

Abstract
Hereditary multiple intestinal atresia (HMIA), a presumed autosomal recessive disorder, is an unusual and rare form of recurrent intestinal atresia which can be associated with severe combined immunodeficiency (SCID). The combination of HMIA and SCID is invariably lethal. The authors describe this fatal association in two siblings. The parents are consanguineous and have three other normal healthy children. Both index cases had abnormal antenatal ultrasounds and were symptomatic after birth. The final diagnosis of HMIA with SCID was confirmed in both siblings. They were never able to receive enteral feeds, remained totally dependent on parenteral nutrition, had repeated episodes of sepsis and died after a very difficult neonatal intensive care course. In this article we have reviewed the clinical course and outcome of both cases. The existing literature on multiple intestinal atresia, HMIA and HMIA with immunodeficiency is also reviewed.
Background
Approximately one third of all cases of neonatal intestinal obstruction are due to intestinal atresia, which can be sporadic or hereditary with a possible autosomal recessive mode of inheritance. The causes, clinical presentation, diagnosis, management and outcome vary considerably according to the location of obstruction.1–3 A co-existing immunodeficiency has been described with familial forms of multiple intestinal atresia (MIA) (hereditary multiple intestinal atresia, HMIA).4 The combination of HMIA and immunodeficiency leads to total parenteral nutrition (TPN) dependence and repeated episodes of sepsis making the association invariably lethal. Both our cases were suspected on antenatal ultrasound scans. Antenatal diagnosis and intervention is an emerging area of experience in this association. Early postnatal immunodeficiency screening in all cases of HMIA may help improve management if a human leucocyte antigen (HLA) compatible donor and bone marrow transplant (BMT) facility are available. The two siblings described in this article could have survived if an HLA compatible donor had been available.
Case presentation
Case A
In 2005, a female baby was born by normal vaginal delivery to a 27-year-old, G2 P1 Yemeni woman at 35 weeks gestation. The baby was in very good condition at birth. The parents are first-degree cousins with no family history of any significant illness. The parent's first male baby was normal and healthy. During this later pregnancy, routine antenatal ultrasound had revealed polyhydramnios, an intra-abdominal cyst and ascites. Antenatal karyotyping was normal. Soon after birth the baby developed repeated bilious vomiting with abdominal distension.
Case B
In 2008, the parents had their fourth female baby born at 36 weeks gestation by an uneventful normal vaginal delivery. During this pregnancy the serial antenatal ultrasound scans reported polyhydramnios with fetal duodenal atresia and an abdominal mass. Antenatal karyotyping was normal. The baby was born in good condition with a birth weight of 3020 g. Her clinical examination was normal with no dysmorphic features.
Investigations
Case A
Plain x-rays showed multiple calcifications throughout the abdomen. Ultrasonographic examination showed a calcified 1.5 cm cyst in the lower abdomen with grossly dilated first and second parts of the duodenum. Upper gastrointestinal tract contrast study revealed a dilated stomach with no contrast passing beyond the gastric outlet (figure 1). Histopathology examination of the resected jejunum showed features suggestive of intestinal atresia (figure 2).
Figure 1.
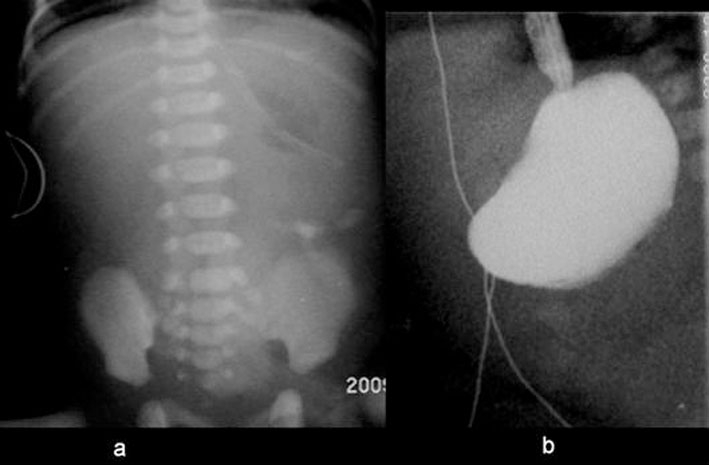
Radiology, case A. (A) Frontal supine radiograph demonstrating paucity of bowel gas and inspissated meconium on the left side of the abdomen. Mineral air is seen in the stomach with a nasogastric tube in situ. (B) Water soluble contrast study shows total gastric outlet obstruction.
