

Abstract
INTRODUCTION
Drug targeting to sites of tissue injury, tumor or infection with limited toxicity is the goal for successful pharmaceutics. Immunocytes (including mononuclear phagocytes (dendritic cells, monocytes and macrophages), neutrophils, and lymphocytes) are highly mobile; they can migrate across impermeable barriers and release their drug cargo at sites of infection or tissue injury. Thus immune cells can be exploited as trojan horses for drug delivery.
AREAS COVERED IN THIS REVIEW
This paper reviews how immunocytes laden with drugs can cross the blood brain or blood tumor barriers, to facilitate treatments for infectious diseases, injury, cancer, or inflammatory diseases. The promises and perils of cell-mediated drug delivery are reviewed, with examples of how immunocytes can be harnessed to improve therapeutic end points.
EXPERT OPINION
Using cells as delivery vehicles enables targeted drug transport, and prolonged circulation times, along with reductions in cell and tissue toxicities. Such systems for drug carriage and targeted release represent a novel disease combating strategy being applied to a spectrum of human disorders. The design of nanocarriers for cell-mediated drug delivery may differ from those used for conventional drug delivery systems; nevertheless, engaging different defense mechanisms into drug delivery may open new perspectives for the active delivery of drugs.
Keywords: cell-carriers, drug delivery, immunocytes, nanoparticles, targeted drug transport
Introduction
The development of targeted drug delivery is amongst the most important goals of pharmaceutical research. Its realization can lead to improved therapeutic efficacy, reductions in drug dosing intervals, and decreased toxicities. However, this is not a simple task as drug homing to pathologically relevant disease sites has only recently been investigated. Obstacles are substantial and include sustained time-based plasma concentrations and local blood flow.
1. Promise and Perils of Cell-Mediated Drug Delivery
Using immunocytes, mononuclear phagocytes (MP; monocytes, macrophages, and dendritic cells) lymphocytes, and neutrophils and stem cells as drug carrier systems offers several advantages over common drug administration regimens. These include targeted drug transport to disease sites; prolonged drug half-lives; time-controlled drug release; and diminished drug immunogenicity and cytoxicity profiles. Immunocytes and stem cells exhibit an intrinsic homing property enabling them to migrate to sites of injury, inflammation, and tumor. In addition, they can act as Trojan horses carrying concealed drug cargoes while migrating across impermeable barriers (for example, the blood brain or blood tumor barriers) to sites of disease.
Despite such advantages, there is as yet limited success for several reasons. First, drug loading in cell carriers is low. Second, there are limitations due to the ability of immunocytes and stem cells to efficiently disintegrate and clear entrapped therapeutic agents. Third, the loaded drug should not be prematurely released, but unloaded, in continuous action, upon the cell's arrival to the site of action or disease. Fourth, the cell carriers should migrate to the disease site in substantial quantities. This should not be compromised during drug loading. Finally, all used formulations must be safe for both the cell carrier and the organism.
Many of these limitations can be addressed by incorporation of drugs into protective polymeric nanocarriers (liposomes and lipid nanoparticles 1-4, micelles 5-8, nanogels 9, 10, nanospheres and nanocapsules 11, 12, solid nanoparticles and nanosuspensions 13-16, block ionomer complexes 17, 18 or nanofibers and nanotubes 19, 20) that preserve drugs inside subcellular organelles (Figure 1). The ideal drug carrier for cell-mediated delivery should have optimal size, shape and surface characteristics; multivalent attachment; controlled drug release; and biocompatibility. All these characteristics are essential for translating cell-mediated drug delivery systems for human use.
Figure 1.
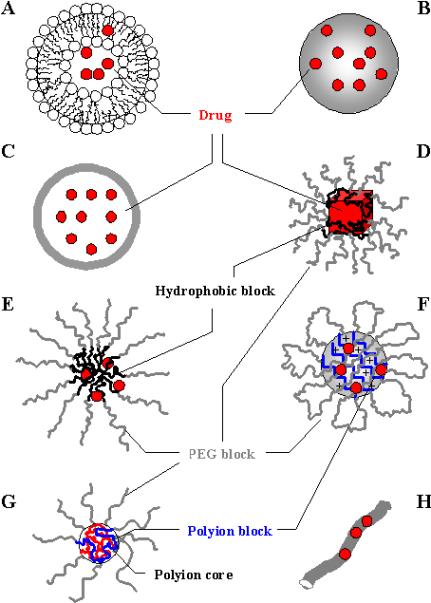
Types of nanocarriers for drug delivery. A: liposomes; B: polymer and lipid nanoparticles; C: nanospheres and nanocapsules; D: nanosuspensions; E: polymer micelles; F: nanogels; G: block ionomer complexes; H: nanofibers and nanotubes. Used with permission 110.
Drug loading into cell-carriers
Nanocarriers commonly have a core-shell structure. Their central part (such as aqueous pool of nanospheres, hydrophobic core of micelles, or polyelectrolyte core of nanogels) permits drug entrapment. It is surrounded with a polymer shell (or lipids in liposomes and lipid nanoparticles), which defines the nanocarrier dispersion stability, circulation time and cell interactions 1-8, 10, 11, 13, 14, 17, 18. Both the core and shell are crucial for successful cell-mediated delivery. For example, the surface coating affects the ability of particles to internalize into the cells and therefore, affects the loading of cell-carriers. In general, charged nanocarriers are rapidly taken up by MP and other immunocytes or stem cells compared to neutral nanoparticles 1, 21-25. The recognition occurs by receptors located on the cell plasma membranes, in particular, mannose, complement, and Fc receptors (MR, CR and FcR) 26-28. In general, MR recognize mannans, as well as integrins (for example CD11b/CD18), CR interact with particles after nonspecific complement opsonization, and FcR recognize particles after specific antibody opsonization 28. Furthermore, positively charged nanoparticles accumulate in MP to a greater extent than negatively charged particles. Thus, positively charged nanoparticles prepared by high pressure homogenization with antiretroviral drugs, indinavir (IDV), ritonavir (RTV), and efavirenz (EFV) accumulate in MP at about two-fold greater than the same size negatively charged carriers 14. Similar effect was reported for nanoparticles (“nanozymes”) comprising of redox enzyme, catalase, and variant synthetic polyelectrolyte block copolymers 29. Loading capacity of nanozymes comprising of positively-charged mono-polymers (polyethyleneimine-(PEI) and poly-L-lysine- (PL) based) is greater than the nanozyme prepared with negatively-charged block poly-L-glutamic acid (PGLU)-based copolymer, This likely occurs due to the greater absorption and internalization of positively-charged nanoparticles to negatively-charged outside plasma membrane of cell-carriers.
An electrostatically neutral and hydrophilic poly(ethylene glycol) (PEG) is perhaps the most common shell-forming polymer currently used in injectable nanocarriers 30-32. Particularly, commercial PEGylated liposome-encapsulated Dox, Doxil, is approved for use in the treatment of recurrent ovarian cancer, AIDS-related Kaposi's sarcoma 33 and metastatic breast cancer 34. However, PEG is by far not the only polymer that should be considered in the context of the cell-mediated delivery, as it can limit cellular uptake as was demonstrated for microspheres bearing high-density surface PEG chains that were resistant to phagocytosis 35. Similar results were obtained with nanoparticles comprised of catalase and positively charged polymers based on PEI 29. Thus, loading capacity of nanozymes without PEG corona in MP was significantly greater than those with PEG. To this end, modification of nanocarrier shell with specific vector moieties can provide for targeted cell delivery of nanocarriers 1, 26, 36-38. In particular, modification of PEG shell of a poly(amidoamine) nanoparticles (PAMAM) with streptavidin facilitates targeting to biotinylated T-cell markers such as anti-CD3 or peptide/ major histocompatibility (MHC) complexes 39 (Figure 2).
Figure 2. Schematic representation of SA-PEGFITC/PAMAM construction.
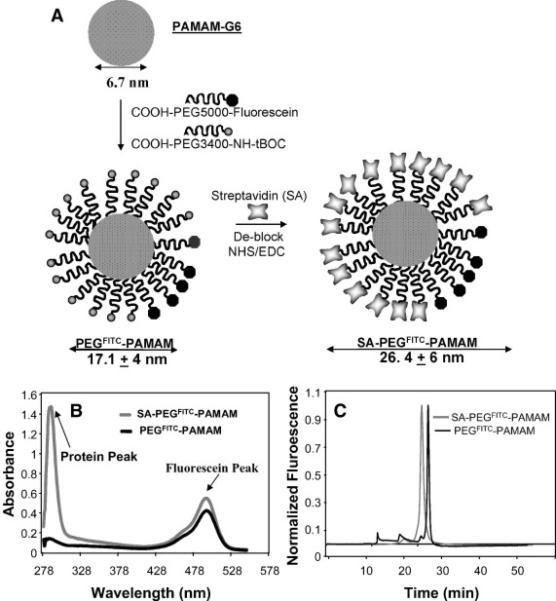
A. The construction of streptavidin-PEGFITC/PAMAM. B. Absorbance profiles of streptavidin-PEGFITC/PAMAM and PEGFITC/PAMAM. C. Reverse-phase HPLC of SAPEGFITC/PAMAM and PEGFITC/PAMAM. Used with permission 39.
Size and shape of nanocarriers are also of importance for cell uptake, although the phagosome may have different sizes depending of the size of the particles, which can range from as little as few hundred nanometers 35, 40, 41 to several of microns 42. For example, murine bone marrow-derived macrophages accumulate IgG-opsonized latex beads greater than 20 μm in diameter 43. Furthermore, particles with the size about 1μm were accumulated at greater extent (2.56 fold) than smaller drug carriers at 500 μm 14.
A recent study by Champion et al. reported striking effects of shape of particles on phagocytosis in alveolar rat macrophages 44. Polystyrene-based particles of more than twenty shapes including spheres, rectangles, rods, worms, oblate ellipses, elliptical disks and UFO-like particles were manufactured. The local particle shape at the point where it was attached to the cell played the crucial role in phagocytosis. For example, a macrophage attached to a sharper side of the ellipse would internalize the particle in a few minutes. In contrast, a macrophage attached to a dull side would not internalize the same ellipse for hours. Although, particle size played a reduced role in the initiation of the phagocytosis it could of course affect its completion especially when the particle volume exceeded that of a cell.
