Sci. USA. 2006;103:4068–4073. doi: 10.1073/pnas.0508295103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wu L, Hickson ID. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- [32].Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, Vogel H, Schultz RA, Bradley A. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat. Genet. 2000;26:424–429. doi: 10.1038/82548. [DOI] [PubMed] [Google Scholar]
- [33].McDaniel LD, Chester N, Watson M, Borowsky AD, Leder P, Schultz RA. Chromosome instability and tumor predisposition inversely correlate with BLM protein levels. DNA Repair. 2003;2:1387–1404. doi: 10.1016/j.dnarep.2003.08.006. [DOI] [PubMed] [Google Scholar]
- [34].Traverso G, Bettegowda C, Kraus J, Speicher MR, Kinzler KW, Vogelstein B, Lengauer C. Hyper-recombination and genetic instability in BLM-deficient epithelial cells. Cancer Res. 2003;63:8578–85781. [PubMed] [Google Scholar]
- [35].Bachrati CZ, Borts RH, Hickson ID. Mobile D-loops are a preferred substrate for the Bloom's syndrome helicase. Nucleic Acids Res. 2006;34:2269–2279. doi: 10.1093/nar/gkl258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].van Brabant AJ, Ye T, Sanz M, German JL, III, Ellis NA, Holloman WK. Binding and melting of D-loops by the Bloom syndrome helicase. Biochemistry. 2000;39:14617–14625. doi: 10.1021/bi0018640. [DOI] [PubMed] [Google Scholar]
- [37].Adams MD, McVey M, Sekelsky JJ. Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science. 2003;299:265–267. doi: 10.1126/science.1077198. [DOI] [PubMed] [Google Scholar]
- [38].Bugreev DV, Yu X, Egelman EH, Mazin AV. Novel pro and antirecombination activities of the Bloom's syndrome helicase. Genes Dev. 2007;21:3085–3094. doi: 10.1101/gad.1609007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McVey M, Larocque JR, Adams MD, Sekelsky JJ. Formation of deletions during double-strand break repair in Drosophila DmBlm mutants occurs after strand invasion. Proc Natl. Acad. Sci. USA. 2004;101:15694–15699. doi: 10.1073/pnas.0406157101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Haber JE, Ira G, Malkova A, Sugawara N. Repairing a double-strand chromosome break by homologous recombination: revisiting Robin Holliday's model. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2004;359:79–86. doi: 10.1098/rstb.2003.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Smith JA, Bannister LA, Bhattacharjee V, Wang Y, Waldman BC, Waldman Accurate homologous recombination is a prominent double-strand break repair pathway in mammalian chromosomes and is modulated by mismatch repair protein Msh2. Mol. Cell. Biol. 2007;27:7816–7827. doi: 10.1128/MCB.00455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Waldman BC, Wang Y, Kilaru K, Yang Z, Bhasin A, Wyatt MD, Waldman AS. Induction of intrachromosomal homologous recombination in human cells by raltitrexed, an inhibitor of thymidylate synthase. DNA Repair. 2008;7:1624–1635. doi: 10.1016/j.dnarep.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophoton. Int. 2004;11:36–42. [Google Scholar]
- [44].Lukacsovich T, Yang D, Waldman AS. Repair of a specific double-strand break generated within a mammalian chromosome by yeast endonuclease I-SceI. Nucleic Acids Res. 1994;22:5649–5657. doi: 10.1093/nar/22.25.5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wagner MJ, Sharp JA, Summers WC. Nucleotide sequence of the thymidine kinase gene of herpes simplex virus type 1. Proc. Natl. Acad. Sci. USA. 1981;78:1441–1445. doi: 10.1073/pnas.78.3.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rajesh C, Baker DK, Pierce AJ, Pittman DL. The splicing-factor related protein SFPQ/PSF interacts with RAD51D and is necessary for homology-directed repair and sister chromatid cohesion. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq738. doi:10.1093/nar/gkq738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Waldman AS, Liskay RM. Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Mol. Cell. Biol. 1988;8:5350–5357. doi: 10.1128/mcb.8.12.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Calin G, Ranzani GN, Amadori D, Herlea V, Matei I, Barbanti-Brodano G, Negrini M. Somatic frameshift mutations in the Bloom syndrome BLM gene are frequent in sporadic gastric carcinomas with microsatellite mutator phenotype. BMC Genet. 2001;2:14. doi: 10.1186/1471-2156-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li HR, Shagisultanova EI, Yamashita K, Piao Z, Perucho M, Malkhosyan SR. Hypersensitivity of tumor cell lines with microsatellite instability to DNA double strand break producing chemotherapeutic agent bleomycin. Cancer Res. 2004;64:4760–4767. doi: 10.1158/0008-5472.CAN-04-0975. [DOI] [PubMed] [Google Scholar]
- [50].Sonoda E, Sasaki MS, Morrison C, Yamaguchi-Iwai Y, Takata M, Takeda S. Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol. Cell. Biol. 1999;19:5166–5169. doi: 10.1128/mcb.19.7.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gaymes TJ, North PS, Brady N, Hickson ID, Mufti GJ, Rassool FV. Increased error-prone non homologous DNA end-joining--a proposed mechanism of chromosomal instability in Bloom's syndrome. Oncogene. 2002;21:2525–2533. doi: 10.1038/sj.onc.1205331. [DOI] [PubMed] [Google Scholar]
- [52].Langland G, Elliott J, Li Y, Creaney J, Dixon K, Groden J. The BLM helicase is necessary for normal DNA double-strand break repair. Cancer Res. 2002;62:2766–2770. [PubMed] [Google Scholar]
- [53].Onclercq-Delic R, Calsou P, Delteil C, Salles B, Papadopoulo D, Amor-Guéret M. Possible anti-recombinogenic role of Bloom's syndrome helicase in double-strand break processing. Nucleic Acids Res. 2003;31:6272–6282. doi: 10.1093/nar/gkg834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].So S, Adachi N, Lieber MR, Koyama H. Genetic interactions between BLM and DNA ligase IV in human cells. J. Biol. Chem. 2004;279:55433–55442. doi: 10.1074/jbc.M409827200. [DOI] [PubMed] [Google Scholar]
- [55].Hu Y, Lu X, Barnes E, Yan M, Lou H, Luo G. Recql5 and Blm RecQ DNA helicases have nonredundant roles in suppressing crossovers. Mol. Cell. Biol. 2005;25:3431–3442. doi: 10.1128/MCB.25.9.3431-3442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Larocque JR, Jasin M. Mechanisms of recombination between diverged sequences in wild-type and BLM-deficient mouse and human cells. Mol. Cell. Biol. 2010;30:1887–1897. doi: 10.1128/MCB.01553-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gruber SB, Ellis NA, Scott KK, Almog R, Kolachana P, Bonner JD, Kirchhoff T, Tomsho LP, Nafa K, Pierce H, Low M, Satagopan J, Rennert H, Huang H, Greenson JK, Groden J, Rapaport B, Shia J, Johnson S, Gregersen PK, Harris CC, Boyd J, Rennert G, Offit K. BLM heterozygosity and the risk of colorectal cancer. Science. 2002;297:2013. doi: 10.1126/science.1074399. [DOI] [PubMed] [Google Scholar]
- [58].Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, Slovek LE, Capobianco AJ, German J, Boivin GP, Groden J. Enhanced tumor formation in mice heterozygous for Blm mutation. Science. 2002;297:2051–2053. doi: 10.1126/science.1074340. [DOI] [PubMed] [Google Scholar]
- [59].Johnson-Schlitz DM, Flores C, Engels WR. Multiple-pathway analysis of double-strand break repair mutations in Drosophila. PLoS Genet. 2007;3:e50. doi: 10.1371/journal.pgen.0030050. [DOI] [PMC free article] [PubMed] [Google Scholar]