Figure 2.

Histopathology, case A. (A, B) Markedly narrow lumen with submucosal fibrosis consistent with stenosis and suggestive of atresia.
Case B
The plain radiograph of the infant's abdomen showed a ‘double bubble’ gas shadow with densities on the left side of the midline (figure 3A), probably due to inspissated meconium. Water soluble contrast examination revealed gastric outlet obstruction (figure 3B). Ultrasound examination of the infant's abdomen carried out at 20 h of age, showed multiple thick-walled cystic lesions in the abdomen consistent with MIA with bowel sequestration (figure 3C,D). One of the cystic lesions was in the second part of the duodenum raising suspicion of duodenal sequestration as well. The rest of the abdominal organs were normal. Histopathology examination of the resected bowel showed features consistent with congenital atresia (stenosed lumina, shortened villi with foci of mucosal ulceration, dystrophic calcification and foreign body giant cell reaction). Dense submucosal fibrosis was noted in the atretic/stenotic bowel segment. Normal ganglion cells were present in adequate numbers. Rare lymphoid aggregates were noted with no evidence of reactive germinal centre in the mucosa or mesenteric lymph nodes (figure 4). Immunological studies revealed T cell lymphocytopenia (CD4 and CD8), B cell lymphocytopenia and very low immunoglobulin (IgG and IgM) levels.
Figure 3.
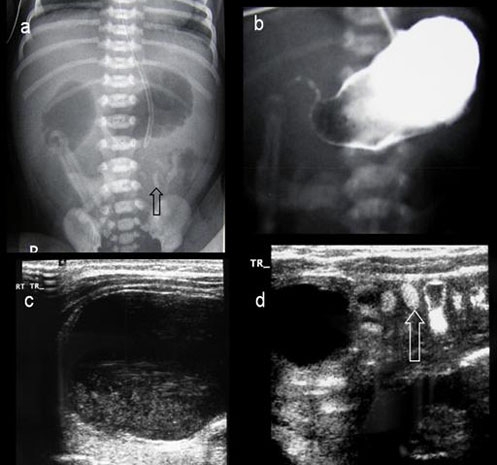
Radiology, case B. (A) Plain radiograph showing the ‘double-bubble’ sign. The arrow points to inspissated meconium in the small bowel. (B) Contrast study showing normal capacity stomach with a trace of contrast in the proximal duodenum. (C) On ultrasound examination, sequestrated fluid distended small bowel is seen in the lower abdomen showing debris in the dependant part. (D) Ultrasonography of the left side of the abdomen showing fluid distended small bowel loop separated by non-distended small bowel containing inspissated meconium (arrow).
Figure 4.
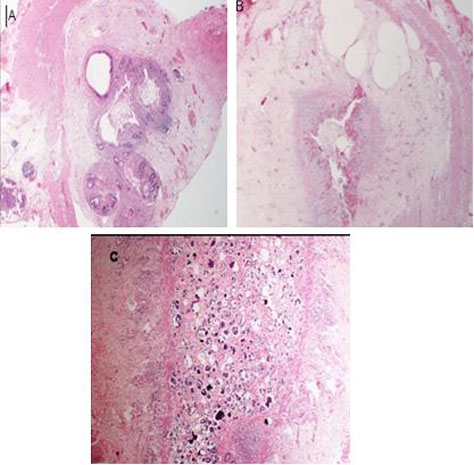
Histopathology, case B. (A, B) Atretic segment with ulceration, granulation tissue and submucosal fibrosis. (C) Mucosal dystrophic calcification in meconium.