Drug preservation inside cells
The stability of the drug loaded into cell carriers among other factors can depend on the intracellular trafficking of the nanocarriers. In general, drug-loaded nanocarriers need to avoid lysosomes to reduce drug disintegration inside cells 14. In this regard, it is noteworthy that cationic and anionic nanoparticles show divergent fates inside MP 38. For example, phagocytosis of cationic polyamine-coated nanoparticles leads to the diminished phagosomal acidification when compared to anionic protein-coated particles. Such cationic nanoparticles protect the incorporated drug against lisosomal degradation 29, 38. Furthermore, loading of “nanozymes” containing positively-charged block copolymers (PEI-PEG and PL-PEG) protected the enzyme in macrophages. Increasing the amount of the block copolymer in the nanozyme formulation improved the stability of the enzyme. In contrast, catalase loaded in a polyion complex with a negatively charged block copolymer (PGLU-PEG) was degraded in macrophages to an even greater extent than catalase loaded alone. Protection of the enzymatic activity inside carrier cells may be, in part, due to a “proton sponge” effect of block copolymers 45, when an excess of amino groups on the surface of the nanoparticles buffers acidification of the cell's endocytic compartments. This serves to inhibit protease activity and decrease drug degradation 38. In other words, a positively charged block-copolymer prevents phagosome-lysosomal clearance functions and as a result, enzyme degradation.
In extreme case, intracellular drug degradation can be avoided by attaching the drug to the surface of cell-carriers. This “back pack” approach would still provide targeted transport and increased blood circulation along with the preserved drug activity. Thus, attaching avidin-coated nanoparticles to the biotinylated plasma membrane can be achieved through the avidin-biotin complex 46. Another possibility is red blood cells (RBCs) with attached drugs on their surface 47. Specifically, glycoprotein A covalently conjugated to the surface of the RBCs may provide extended half-life, controlled volume of distribution, and multivalent therapeutic interactions. However, general limitations of the “back pack” approach may include decreased loading of cell carriers, impeded drug release at the disease site, as well as increased immunogenicity and toxicity.
Drug release from cell-carriers
Mechanism of nanoparticle unloading at the site of action remains an active area of investigation. Controlled release of drugs from the cell-carriers modulates the rate of drug appearance, dose and duration of exposure at the diseased sites. To this end, utilizing cellular responses to various conditions could provide desired triggered release. Obviously, targeting of cell-carriers and their prolonged residence at the disease site should provide opportunities for drug unloading. In addition, MP, in particular, are known to produce and store various compounds in intracellular vesicles, and liberate them via exocytosis at the site of the disease. A similar mechanism can trigger release of drug-loaded nanocarriers, when macrophages serve as drug delivery vehicles 48. Furthermore, the drug release can be also triggered by the increase of intracellular concentration of Ca2+ 49. Finally, mild hypothermia was shown to facilitate controlled release of drug-loaded liposomes from macrophages in anti-cancer therapy 50.
Overall the structure and composition of protective nanocontainers play a crucial role in the effectiveness of formulations for cell-based drug delivery systems. For example, our recent studies indicate that structure of block copolymer used for catalase nanoformulation, nanozyme, affect its cytotoxicity, loading and release capacities, as well as preservation of catalase enzymatic activity inside cell-carriers 29. Thus, nanozymes containing a negatively-charged block copolymer (PGLU-PEG/catalase) demonstrated low toxicity, high loading capacity and effective release from macrophages. However, the polymer provided limited protection of the enzyme against cell-associated protease degradation. In contrast, nanozymes based on positively charged block copolymers, especially the PLs, showed increased cytotoxicity and low loading and release rates, but were protective of the catalase. Increasing the amount of positively-charged block-copolymer in nanozyme leaded to protection of catalase enzymatic activity but substantially reduced loadings and release. Importantly, nanozymes with PEG corona show water stability, limited cytoxicity and efficient catalase protection. Nonetheless, these formulations also demonstrated decreased loading capacity and release rates. In addition, nanozymes based on mono-polymer (without PEG corona) have higher loading and release levels, but did not protect catalase inside the cell-carriers. Taken together, the most optimal nanozyme formulation was the one based on positively charged block copolymers (PEI-PEG/catalase and PL-PEG/catalase) that demonstrate the most efficient protection of catalase enzymatic activity along with relatively high loading and release rates with limited cytotoxicity (Figure 3).
Figure 3. Optimization of nanocarriers structure for catalase delivery in macrophages.
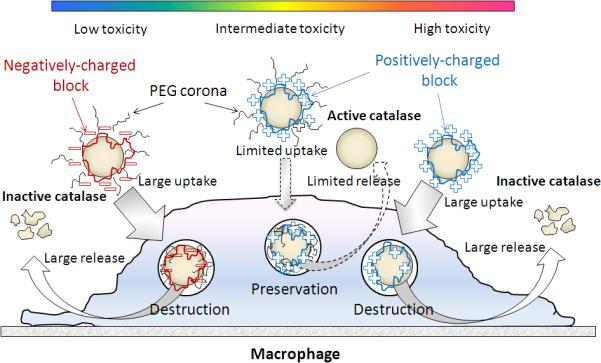
Nanozymes consisting of catalase and a negatively charged block copolymer demonstrate low toxicity, high loading capacity and release from BMM, however provide limited protection of the enzyme against protease degradation inside the cell-carriers. Nanozymes based on positively charged block copolymers, especially the PLs, showed increased cytotoxicity and low loading and release rates, but were highly protective of catalase. Furthermore, nanozymes with PEG corona show good stability in water, limited cytoxicity and efficient protection of catalase, but decreased loading capacity and cell release. The optimal nanozyme formulation is one based on positively charged block copolymers (PEI-PEG/catalase and PL-PEG/catalase). Used with permission 29.
Homing of drug-loaded cell-carriers at disease sites
The numbers of cell-carriers that can reach the disease site is especially crucial in the case of CNS disorders when drug-loaded cells need to penetrate the blood brain barrier (BBB) to mediate therapeutic effect. Many neurological diseases, such as Alzheimer's and Parkinson's diseases (AD and PD), Prion disease, meningitis, encephalitis and HIV-associated neurocognitive disorders (HAND), have in common an inflammatory component 51. The process of inflammation is characterized by extensive MP recruitment. Notably, MP migrate toward the inflammation site via the processes known as diapedesis and chemataxis 52. Such cells efficiently cross the BBB due to their margination and extravasation properties causing barrier breakdown as a consequence of brain inflammation53-57. Many reports in the literature indicate that blood borne monocytes traffic primarily between adjacent endothelial cells, i.e. paracellularly through the junctional complexes58, 59. Therefore, these cells can be loaded with a required drug and administered intravenously to reach the brain. For example in an experimental model of PD, considerable levels of catalase loaded into BMM were reported in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-intoxicated brains48. Approximately 2.1% of the injected catalase dose was delivered to the brain during MPTP-associated inflammation. Furthermore, several studies confirmed the migration of inflammatory-response cells, in particular, bone-marrow-derived mesenchymal stromal cells toward injury sites such as infarcted myocardium60, 61, spinal cord injury62, and cerebral ischemia63.
Neural stem cells (NSCs) were also suggested as drug delivery vehicles for gene therapy in the CNS 64-66. Indeed, these cells are highly migratory and migrate to areas of brain pathology including ischemic and neoplastic brain lesions that are commonly present in AD, PD, brain cancer, stroke, and multiple sclerosis. How the mobility of stem cells are directed is not understood, although NSCs express a wide variety of receptors that should enable them to respond to many chemotactic signals present in brain pathologies (Figure 4) 66. In particular, activated microglia induced NSCs migration to the brain. Delivery of various neurotropic factors is in the focus of cell-mediated strategies for neurological diseases treatment.
Figure 4. Using neural stem cells for drug delivery to CNS.
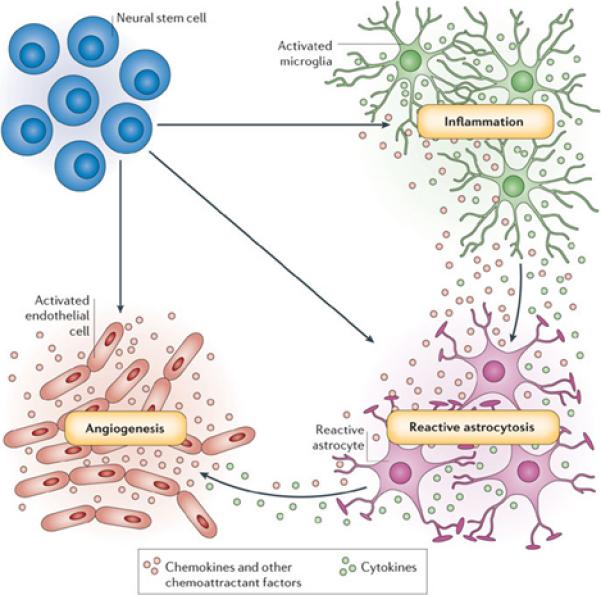
Neural stem cells are attracted by at least three physiological processes that are common to many brain pathologies: inflammation, reactive astrocytosis and angiogenesis. Pathology-induced CNS inflammation is mediated by activated microglia that release cytokines and chemokines, which, in turn, increases the inflammatory reaction. The brain lesion and subsequent inflammation trigger reactive astrocytosis. The lesion-induced angiogenesis and inflammation-activated endothelial cells enhance neural stem cell homing to brain pathology by secreting chemoattractant factors, and also offer an atypical, perivascular niche for support of immigrating neural stem cells. Used with permission 66.
In regards to cell-mediated drug delivery of antineoplastic agents, MP are known to accumulate in large numbers in vascular, hypoxic sites in cancer tumors, for example, in breast and prostate carcinomas 67, 68. Hypoxia is widespread in malignant human tumors due to their poorly organized vasculature. The cytokines released by tumor cells in response to hypoxia and other physiological stresses usually attract macrophages and monocytes, which should facilitate anticancer drug delivery using these cells as vehicles, avoiding indiscriminate drug distribution and decreasing severe toxicity.
Finally, targeting to disease sites can be improved by drug-incorporated magnetic nanoparticles loaded into the cell-carriers and followed by application of local magnetic fields 37. Thus, albino rats with brain inflammation (induced by intrastratial microinjection of human recombinant IL-1β) received intravenous injections of RGD-coated magnetic liposomes and non-magnetic liposomes as a control. RGD peptide (i.e. small peptide domane Arg-Gly-Asp) was used for selective binding by monocytes and neutrophils that express integrin receptors on their surface. Magnetic liposomes demonstrated about a ten-fold increase in brain levels compared with non-magnetic controlled carriers when local magnetic field was applied. In addition, magnetic neutrophils prepared in vitro target lungs under magnetic guidance following intravenous injection 69. Moreover, drug loaded magnetic liposomes can be targeted for selective and preferential presentation to blood monocytes/neutrophils that result in both drug and magnetic incorporation into these cells and can be guided to target tissue sites.
Safety of cell-mediated drug delivery systems
An obvious concern for inflammatory-response cell based drug delivery relates to possible cytotoxic effects of cell-carriers. MP attracted to the site of pathology by cytokines release reactive oxygen species (ROS) that cause cell damage. Moreover, a number of therapeutic strategies for CNS neurodegenerative disorders are based on the prevention of monocyte-macrophage infiltration 70. Therefore, precluding cytotoxicities for cell-based drug formulations is a requirement for formulation developments. Furthermore, genetically-modified and immortalized cell-carriers show atypical characteristics, such as higher degree of multipotency, which may increase the probability of tumor formation. Nevertheless, studies so far report no cytotoxic effects after macrophage-mediated drug delivery in the brain of healthy C57BL/6 mice adoptively transferred with macrophages carrying nanoformulated catalase 71. In addition, propagation and expansion of immortalized and genetically-modified cell-carriers should allow improve quality controls 66.
Cell-mediated drug delivery systems in clinical settings
Finally, developing such methods in a clinical setting may be successful when cell-carriers are harvested from peripheral blood by apheresis, then loaded with particles and re-infused into the patient. An alternative approach may be harvesting stem cells from bone marrow, propagation them in culture to obtain monocytes, and then loading the cells with a drug and adoptively transferring them. This procedure will allow for expansion of the cell population, although this would require a more invasive procedure. To this end, immortalization of cell-carriers can allow propagation of cells with definable properties so that clonal populations with particular traits can be established. Furthermore, drug loading into cell-carriers can be achieved directly in the patient's peripheral blood, when nanoformulated drugs would be injected and selectively taken up by circulating cell carriers. Targeting of such nanocarriers could be achieved by coating the nanocarrier surface with the receptor-specific moieties (e.g., folate, gelatin, fibronectin, A-protein, mannose, or RGD peptide), which is recognized by specific MP surface receptors. Overall, chronic diseases are more suitable for this type of drug delivery than pathologies that require acute intervention. Based on such considerations, we now present some successful investigations using living cells as drug delivery vehicles for therapeutics (Table 1).
Table 1.
Examples of cell-mediated drug delivery systems
| Type of cell-carrier | Therapeutic agent | Rote of administration1 | References |
|---|---|---|---|
| Alziemer's disease | |||
| Neural stem cells (NSCs) | ciliary neurotrophic factor (NTFs) | i.c. | 73 |
| NSCs | nerve growth factor (NGF) | i.c. | 65 |
| NSCs | brain-derived neurotropic factor (BDNF) | i.c. | 64, 75 |
| NSCs | choline acetyltransferase (ChAT) | i.c. | 72, 74 |
| Parkinson's disease | |||
| Monocytes, macrophages | catalase | i.v. | 29, 48, 71 |
| Fetal dopamine neurons | dopamine | i.c. | 76 |
| NSCs | glial cell-line derived neurotropic factor (GDNF) | i.c. | 77, 80 |
| NSCs | vascular endothelial growth factor (VGEF) | i.c. | 78, 79 |
| HIV-1 infection and neuroAIDS | |||
| Monocytes, macrophages | atazanavir, ritonavir, indinavir | i.v. | 14, 81-83 |
| Cancer | |||
| T-cells | doxorubicin | In vitro | 94 |
| Mesenchymal stem cells (MSCs) | tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) | ipsilateral | 93 |
| MSCs | interferon-β | i.v. | 86, 90, 95 |
| MSCs | oncolytic adenovirus | i.v. | 91, 92 |
| MSCs | antineoplastic agent | In vitro, in 3D collagen gels | 46 |
| Erythrocytes | 5-fluorouracil | i.v. | 96 |
| Monocytes and macrophages | Au-containing nanoparticles | In vitro, in spheroids | 85, 89 |
| Introperitoneal macrophages | 5-fluorouracil | i.p. | 50 |
| Bacterially-derived mini-cells | small interfering RNA (siRNA), cytotoxic drugs | i.v., i.t. | 97, 98 |
| Bacterially-derived mini-cells | doxorubicin | i.v., i.t. | 98 |
| Macrophages | imaging agents, quantum dots | i.v. | 103 |
| Macrophages | N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer-gadolinium (Gd) chelates | i.v. | 104 |
| Hybridoma cell lines 2A and OX86 | anti-CD137 and anti-OX40 mAb | s.c. | 102 |
| Inflammation | |||
| Macrophages | betamethasone phosphate | in situ | 25 |
| Erythrocytes | glycoprotein A | 25 | |
| Sertoli cells | anti-inflammatory agent, curcumin | i.v. | 105 |
| Microbial Infections | |||
| Erythrocytes | antifungal agent, amphotericin B | in viro | 106 |
| Macrophages | chloroquine | i.v. | 107 |
| Epilepsy | |||
| Human MSCs and human embryonic stem cells | adenosine | i.c. in cell-encapsulation device | 108, 109 |
i.c., intracranial; i.v., intravenous; i.p., intraperitoneal; i.t., intratumoral; s.c., subcutaneous injections.
2. Neurodegenerative disorders and cell-based carriage of therapeutic nanoparticles
AD and PD
The progressive impairment of short-term memory and emotional disturbances that typify AD results from synaptic dysfunction and neuronal death in the hippocampus and linked regions in the cerebral cortex and limbic system. Because of the increasing numbers of affected people, there is an urgent need to develop strategies able to interfere with disease progression and protect neurons. In this respect, delivery of neurotrophic factors (NTFs) including nerve growth factor (NGF) 65, brain-derived neurotropic factor (BDNF) 64, and choline acetyltransferase (ChAT) 72 is of urgent need. To overcome limited drug transport across the BBB, direct brain implantation of cells engineered to produce NTFs was developed 73. In particular, ciliary neurotrophic factor delivered in cells implanted in the brain prevents Aβ oligomer-induced neuronal damage and neurobehavioral impairments in mouse models of AD. In another study NSCs overexpressing ChAT cDNA targeted acetylcholine deficits 74. The ability cell-carriers, placed by cortical transplants, to improve cognition was tested after induced cholinergic lesions in rats. Significant improvements were recorded in Water maze acquisition as well as in the retention and spatial probe trials 74. A robust enhancement of hippocampal synaptic density, mediated by BDNF and delivered by NSCs was seen 75. NSCs ameliorated complex behavioral deficits associated with AD pathology by BDNF. In these experiments the intracranial transplantation of the cell-carriers were utilized.
Due to specific nigrostriatal degeneration, PD was one of the first targets for cell therapy transplants. Fetal dopamine neurons were inoculated into the putamen, where the cells worked as dopamine pumps as seen during systemic administration of L-DOPA 76. It was suggested that the transplanted cells were gene therapy vehicles for dopamine delivery rather than replacement neurons 76. Delivery of neurotropic factors such as glial cell-line derived neurotropic factor (GDNF) or vascular endothelial growth factor (VGEF) in neural stem cell transplants were tested in PD mouse models 77-79. GDNF was delivered in bone marrow-derived macrophages to the effected brain 80. The cells were transduced ex vivo with lentivirus expressing a GDNF gene driven by a synthetic macrophage-specific promoter and then transplanted into recipient mice. Eight weeks after transplantation, the mice were injected with MPTP for seven days to induce nigrostriatal neurodegeneration. Macrophage GDNF treatments dramatically ameliorated MPTP-induced degeneration of tyrosine hydroxylase (TH)-positive neurons seen in the substantia nigra and TH(+) terminals in the striatum. This resulted in axon regeneration and reversed hypoactivity in the open field test.
Development of novel CNS drug delivery using macrophages carriers for delivery of the antioxidant enzyme, catalase, in MPTP mice was reported by our laboratories 48, 71. In this system, nanoformulated catalase (nanozyme) was obtained by coupling the enzyme to a cationic block copolymer, PEI-PEG, leading to a polyion complex micelle. Bone marrow-derived macrophages (BMM) carried significant amounts of catalase then slowly released the active enzyme over several days 48. The enzyme released upon stimulation of nanozyme-loaded cell-carriers decomposed microglial hydrogen peroxide produced upon nitrated alpha-synuclein (N-α-syn) or tumor necrosis factor alpha (TNF-α) induced activation in vitro. Subsequent studies examined relationships between the composition of catalase nanozyme, their physicochemical characteristics (morphology, size, and ξ-potential), and cell loadings, release, and enzymatic activities for macrophage carriage 29.
Significant amount of catalase was detected in brains of mice after transfer of BMM loaded with nanoformulated catalase following MPTP intoxication. It was demonstrated that such nanozyme-loaded BMM injected into MPTP-intoxicated mice reduce neuroinflammation and attenuate nigrostriatal degeneration 71. In particular, MPTP intoxication decreased the number of TH-positive nigral dopaminergic neurons (32 % survival) compared to saline-treated controls (Figure 5). In contrast, the number of surviving dopaminergic neurons in MPTP-intoxicated mice treated with nanozyme-loaded BMM was greater than the total number of neurons in MPTP-treated mice (62.4 % survival). Furthermore, treatment with nanozyme alone (without cell carriers) also produced some neuroprotection effect, although with fewer neurons (41.3 % survival) compared to the mice treated with cell/nanozyme formulation. Finally, treatment with empty monocytes did not preserve neurons in MPTP-intoxicated animals (31.1 % survival). This signified a neuroprotective effect of nanozyme-loaded monocytes in MPTP-induced neurodegeneration.