Treatment
Case A
Explorative laparotomy showed evidence of meconium peritonitis with multiple atresias extending over a segment of approximately 31 cm starting at the proximal jejunum. The atretic segments were resected. Bowel continuity was restored by five end-to-end anastomoses.
Case B
Urgent laparotomy revealed type I duodenal atresia and type II multiple jejuno-ileo-colic atresias. Multiple atretic segments were resected with end-to-end anastomoses and a terminal loop ileostomy was created.
Outcome and follow-up
Case A
The baby developed recurrent episodes of sepsis with different organisms including Escherichia coli, Enterococcus faecalis, Staphylococcus hemolyticus and Staphylococcus epidermidis. The septic episodes were difficult to clear with the antibiotics to which these organisms were sensitive. She was transferred to Great Ormond Street Hospital, London for immunological investigations. The baby was found to have severe combined immunodeficiency (SCID). She had a very difficult intensive care course and ultimately died at the age of 6 months following failure to find an HLA compatible donor for BMT.
Case B
The baby developed mixed bacterial sepsis due to Enterobacter cloacae and E faecalis. Appropriate antibiotic therapy was commenced. Enteral feeding could not be started at any time. The baby remained TPN dependent. She died at the age of 3 months due to unresponsive mixed severe sepsis and a very difficult period of intensive care.
Discussion
Historical evolution of MIA and HMIA
Intestinal atresia accounts for approximately one third of all cases of neonatal intestinal obstruction.1 The causes, clinical presentation, diagnosis, operative management, postoperative care and outcome vary considerably according to the location of obstruction.2
HMIA is an unusual form of intestinal atresia with a possible autosomal recessive mode of inheritance.3 Immune defects have been described in several patients with various types of familial bowel atresia. When an immune deficiency coexists, it is either a separate primary abnormality or possibly the result of deficient development of the immune function of the gut as a consequence of multiple atresias.4 The combination of HMIA and immunodeficiency is invariably lethal.
Historically, ileal atresia was first described by Goeller in 1684.1 The condition of duodenal atresia was first described by Calder in 1733, but it was not until 1916 that Ernst of Copenhagen reported the first successful operation leading to cure.5 The aetiology of intestinal atresia was first speculated by Meckel in a review of the topic published in 1812.1 In 1889, Bland Sutton proposed a classification of the types of atresia and suggested that they occurred at sites of obliterative embryonic events such as atrophy of the vitelline duct. In 1900, Tandler proposed the first theory to explain the aetiology of intestinal atresia. He postulated that the failure of normal recanalisation of the solid cord stage of the fetal bowel led to atresia. The 1912 theory of an underlying fetal vascular accident proposed by Spriggs further expanded the aetiological possibilities.1 In 1955, the theory of vascular accident was further strengthened by the initial animal model studies of Louw and Barnard,6 followed by similar studies by Curtois in 1959, Santulli and Blanc in 1961, Abrams in 1968 and Koga in 1975.1
Familial intestinal atresia was first described by Winter in 1956 when he reported the cases of two brothers in a Polish family of three children.7 Blank in 1965 described familial congenital atresia in two brothers of a family of four, all of whom had cystic fibrosis.8 Mishalany and Najjar in 1968 reported three cases of familial jejunal atresia in a Lebanese family.8 In 1971, Mishalany and Der Kaloustian9 reported multiple-level intestinal atresia in two siblings of distantly related parents. In 1988, in Dublin, Ireland, Puri and Fujimoto found that 2% of neonates with intestinal atresia had multiple atresia; 30% of these infants (seven cases) belonged to three families.10 In 1999, Nawaz et al11 reported the results of a retrospective study in the United Arab Emirates to evaluate patients with neonatal intestinal atresia. Between 1982 and 1997 they identified 21 consecutive newborns with intestinal atresia. Eleven patients (eight males and three females) had small bowel atresias. Two of them had MIAs, while in one case there was an associated colonic atresia.
In 1990, Moreno et al reported the first association of multiple gastrointestinal atresias and immunodeficiency. He described three siblings with multiple-level intestinal atresias. One sibling also had SCID syndrome. The clinical histories of the other two siblings strongly suggested a congenital immunodeficiency syndrome. The parents of these siblings were non-consanguineous and healthy.12 Walker et al in 199513 and Rothenberg et al in 199814 reported two cases of a syndrome involving immunodeficiency and MIAs.