Figure 5. Neuroprotective effect of nanozyme-loaded BMM in an MPTP model of PD.

MPTP-intoxicated C57Bl/6 mice (18 mg/kg) were intravenously injected with saline (second bar), nanozyme alone (third bar), macrophages loaded with nanozyme (5×106 cells/mouse/100μl) (fourth bar), or empty macrophages (fifth bar). Healthy non-intoxicated animals were used as control group (first bar). Seven days after treatment the animals were sacrificed; brain slices were stained for tyrosine hydroxylase-positive nigral dopaminergic neurons (A). Results from N = 5 animals per group demonstrate a significant loss of nigrostratial neurons in MPTP treated mice, which is prevented by adoptive transfer of macrophages loaded with nanozyme (B). No significant neuroprotective effect was detected after treatment with nanozyme alone, or empty macrophages. Values are means ± SEM, and P < 0.05 compared with asaline; bMPTP; cMPTP+macrophage/nanozyme. Used with permission 71.
The possible means by which BMM-mediated therapeutic effects of the nanozymes were suggested (Figure 6) : 1) nanozyme-loaded BMM cross the BBB and release catalase at the site of inflammation (particularly, in the substantia nigra pars compacta, SNpc); 2) a “depot” was established such that nanozyme is slowly released from BMM to the blood stream and bypasses the BBB independently of cell-carriers; and 3) catalase nanozyme released from BMM in the liver and spleen suppresses peripheral leukocyte activation that results in significant protection of SNpc neurons against MPTP-induced neurodegeneration 71. Overall, as few therapeutic modalities exist which affect progression of PD and aimed at neuroregenerative therapies, the cell-mediated delivery of catalase and GDNF may represent efficacious strategies that attenuate neuroinflammation and provide neuroprotection for disease.
Figure 6. A pictorial scheme for cell-based nanoformulated drug delivery.
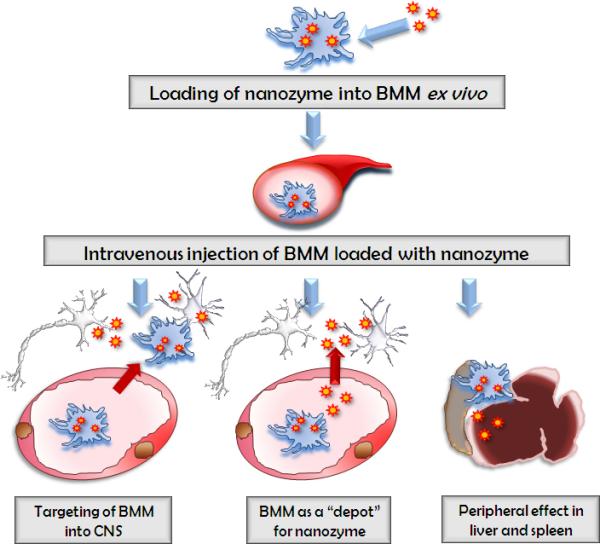
Three possible ways of BMM-mediated therapeutic effects of catalase nanozyme in PD mouse model: Pathway I: BMM loaded with nanozyme cross the BBB and release catalase in the SNpc; Pathway II: nanozyme is released from BMM to the blood stream and bypasses the BBB independently of cell-carriers; Pathway III: catalase nanozyme released from BMM in the liver and spleen suppresses peripheral leukocyte activation that results in significant protection of nigrostratial neurons against MPTP-induced neurodegeneration. Used with permission 71.
HIV-1 infections and neuroAIDS
NeuroAIDS is a clinical disorder that encompasses neurologic disorders seen primarily as a consequence of damage to the central nervous system by HIV. The clinical syndromes include cognitive, motor and behavioral disorders of varying severity. Such syndromes affect 30 to 40% of adults and children with AIDS and, despite the advent of potent combination antiretroviral therapy (cART), incidence rates remain constant although disease severity is less. MP carriage has been utilized as a delivery system for antiretroviral therapy of HIV in mice 14, 57, 81-83. To protect the drug against degradation, as well as the cell-carriers against the drug, it was incorporated into nanosized drug crystals (termed nano antiretroviral therapy or nanoART, Figure 7). NanoART were made by high-pressure homogenization of crystalline drug with various poloxamer and/or phospholipid surfactants, or by wet-milling homogenization, which produced nanoformulations with high loading capacities (over 80%) and relatively small particle sizes (200 to 350 nm). Scanning electron microscopy analysis revealed particles of various sizes and morphologies, which were either polygonal, rod, cuboidal, or spherical in shape with smooth-surfaces.
Figure 7. NanoART.

(A) NanoART of antiretroviral agents were prepared by high-pressure homogenization using an AvestinC-5 homogenizer; (B) scanning-electron microscopy demonstrated different nanoparticle morphology; (C) uptake of drug-loaded nanoART (red) in monocyte-derived macrophages (green); (D) cells treated with nanoART showed complete or near complete suppression of HIV1 p24 antigen production; (E) biodistribution of nanoART in mice following intravenious injections; (F) patient treatment with nanoART in clinical settings. Used with permission 111.
Simultaneous uptake of nanoformulated cART into the cell-carriers was shown 83. It was also demonstrated that size, charge, coating, and shape of the nanoparticles are crucial for efficient MP uptake, drug release, and cell migration 14. Coating of nanoART greatly affected drug accumulation in cell-carriers. Positively-charged nanoART are taken up better than negatively charged ones. To determine whether any of the wet-milled nanoformulations could induce long-term antiretroviral effects, macrophages were pretreated with individual nanoART preparations and then challenged with HIV-1ADA up to 15 days after drug treatment. Nanoparticles loaded with atazanavir, ritonavir and indinavir were released from macrophages and demonstrated >85, 80 and 40% inhibition, respectively, of progeny virion production and HIV-1p24 antigen through challenge day 15. All formulations of EFV showed almost complete suppression of viral infection 83. These results supported the continued development of macrophage-mediated nanoART carriage for treating HIV-1 disease. In addition, delivery of antisense oligodeoxynucleotides, ribozymes, incorporated into liposomes to HIV-1-infected cells was reported in human monocyte-derived macrophages 84. Thus, in addition to nanoART a ribozyme complementary to HIV-1 5'-long terminal repeat delivered in pH-sensitive liposomes inhibited virus production by 90%, while the free ribozyme caused limited viral inhibitions.
3. Cancer
Cell-mediated delivery is also a potential therapeutic and diagnostic strategy for cancer 46, 50, 85-93. The advantage herein is that cytotoxic activity of the cell-carriers can be used in tandem with site-specific delivery of antineoplastic agents. In addition, antineoplastic agents are safely packaged into cell-carriers to reduce secondary cytotoxicities. However, adverse effects of the cytotoxic drugs on the cell-carriers themselves need be considered. For example, when nanoparticles coated with cytotoxic antibiotic, doxorubicin, were loaded into T-cells, the release of drug inside the cell-carrier caused cell damage 94. As a result over 60% T cells loaded with the nanoparticles were dead over 15 hours after the loading.
To overcome these limitations, mesenchymal stem cells (MSCs) were genetically engineered to produce antitumor proteins 86, 90-93, 95. MSCs possess a set of several unique properties, which make them ideally suited for both cellular therapies in regenerative medicine and as vehicles for gene and drug delivery to treat cancer. These properties include: 1) relative ease of isolation; 2) the ability to differentiate into a wide variety of functional cell types of both mesenchymal and non-mesenchymal origin; 3) the ability to be extensively expanded in culture without loss of differentiative capacity; 4) the hypoimmunogenicity and even ability to produce immunosuppression upon transplantation; 5) the pronounced anti-inflammatory properties; and last but not the least, 6) the ability to home to damaged tissues, tumors, and metastases following in vivo administration. Thus, MSCs of human origins were genetically modified to express TNF related apoptosis-inducing ligand (TRAIL) at the glioma tumor site. First, the retention of tumor tropic ability of hMSC S-TRAIL cells by in vitro and in vivo migration assays was clearly demonstrated 93. Next, for the in vivo assessment of therapeutic efficacy, hMSCs were injected ipsilateral to an established intracranial glioma tumor in a mouse xenograft model. Genetically engineered hMSC S-TRAIL cells were effective in inhibiting intracranial U87 glioma tumor growth (81.6%) in vivo and resulted in significantly longer animal survival. Immunohistochemical studies demonstrated a significant, 8-fold greater tumor cell apoptosis in the hMSC S-TRAIL-treated group compared to controls. Overall, the study demonstrated the therapeutic efficacy of hMSC S-TRAIL cells and confirmed that hMSCs can serve as a powerful cell-based delivery vehicle for the site-specific release of therapeutic proteins. Furthermore, reduction of tumor growth by genetically-modified hMSCs expressing interferon-β was reported in models of metastatic breast cancer, melanoma 86, 90, and gliomas 95. The hMSC were also used as carriers for conditionally replicating adenoviruses in a breast cancer metastasis model 91 and in a model of intracranial malignant glioma 92.
Attachment of nanoparticles with cytotoxic agent to the cell surface allowed cell carriers to remain intact. Thus, NeutrAvidin-coated nanoparticles were anchored on biotinylated plasma membrane of hMSC where they remained attached for up to two days 46. The hMSC with such nanoparticulate patches retained their inherent tumoritropic properties in a tumor model with a 3D-extracellular matrix. These results provide a novel strategy to actively deliver nanostructures and therapeutics to tumors utilizing stem cells as carriers.
Besides MSCs, erythrocytes, MP, and bacterially-derived minicells were also evaluated as carriers for delivery of antineoplastic agents. In particular, erythrocytes were used as carriers for anticancer agent, 5-fluorouracil (5-FU), to treat malignant ascites 96. Intravenous injections of the cell-carriers loaded with 5-FU resulted in significant regression of the quantity of malignant ascites and the increase of survival time in mice. Notably, MP can serve as “Trojan Horses” for nanoparticle transport into tumor regions 85, 89. Thus, infiltration of monocytes and active delivery of Au-containing nanoparticles to the center of tumor spheroids (which contain hypoxic centers) was reported 89. Following the delivery the tumor spheroids were destroyed by irradiation with near-infrared light, which actuated the delivered Au-containing nanoparticles. In another study, macrophages acted as a cellular vehicle for 5-FU encapsulated in oligomannose-coated liposomes (OMLs) 50. The successful decrease of tumor growth by co-administration of OML-encased 5-FU and OML-encased magnetic nanoparticles, followed by treatment with an alternating magnetic field was reported in a mice. These OMLs were injected into the peritoneal cavity, and then gradually accumulated in the omentum and the other lymphoid tissues within 24 hours. Treatment of macrophages at 39 °C for 30 min. led to the release of 5-FU from the macrophages, suggesting that controlled release from macrophages could be achieved by mild hyperthermia. The encased magnetic nanoparticles, which are known to convert electromagnetic energy to heat in the OMLs allowed achieving in vivo hyperthermia. Finally, MP were utilized for delivery of therapeutic DNA constructs into tumors 85.
Bacterially-derived minicells with diameters of 400 nm were suggested for targeted delivery of different chemotherapeutics and small interfering RNA (siRNA) duplexes that cause stabilization and regression of tumor size in human cancer xenografts in mice 97, 98. The cell-carriers were specifically targeted to tumor cell-surface proteins with biospecific antibodies (BsAb). About 80% efficiency of packing minicells with siRNA was reported. Thus, minicells carrying at least 12,000 siRNA molecules and 100 copies of short hairpin RNA (shRNA)-encoding plasmid can specifically and sequentially deliver to tumor xenografts these therapeutics and compromise drug resistance by knocking down a drug resistance protein. This cell-mediated drug delivery system enables to use several thousand-fold less drug than needed for conventional systemic administration of cancer therapies. Evaluation of uptake and intracellular kinetics of minicell-delivered siRNA indicates that after endocytosis, the minicells traverse the well-established early and late endosomal pathways, terminating into acidified organelles, the lysosomes, where mini-cells are degraded and release their cargo98. In addition, several methods of packaging in minicells of a range of anticancer chemotherapeutic drugs, such as doxorubicin, paclitaxel, irinotecan, 5-FLU, cisplatin, carboplatin monastrol, and vinblastine despite their disparate structure, charge, hydrophobicity, and solubility were developed 99-101.
Antibody-producing hybridoma cells were used for delivery of anti-CD137 and anti-OX40 mAb 102. Microcapsules containing viable cells that secrete antibody were implanted by injection into the subcutaneous tissue of Balb/C mice bearing CT26 colon carcinomas. CD137 and OX40 are members of the TNF receptor family that provide costimulatory effect on T lymphocytes. The treatment resulted in complete tumor eradication in an elevated fraction of cases and strong tumor-specific cytotoxic T lymphocyte responses with either anti-CD137 or anti-OX40 producing hybridomas.
Besides cytotoxic agents, delivery of imaging agents is of importance for cancer therapy. MP-mediated delivery of quantum dots to brain tumors was explored for this purpose 103. Thus, the surgical management of brain tumors requires the precise localization of tumor tissues within normal brain parenchyma in order to achieve accurate diagnostic biopsy and complete surgical resection. To this end, quantum dots are optical semiconductor nanocrystals that exhibit stable, bright fluorescence. It was demonstrated that the intravenous injection of quantum dots is accompanied by reticuloendothelial system and macrophage sequestration. Macrophages infiltrate brain tumors and phagocytize intravenously injected quantum dots, optically labeling the tumors.
Another class of imaging agents, N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer-gadolinium (Gd) chelates, were used for enhanced magnetic resonance imaging (MRI) of macrophage-mediated malignancies 104. To increase the nanoparticles uptake in macrophages, copolymers were vectorized with mannosamine, which was shown to increase mannose receptor mediated uptake in human MP. It was suggested that intravenously administered GD-nanoparticles will be accumulated in macrophages and then transported to the pathological sites (e.g., solid tumor). Overall, the tumor recruitment of inflammatory-response cells may be exploited for cell-mediated delivery of antineoplastic and imaging agents.
4. Lung Injury
Drug delivery to small airways, terminal bronchioles and alveoli is complicated due to the methodological limitations in targeting the deep lung with high efficiency drug distribution to the site of pathology. To overcome these limitations, chitosan nanoparticles with entrapped curcumin (an anti-inflammatory compound) was loaded into Testis-derived Sertoli cells and then injected intravenously into mice with bronchial or alveolar injuries105. By 24-hours post-injection, most of the curcumin load (~90%) delivered in the injected Sertoli cells was present and distributed throughout the lungs including the perialveloar sac area in the lower lungs. These results identify a novel and efficient method for targeted delivery of drugs for treatment of bronchitis and alveolitis. In addition, lungs are suitable for both local and systemic drug delivery, therefore this route may be used broadly for other diseases.
5. Microbial Infections
The efficiency of antifungal agents has been diminished by their severe side effects and poor pharmacokinetics. To this end, erythrocytes were suggested as delivery vehicles for antifungal agent, amphotericin B (AmB). In order to avoid toxic effects and achieve efficient drug loading, AmB was encapsulated in nanosuspension (AmB-NS) by high-pressure homogenization 106. AmB-NS was loaded into RBC by using hypotonic hemolysis, leading to intracellular AmB amounts of 3.81 +/- 0.47 pg RBC-1 and an entrapment efficacy of 15-18%. Hence, RBC served as primary carriers. Then, the uptake of RBC loaded with AmB-NS by leucocytes was studied by flow cytometry. More than 98% of the phagocyte population (granulocytes and peripheral monocytes accumulated AmB-NS after four hours of incubation, and then showed a slow AmB release over ten days without any alteration in cell viability. This results in an immediate, permanent inhibition of intra- and extracellular fungal activity. AmBNS-RBC-leukocyte-mediated delivery of AmB was efficient in amounts 1000 times lower than the toxic dose.
Another antifungal agent, chloroquine, loaded into phosphatidylserine-containing negatively charged liposomes was used in MP-mediated delivery against C. neoformans infection in the mouse brain 107. Administration of chloroquine-loaded liposomes accumulated inside macrophage phagolysosomes resulted in a remarkable reduction in fungal load in the brain even in low doses compared to free drug in high doses thus increasing antifungal activity of macrophages. Therefore, this drug-delivery method is effective for the transport of water-insoluble substances, such as AmB, and this warrants consideration for further testing.
6. Epilepsy
Relatively straightforward method of engineered adenosine-releasing cells was developed for treatment of epilepsy 108. Lack of adenosine, a modulator of neuronal activity with anticonvulsant and neuroprotective properties, was found to contribute to ictogenesis. Therefore, focal reconstitution of adenosine within an epileptogenic brain region constitutes a rational therapeutic approach, whereas systemic augmentation of adenosine is precluded by side effects. To this end, human mesenchymal stem cells and human embryonic stem cells were embedded in a cell-encapsulation device to release adenosine and reduce acute injury and seizures, as well as chronic seizures 108. Human embryonic stem cells (hESCs) have a high proliferative capacity and can be subjected to specific cellular differentiation pathways. hESCs, differentiated in vitro into neuroepithelial cells and grafted into the mouse brain, displayed intrahippocampal location and neuronal morphology. Overall, this therapeutic approach demonstrated antiepileptic and neuroprotective properties when grafted into the mouse hippocampus. The therapeutic potential of this approach suggests the feasibility to engineer autologous adenosine-releasing stem cells derived from a patient 109.
7. Expert Opinion
The concept of using cells as drug delivery vehicles is under development. Challenges include sufficient loading capacity and triggered drug release, preservation of therapeutic agents against degradation inside the cells, protection of cell-carriers from drug cytotoxic effects, and efficient homing of cells to the disease site. In addition, harvesting of cells sufficient quantities or their expansion is also important issue.
Two different approaches are utilized in cell-mediated drug delivery. First, cell-carriers (predominantly MP) are genetically modified to produce therapeutically active molecules. Second, host cells are loaded with a drug usually incorporated into protective container, and then used as “Trojan horses” to deliver the drug to the disease side (Figure 8). Most of cell-based delivery systems suggest the unloading the therapeutic agent at the disease site. However, a few utilized another approach, when the targeted cells accumulate the carrier along with the drug (mini-cells with anticancer agents and red blood cells with antifungal agents).
Figure 8.

Cell-based Drug Delivery Systems.
Regarding the first approach, the ideal cell delivery vehicle would be stable in tissue culture and capable of sustained, preferably regulated expression of therapeutic molecules. The cells should have appropriate and predictable differentiation pattern and survive long time in vivo after transplantation. Obviously, for both approaches, the cell carriers should demonstrate responsiveness to the chemotactic signals produced by the type of pathology that they are used to treat. Regarding the second approach, loading cell-carriers with drug in the blood stream by injecting nanoparticlular drug targeted to these cells is the most attractive strategy. To this end, it is important to note that these cells are moving targets. Nanosystems that bind to these targets are potentially a powerful approach for imaging their movement and delivering a therapeutic drug dose to the disease site. Clearly, a multifunctional molecular scaffold that is aimed at delivery of drug specifically to the cell-carrier, but in addition could be engineered for imaging purposes would offer substantial benefits over current approaches.
Such strategy is opposite to the common approach when drug-loaded nanoparticles are designed to minimize their entrapment in reticulo-endothelail system and avoid drug decomposition and clearance. Consequently, design of nanocarriers for cell-mediated drug delivery (size, shape, surface charge, etc.) may differ from those used for conventional drug delivery systems. Nevertheless, engaging different defense mechanisms into drug delivery may open a new perspective of active delivery of drugs for treatment of various devastating diseases.
Acknowledgments
Declaration of interest
This work was supported by the United States National Institutes of Health grants 1RO1 NS057748 (to EVB); 2R01 NS034239, 2R37 NS36126, P01 NS31492, P20RR 15635, P01 MH64570, 5 P01 DA026146, 1P01 DA028555 and P01 NS43985 (to HEG); RO1 NS051334, 2RO1 CA89225, 1 R01 CA116591, UO1 CA151806, 1P20 RR021937, the Unites States Department of Defense grants W81XWH-09-1-0386 and DoD USAMRMC 06108004, and the Government of Russian Federation grants 02.740.11.523 and 11.G4.31.0004 (to AVK).
Footnotes
- Using cell-mediated drug delivery systems offers several advantages including: a) targeted drug transport to disease sites; b) prolonged drug half-lives; c) time-controlled release of loaded drugs; and d) diminished drug immunogenicity and cytoxicity profiles.
- The goals need to be achieved for successful cell-mediated drug delivery include: a) high drug loading into cell-carriers; b) efficient preservation of entrapped therapeutic agents against disintegration and clearance in the host cells; c) drug triggered release at the site of action; d) efficient homing of cell-carriers to a disease site; e) safety of cell-based drug formulations for the whole organism.
- Two different approaches are utilized in cell-mediated drug delivery: genetically modified cell-carriers producing therapeutically active molecules; and drug loaded cell-carriers used as “Trojan horses” to deliver the drug to the disease side.
- Living cells for drug carriage and release represent a novel disease combating strategy that can be applied to a spectrum of human infectious, cancerous, and degenerative disorders.
REFERENCES
- 1.Fujiwara M, Baldeschwieler JD, Grubbs RH. Receptor-mediated endocytosis of poly(acrylic acid)-conjugated liposomes by macrophages. Biochim Biophys Acta. 1996 Jan 12;1278(1):59–67. doi: 10.1016/0005-2736(95)00183-2. [DOI] [PubMed] [Google Scholar]
- 2.Torchilin VP. Drug targeting. Eur J Pharm Sci. 2000 Oct;11(Suppl 2):S81–91. doi: 10.1016/s0928-0987(00)00166-4. [DOI] [PubMed] [Google Scholar]
- 3.Mora M, Sagrista ML, Trombetta D, et al. Design and characterization of liposomes containing long-chain N-acylPEs for brain delivery: penetration of liposomes incorporating GM1 into the rat brain. Pharm Res. 2002 Oct;19(10):1430–8. doi: 10.1023/a:1020440229102. [DOI] [PubMed] [Google Scholar]
- 4.Aoki H, Kakinuma K, Morita K, et al. Therapeutic efficacy of targeting chemotherapy using local hyperthermia and thermosensitive liposome: evaluation of drug distribution in a rat glioma model. Int J Hyperthermia. 2004 Sep;20(6):595–605. doi: 10.1080/02656730410001703186. [DOI] [PubMed] [Google Scholar]
- 5.Kabanov AV, Chekhonin VP, Alakhov V, et al. The neuroleptic activity of haloperidol increases after its solubilization in surfactant micelles. Micelles as microcontainers for drug targeting. FEBS Lett. 1989 Dec 4;258(2):343–5. doi: 10.1016/0014-5793(89)81689-8. [DOI] [PubMed] [Google Scholar]
- 6.Kabanov AV, Vinogradov SV, Suzdaltseva YG, et al. Water-soluble block polycations as carriers for oligonucleotide delivery. Bioconjug Chem. 1995 Nov-Dec;6(6):639–43. doi: 10.1021/bc00036a001. [DOI] [PubMed] [Google Scholar]
- 7.Kabanov A, Alakhov V. Pluronic block copolymers in drug delivery: from micellar nanocontainers to biological response modifiers. Crit Rev Ther Drug Carrier Syst. 2002;19(1):1–72. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.10. [DOI] [PubMed] [Google Scholar]
- 8.Kwon GS. Polymeric micelles for delivery of poorly water-soluble compounds. Crit Rev Ther Drug Carrier Syst. 2003;20(5):357–403. doi: 10.1615/critrevtherdrugcarriersyst.v20.i5.20. [DOI] [PubMed] [Google Scholar]
- 9.Vinogradov S, Batrakova E, Kabanov A. Poly(ethylene glycol)-polyethyleneimine NanoGel (TM) particles: novel drug delivery systems for antisense oligonucleotides. Colloids and Surfaces B-Biointerfaces. 1999 Nov;16(1-4):291–304. [Google Scholar]
- 10.Vinogradov SV, Batrakova EV, Kabanov AV. Nanogels for oligonucleotide delivery to the brain. Bioconjug Chem. 2004 Jan-Feb;15(1):50–60. doi: 10.1021/bc034164r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gref R, Minamitake Y, Peracchia M, et al. Biodegradable long-circulating polymeric nanospheres. Science. 1994 Mar 18;263(5153):1600–3. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 12.Hyuk IS, Jeong U, Xia Y. Polymer hollow particles with controllable holes in their surfaces. Nat Mater. 2005 Sep;4(9):671–5. doi: 10.1038/nmat1448. [DOI] [PubMed] [Google Scholar]
- 13.Calvo P, Gouritin B, Chacun H, et al. Long-circulating PEGylated polycyanoacrylate nanoparticles as new drug carrier for brain delivery. Pharm Res. 2001 Aug;18(8):1157–66. doi: 10.1023/a:1010931127745. [DOI] [PubMed] [Google Scholar]
- 14**.Nowacek AS, Miller RL, McMillan J, et al. NanoART synthesis, characterization, uptake, release and toxicology for human monocyte-macrophage drug delivery. Nanomed. 2009 Dec;4(8):903–17. doi: 10.2217/nnm.09.71. [This study described characterization of nanoformulated antiretroviral drugs for HIV-1 infection and its application for human cell-mediated drug delivery.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001 Mar 23;47(1):3–19. doi: 10.1016/s0169-409x(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 16.Friedrich I, Reichl S, Muller-Goymann CC. Drug release and permeation studies of nanosuspensions based on solidified reverse micellar solutions (SRMS). Int J Pharm. 2005 Nov 23;305(1-2):167–75. doi: 10.1016/j.ijpharm.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 17.Harada A, Kataoka K. Chain length recognition: core-shell supramolecular assembly from oppositely charged block copolymers. Science. 1999 Jan 1;283(5398):65–7. doi: 10.1126/science.283.5398.65. [DOI] [PubMed] [Google Scholar]
- 18.Jaturanpinyo M, Harada A, Yuan X, et al. Preparation of bionanoreactor based on core-shell structured polyion complex micelles entrapping trypsin in the core cross-linked with glutaraldehyde. Bioconjug Chem. 2004 Mar-Apr;15(2):344–8. doi: 10.1021/bc034149m. [DOI] [PubMed] [Google Scholar]
- 19.Bull SR, Guler MO, Bras RE, et al. Self-assembled peptide amphiphile nanofibers conjugated to MRI contrast agents. Nano Lett. 2005 Jan;5(1):1–4. doi: 10.1021/nl0484898. [DOI] [PubMed] [Google Scholar]
- 20.Guler MO, Pokorski JK, Appella DH, et al. Enhanced oligonucleotide binding to self-assembled nanofibers. Bioconjug Chem. 2005 May-Jun;16(3):501–3. doi: 10.1021/bc050053b. [DOI] [PubMed] [Google Scholar]
- 21.Juliano R, Stamp D. The effect of particle size and charge on the clearance rates of liposomes and liposome encapsulated drugs. Biochem Biophys Res Commun. 1975;63:651–8. doi: 10.1016/s0006-291x(75)80433-5. [DOI] [PubMed] [Google Scholar]
- 22.Lee KD, Hong K, Papahadjopoulos D. Recognition of liposomes by cells: in vitro binding and endocytosis mediated by specific lipid headgroups and surface charge density. Biochim Biophys Acta. 1992 Jan 31;1103(2):185–97. doi: 10.1016/0005-2736(92)90086-2. [DOI] [PubMed] [Google Scholar]
- 23.Nishikawa K, Arai H, Inoue K. Scavenger receptor-mediated uptake and metabolism of lipid vesicles containing acidic phospholipids by mouse peritoneal macrophages. J Biol Chem. 1990 Mar 25;265(9):5226–31. [PubMed] [Google Scholar]
- 24.Miller CR, Bondurant B, McLean SD, et al. Liposome-cell interactions in vitro: effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry. 1998 Sep 15;37(37):12875–83. doi: 10.1021/bi980096y. [DOI] [PubMed] [Google Scholar]
- 25.Ishihara T, Izumo N, Higaki M, et al. Role of zinc in formulation of PLGA/PLA nanoparticles encapsulating betamethasone phosphate and its release profile. J Control Release. 2005 Jun 20;105(1-2):68–76. doi: 10.1016/j.jconrel.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 26.Tempone AG, Perez D, Rath S, et al. Targeting Leishmania (L.) chagasi amastigotes through macrophage scavenger receptors: the use of drugs entrapped in liposomes containing phosphatidylserine. J Antimicrob Chemother. 2004 Jul;54(1):60–8. doi: 10.1093/jac/dkh281. [DOI] [PubMed] [Google Scholar]
- 27.Krieger M. Molecular flypaper and atherosclerosis: structure of the macrophage scavenger receptor. Trends Biochem Sci. 1992 Apr;17(4):141–6. doi: 10.1016/0968-0004(92)90322-z. [DOI] [PubMed] [Google Scholar]
- 28.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Y, Haney MJ, Klyachko NL, et al. Polyelectrolyte complex optimization for macrophage delivery of redox enzyme nanoparticles. Nanomedicine (Lond) 2011 Jan;6(1):25–42. doi: 10.2217/nnm.10.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harada A, Kataoka K. Pronounced activity of enzymes through the incorporation into the core of polyion complex micelles made from charged block copolymers. J Control Release. 2001 May 14;72(1-3):85–91. doi: 10.1016/s0168-3659(01)00264-4. [DOI] [PubMed] [Google Scholar]
- 31.Harada A, Kataoka K. Switching by pulse electric field of the elevated enzymatic reaction in the core of polyion complex micelles. J Am Chem Soc. 2003 Dec 17;125(50):15306–7. doi: 10.1021/ja038572h. [DOI] [PubMed] [Google Scholar]
- 32.Knop K, Hoogenboom R, Fischer D, et al. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed Engl. Aug 23;49(36):6288–308. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- 33.Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42(5):419–36. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 34.Papaldo P, Fabi A, Ferretti G, et al. A phase II study on metastatic breast cancer patients treated with weekly vinorelbine with or without trastuzumab according to HER2 expression: changing the natural history of HER2-positive disease. Ann Oncol. 2006 Apr;17(4):630–6. doi: 10.1093/annonc/mdj110. [DOI] [PubMed] [Google Scholar]
- 35.Gbadamosi JK, Hunter AC, Moghimi SM. PEGylation of microspheres generates a heterogeneous population of particles with differential surface characteristics and biological performance. FEBS Lett. 2002 Dec 18;532(3):338–44. doi: 10.1016/s0014-5793(02)03710-9. [DOI] [PubMed] [Google Scholar]
- 36.Daleke DL, Hong K, Papahadjopoulos D. Endocytosis of liposomes by macrophages: binding, acidification and leakage of liposomes monitored by a new fluorescence assay. Biochim Biophys Acta. 1990 May 24;1024(2):352–66. doi: 10.1016/0005-2736(90)90365-u. [DOI] [PubMed] [Google Scholar]
- 37.Jain S, Mishra V, Singh P, et al. RGD-anchored magnetic liposomes for monocytes/neutrophils-mediated brain targeting. Int J Pharm. 2003 Aug 11;261(1-2):43–55. doi: 10.1016/s0378-5173(03)00269-2. [DOI] [PubMed] [Google Scholar]
- 38.Thiele L, Merkle HP, Walter E. Phagocytosis and phagosomal fate of surface-modified microparticles in dendritic cells and macrophages. Pharm Res. 2003 Feb;20(2):221–8. doi: 10.1023/a:1022271020390. [DOI] [PubMed] [Google Scholar]
- 39.Fahmy TM, Schneck JP, Saltzman WM. A nanoscopic multivalent antigen-presenting carrier for sensitive detection and drug delivery to T cells. Nanomedicine. 2007 Mar;3(1):75–85. doi: 10.1016/j.nano.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 40.Tabata Y, Ikada Y. Effect of the size and surface charge of polymer microspheres on their phagocytosis by macrophage. Biomaterials. 1988 Jul;9(4):356–62. doi: 10.1016/0142-9612(88)90033-6. [DOI] [PubMed] [Google Scholar]
- 41.Mizushima Y, Hamano T, Yokoyama K. Tissue distribution and anti-inflammatory activity of corticosteroids incorporated in lipid emulsion. Ann Rheum Dis. 1982 Jun;41(3):263–7. doi: 10.1136/ard.41.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hillaireau H, Couvreur P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009 Sep;66(17):2873–96. doi: 10.1007/s00018-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cannon GJ, Swanson JA. The macrophage capacity for phagocytosis. J Cell Sci. 1992 Apr;101(Pt 4):907–13. doi: 10.1242/jcs.101.4.907. [DOI] [PubMed] [Google Scholar]
- 44.Champion JA, Katare YK, Mitragotri S. Making polymeric micro- and nanoparticles of complex shapes. Proc Natl Acad Sci U S A. 2007 Jul 17;104(29):11901–4. doi: 10.1073/pnas.0705326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Behr J-P. The Proton Sponge: a Trick to Enter Cells the Viruses Did Not Exploit CHIMIA. International Journal for Chemistry. 1997;51(1):34–6. [Google Scholar]
- 46.Cheng H, Kastrup CJ, Ramanathan R, et al. Nanoparticulate cellular patches for cell-mediated tumoritropic delivery. ACS Nano. 2010 Feb 23;4(2):625–31. doi: 10.1021/nn901319y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krantz A. Red cell-mediated therapy: opportunities and challenges. Blood Cells Mol Dis. 1997;23(1):58–68. doi: 10.1006/bcmd.1997.0119. [DOI] [PubMed] [Google Scholar]
- 48*.Batrakova EV, Li S, Reynolds AD, et al. A macrophage-nanozyme delivery system for Parkinson's disease. Bioconjug Chem. 2007 Sep-Oct;18(5):1498–506. doi: 10.1021/bc700184b. [This manuscript reported development and characterization of cell-mediated delivery system of antioxidant enzyme, catalase. Loading, release, and protection of enzymatic activity of catalase in bone marrow-derived monocytes (BMM) are evaluated in in vitro model of PD.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sollner T, Bennett MK, Whiteheart SW, et al. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993 Nov 5;75(3):409–18. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 50.Ikehara Y, Niwa T, Biao L, et al. A carbohydrate recognition-based drug delivery and controlled release system using intraperitoneal macrophages as a cellular vehicle. Cancer Res. 2006 Sep 1;66(17):8740–8. doi: 10.1158/0008-5472.CAN-06-0470. [DOI] [PubMed] [Google Scholar]
- 51.Perry VH, Bell MD, Brown HC, et al. Inflammation in the nervous system. Curr Opin Neurobiol. 1995 Oct;5(5):636–41. doi: 10.1016/0959-4388(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 52.Kuby J. Immunology. Freeman, WH. and Co.; New York: 1994. [Google Scholar]
- 53.Anthony DC, Bolton SJ, Fearn S, et al. Age-related effects of interleukin-1 beta on polymorphonuclear neutrophil-dependent increases in blood-brain barrier permeability in rats. Brain. 1997 Mar;120(Pt 3):435–44. doi: 10.1093/brain/120.3.435. [DOI] [PubMed] [Google Scholar]
- 54.Anthony DC, Blond D, Dempster R, et al. Chemokine targets in acute brain injury and disease. Prog Brain Res. 2001;132:507–24. doi: 10.1016/s0079-6123(01)32099-x. [DOI] [PubMed] [Google Scholar]
- 55.Blamire AM, Anthony DC, Rajagopalan B, et al. Interleukin-1beta -induced changes in blood-brain barrier permeability, apparent diffusion coefficient, and cerebral blood volume in the rat brain: a magnetic resonance study. J Neurosci. 2000 Nov 1;20(21):8153–9. doi: 10.1523/JNEUROSCI.20-21-08153.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Persidsky Y, Ghorpade A, Rasmussen J, et al. Microglial and astrocyte chemokines regulate monocyte migration through the blood-brain barrier in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999 Nov;155(5):1599–611. doi: 10.1016/S0002-9440(10)65476-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dou H, Destache CJ, Morehead JR, et al. Development of a macrophage-based nanoparticle platform for antiretroviral drug delivery. Blood. 2006 Oct 15;108(8):2827–35. doi: 10.1182/blood-2006-03-012534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pawlowski NA, Kaplan G, Abraham E, et al. The selective binding and transmigration of monocytes through the junctional complexes of human endothelium. J Exp Med. 1988 Nov 1;168(5):1865–82. doi: 10.1084/jem.168.5.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lossinsky AS, Shivers RR. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Review. Histol Histopathol. 2004 Apr;19(2):535–64. doi: 10.14670/HH-19.535. [DOI] [PubMed] [Google Scholar]
- 60.Orlic D, Kajstura J, Chimenti S, et al. Bone marrow cells regenerate infarcted myocardium. Nature. 2001 Apr 5;410(6829):701–5. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 61.Orlic D, Kajstura J, Chimenti S, et al. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc Natl Acad Sci U S A. 2001 Aug 28;98(18):10344–9. doi: 10.1073/pnas.181177898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hofstetter CP, Schwarz EJ, Hess D, et al. Marrow stromal cells form guiding strands in the injured spinal cord and promote recovery. Proc Natl Acad Sci U S A. 2002 Feb 19;99(4):2199–204. doi: 10.1073/pnas.042678299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mahmood A, Lu D, Lu M, et al. Treatment of traumatic brain injury in adult rats with intravenous administration of human bone marrow stromal cells. Neurosurgery. 2003 Sep;53(3):697–702. doi: 10.1227/01.neu.0000079333.61863.aa. discussion -3. [DOI] [PubMed] [Google Scholar]
- 64.Martinez-Serrano A, Hantzopoulos PA, Bjorklund A. Ex vivo gene transfer of brain-derived neurotrophic factor to the intact rat forebrain: neurotrophic effects on cholinergic neurons. Eur J Neurosci. 1996 Apr;8(4):727–35. doi: 10.1111/j.1460-9568.1996.tb01258.x. [DOI] [PubMed] [Google Scholar]
- 65.Martinez-Serrano A, Bjorklund A. Ex vivo nerve growth factor gene transfer to the basal forebrain in presymptomatic middle-aged rats prevents the development of cholinergic neuron atrophy and cognitive impairment during aging. Proc Natl Acad Sci U S A. 1998 Feb 17;95(4):1858–63. doi: 10.1073/pnas.95.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muller FJ, Snyder EY, Loring JF. Gene therapy: can neural stem cells deliver? Nat Rev Neurosci. 2006 Jan;7(1):75–84. doi: 10.1038/nrn1829. [DOI] [PubMed] [Google Scholar]
- 67.Leek RD, Lewis CE, Whitehouse R, et al. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996 Oct 15;56(20):4625–9. [PubMed] [Google Scholar]
- 68.Lewis JS, Landers RJ, Underwood JC, et al. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J Pathol. 2000 Oct;192(2):150–8. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH687>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 69.Ranney DF, Huffaker HH. Magnetic microspheres for the targeted controlled release of drugs and diagnostic agents. Ann N Y Acad Sci. 1987;507:104–19. doi: 10.1111/j.1749-6632.1987.tb45795.x. [DOI] [PubMed] [Google Scholar]
- 70.Hendriks JJ, Teunissen CE, de Vries HE, et al. Macrophages and neurodegeneration. Brain Res Brain Res Rev. 2005 Apr;48(2):185–95. doi: 10.1016/j.brainresrev.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 71**.Brynskikh AM, Zhao Y, Mosley RL, et al. Macrophage delivery of therapeutic nanozymes in a murine model of Parkinson's disease. Nanomedicine (Lond) 2010 Apr;5(3):379–96. doi: 10.2217/nnm.10.7. [This study supports the feasibility of cell-mediated drug delivery to the brain by examining i) the nanozyme loading capacity for cell carriers; ii) the effect of nanozymes on cell viability and function; and iii) the neuroprotective activities of BMM-carried nanozyme against MPTP intoxication in in vivo model of PD.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Low WC, Lewis PR, Bunch ST, et al. Function recovery following neural transplantation of embryonic septal nuclei in adult rats with septohippocampal lesions. Nature. 1982 Nov 18;300(5889):260–2. doi: 10.1038/300260a0. [DOI] [PubMed] [Google Scholar]
- 73.Garcia P, Youssef I, Utvik JK, et al. Ciliary neurotrophic factor cell-based delivery prevents synaptic impairment and improves memory in mouse models of Alzheimer's disease. J Neurosci. 2010 Jun 2;30(22):7516–27. doi: 10.1523/JNEUROSCI.4182-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pizzo DP, Coufal NG, Lortie MJ, et al. Regulatable acetylcholine-producing fibroblasts enhance cognitive performance. Mol Ther. 2006 Jan;13(1):175–82. doi: 10.1016/j.ymthe.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 75.Blurton-Jones M, Kitazawa M, Martinez-Coria H, et al. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A. 2009 Aug 11;106(32):13594–9. doi: 10.1073/pnas.0901402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zurn AD, Tseng J, Aebischer P. Treatment of Parkinson's disease. Symptomatic cell therapies: cells as biological minipumps. Eur Neurol. 1996;36(6):405–8. [PubMed] [Google Scholar]
- 77.Akerud P, Canals JM, Snyder EY, et al. Neuroprotection through delivery of glial cell line-derived neurotrophic factor by neural stem cells in a mouse model of Parkinson's disease. J Neurosci. 2001 Oct 15;21(20):8108–18. doi: 10.1523/JNEUROSCI.21-20-08108.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Casper D, Engstrom SJ, Mirchandani GR, et al. Enhanced vascularization and survival of neural transplants with ex vivo angiogenic gene transfer. Cell Transplant. 2002;11(4):331–49. [PubMed] [Google Scholar]
- 79.Yasuhara T, Shingo T, Muraoka K, et al. Neurorescue effects of VEGF on a rat model of Parkinson's disease. Brain Res. 2005 Aug 16;1053(1-2):10–8. doi: 10.1016/j.brainres.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 80.Biju K, Zhou Q, Li G, et al. Macrophage-mediated GDNF delivery protects against dopaminergic neurodegeneration: a therapeutic strategy for Parkinson's disease. Mol Ther. 2010 Aug;18(8):1536–44. doi: 10.1038/mt.2010.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dou H, Morehead JR, Destache C, et al. Laboratory investigations for the morphologic, pharmacokinetic, and anti-retroviral properties of indinavir nanoparticles in human nomocyte-derived macrophages. Virology. 2007 Feb;358(1):148–58. doi: 10.1016/j.virol.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 82.Dou H, Grotepas CB, McMillan JM, et al. Macrophage delivery of nanoformulated antiretroviral drug to the brain in a murine model of neuroAIDS. J Immunol. 2009 Jul 1;183(1):661–9. doi: 10.4049/jimmunol.0900274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nowacek AS, McMillan J, Miller R, et al. Nanoformulated Antiretroviral Drug Combinations Extend Drug Release and Antiretroviral Responses in HIV-1-Infected Macrophages: Implications for NeuroAIDS Therapeutics. J Neuroimmune Pharmacol. 2010 Mar 17; doi: 10.1007/s11481-010-9198-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Duzgunes N, Pretzer E, Simoes S, et al. Liposome-mediated delivery of antiviral agents to human immunodeficiency virus-infected cells. Mol Membr Biol. 1999 Jan-Mar;16(1):111–8. doi: 10.1080/096876899294832. [DOI] [PubMed] [Google Scholar]
- 85.Chokri M, Lopez M, Oleron C, et al. Production of human macrophages with potent antitumor properties (MAK) by culture of monocytes in the presence of GM-CSF and 1,25-dihydroxy vitamin D3. Anticancer Res. 1992 Nov-Dec;12(6B):2257–60. [PubMed] [Google Scholar]
- 86.Studeny M, Marini FC, Champlin RE, et al. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002 Jul 1;62(13):3603–8. [PubMed] [Google Scholar]
- 87.Stagg J, Lejeune L, Paquin A, et al. Marrow stromal cells for interleukin-2 delivery in cancer immunotherapy. Hum Gene Ther. 2004 Jun;15(6):597–608. doi: 10.1089/104303404323142042. [DOI] [PubMed] [Google Scholar]
- 88.Nakamura K, Ito Y, Kawano Y, Kurozumi K, et al. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004 Jul;11(14):1155–64. doi: 10.1038/sj.gt.3302276. [DOI] [PubMed] [Google Scholar]
- 89.Choi MR, Stanton-Maxey KJ, Stanley JK, et al. A cellular Trojan Horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007 Dec;7(12):3759–65. doi: 10.1021/nl072209h. [DOI] [PubMed] [Google Scholar]
- 90.Studeny M, Marini FC, Dembinski JL, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst. 2004 Nov 3;96(21):1593–603. doi: 10.1093/jnci/djh299. [DOI] [PubMed] [Google Scholar]
- 91.Stoff-Khalili MA, Rivera AA, Mathis JM, et al. Mesenchymal stem cells as a vehicle for targeted delivery of CRAds to lung metastases of breast carcinoma. Breast Cancer Res Treat. 2007 Oct;105(2):157–67. doi: 10.1007/s10549-006-9449-8. [DOI] [PubMed] [Google Scholar]
- 92.Sonabend AM, Ulasov IV, Tyler MA, et al. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem Cells. 2008 Mar;26(3):831–41. doi: 10.1634/stemcells.2007-0758. [DOI] [PubMed] [Google Scholar]
- 93.Menon LG, Kelly K, Yang HW, et al. Human bone marrow-derived mesenchymal stromal cells expressing S-TRAIL as a cellular delivery vehicle for human glioma therapy. Stem Cells. 2009 Sep;27(9):2320–30. doi: 10.1002/stem.136. [DOI] [PubMed] [Google Scholar]
- 94.Steinfeld U, Pauli C, Kaltz N, et al. T lymphocytes as potential therapeutic drug carrier for cancer treatment. Int J Pharm. 2006 Mar 27;311(1-2):229–36. doi: 10.1016/j.ijpharm.2005.12.040. [DOI] [PubMed] [Google Scholar]
- 95.Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005 Apr 15;65(8):3307–18. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 96.Wang GP, Guan YS, Jin XR, et al. Development of novel 5-fluorouracil carrier erythrocyte with pharmacokinetics and potent antitumor activity in mice bearing malignant ascites. J Gastroenterol Hepatol. 2010 May;25(5):985–90. doi: 10.1111/j.1440-1746.2009.06155.x. [DOI] [PubMed] [Google Scholar]
- 97.MacDiarmid JA, Mugridge NB, Weiss JC, et al. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell. 2007 May;11(5):431–45. doi: 10.1016/j.ccr.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 98.MacDiarmid JA, Amaro-Mugridge NB, Madrid-Weiss J, et al. Sequential treatment of drug-resistant tumors with targeted minicells containing siRNA or a cytotoxic drug. Nat Biotechnol. 2009 Jul;27(7):643–51. doi: 10.1038/nbt.1547. [DOI] [PubMed] [Google Scholar]
- 99.Nikaido H. Multidrug efflux pumps of gram-negative bacteria. J Bacteriol. 1996 Oct;178(20):5853–9. doi: 10.1128/jb.178.20.5853-5859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003 Dec;67(4):593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Poole K. Outer membranes and efflux: the path to multidrug resistance in Gram-negative bacteria. Curr Pharm Biotechnol. 2002 Jun;3(2):77–98. doi: 10.2174/1389201023378454. [DOI] [PubMed] [Google Scholar]
- 102.Dubrot J, Portero A, Orive G, et al. Delivery of immunostimulatory monoclonal antibodies by encapsulated hybridoma cells. Cancer Immunol Immunother. 2010 Nov;59(11):1621–31. doi: 10.1007/s00262-010-0888-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Popescu MA, Toms SA. In vivo optical imaging using quantum dots for the management of brain tumors. Expert Rev Mol Diagn. 2006 Nov;6(6):879–90. doi: 10.1586/14737159.6.6.879. [DOI] [PubMed] [Google Scholar]
- 104.Zarabi B, Nan A, Zhuo J, et al. Macrophage targeted N-(2-hydroxypropyl)methacrylamide conjugates for magnetic resonance imaging. Mol Pharm. 2006 Sep-Oct;3(5):550–7. doi: 10.1021/mp060072i. [DOI] [PubMed] [Google Scholar]
- 105.Kumar A, El-Badri N, Glaum M, et al. Initial Observations of Cell Mediated Drug Delivery to the Deep Lung. Cell Transplant. 2010 Nov 5; doi: 10.3727/096368910X536491. Nov 5, (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Staedtke V, Brahler M, Muller A, et al. In vitro inhibition of fungal activity by macrophage-mediated sequestration and release of encapsulated amphotericin B nanosupension in red blood cells. Small. 2010 Jan;6(1):96–103. doi: 10.1002/smll.200900919. [DOI] [PubMed] [Google Scholar]
- 107.Khan MA, Jabeen R, Nasti TH, et al. Enhanced anticryptococcal activity of chloroquine in phosphatidylserine-containing liposomes in a murine model. J Antimicrob Chemother. 2005 Feb;55(2):223–8. doi: 10.1093/jac/dkh522. [DOI] [PubMed] [Google Scholar]
- 108.Boison D. Engineered adenosine-releasing cells for epilepsy therapy: human mesenchymal stem cells and human embryonic stem cells. Neurotherapeutics. 2009 Apr;6(2):278–83. doi: 10.1016/j.nurt.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Boison D. Adenosine augmentation therapies (AATs) for epilepsy: prospect of cell and gene therapies. Epilepsy Res. 2009 Aug;85(2-3):131–41. doi: 10.1016/j.eplepsyres.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kabanov AV, Batrakova EV. Polymer nanomaterials. In: Gendelman HE, Ikezu T, editors. Neuroimmune Pharmacology. Springer; Omaha: 2008. pp. 691–707. [Google Scholar]
- 111.Nowacek A, Gendelman HE. NanoART, neuroAIDS and CNS drug delivery. Nanomedicine (Lond) 2009 Jul;4(5):557–74. doi: 10.2217/nnm.09.38. [DOI] [PMC free article] [PubMed] [Google Scholar]