In 1998, Lambrecht and Kluth, in addition to their own case, reviewed 35 cases of HMIA, five of whom had immunodeficiency. They concluded that: (1) the abdominal x-rays showed signs of gastric or duodenal atresia combined with typical large rounded or oval homogeneous calcifications in the abdominal cavity; (2) intraoperative findings demonstrated widespread atresias (exclusively type I and II) extending from the stomach to the rectum; (3) there was cystic dilatation of the bile ducts, in some cases with both complete pyloric and duodenal or proximal jejunal atresias; (4) the pathogenesis was still speculative although a combined immunodeficiency should be excluded; and (5) a fatal outcome is invariable.15 These initial case reports of MIAs with immunodeficiency were followed by a number of case reports from other parts of the world. We have summarised the salient features of all these case reports in table 1.
Table 1.
Case reports of HMIA with immunodeficiency
| Author (year) | Number of cases | Gestational age | Polyhydramnios | Antenatal diagnosis | Major immunodeficiency features | Outcome |
|---|---|---|---|---|---|---|
| Moreno et al12 | 3 Siblings in one family | 37 Weeks | Positive | NA | ALC 600–900 cells/ µl | Died at 56 days, Streptococcus faecalis sepsis |
| 37 Weeks | NA | Suspected | ALC 360, γ globin 2 g/l | Died at 3.5 months. GVHD after transfusion | ||
| 38 Weeks | Positive | Dilated small bowel | IgG>42, IgM>20, IgA>7. ALC 400–1100. Decreased T3, 4 and 8 cells (%). | Died at 7 months | ||
| Walker et al13 | 1 | 36 Weeks | NA | NA | ALC 370–2000, absent (0) B cells, low T cells, IgG 111, IgM 13, IgA>7 | Died at 60 days. GVHD after transfusion and bacterial sepsis |
| Rothenberg et al14 | 1 | NA | NA | NA | ALC 300–3000, low T cells, low B cells, IgG 47, IgM 12, IgA 12, IgE 249 | Died at 8 months. Sepsis, cholestasis, cerebral atrophy |
| Moore et al24 | Five patients in two families | 23 Weeks | Positive | Positive | All with Fanconi pancytopenia | Died at 16 years from fungal sepsis |
| NA | NA | NA | Died from sepsis | |||
| NA | NA | NA | Pregnancy terminated at 23 weeks | |||
| NA | NA | NA | Suspected, not proven | Died of sepsis | ||
| 38 Weeks | Positive | Positive | T and B cell dysfunction | T and B cell dysfunction | ||
| Kim et al25 | 2 | 30 Weeks | NA | NA | Neutropenia | Discharged |
| Bass26 | 1 | 38 Weeks | Positive | Distended, echogenic bowel | Mixed T and B cell defect | Died at 2 months of Pneumocistis carinii sepsis |
| Bilodeau et al3* | 2 | 36 Weeks | Positive for both | Bowel dilated and calcified | Low IgM | Died at 42 days |
| 38 Weeks | Died at 64 days | |||||
| Gilroy et al21 | 1 | 35 Weeks | NA | NA | ALC 880, IgG 167, IgM 21, IgA not detected | Liver-small bowel transplant |
| Jung et al16 | 4 | NA | NA | NA | Low IgG, combined B and T cell immunodeficiency | Died from GVHD |
| Al-Salem27 | 1 | Term | NA | NA | ? Immunodeficiency | Died at 38 days from recurrent sepsis |
| Chen28 | 1 | 37 Weeks | Positive | Intestinal obstruction by ULS | Combined immunodeficiency, IgG 173, IgM>5, IgA>7 | Died at 13 months from parainfluenza-2 infection |
| Our cases (Ali et al, 2010) | 2 | 35 Weeks | Positive | Polyhydramnios, abdominal cyst | Combined immunodeficiency | Died at 6 months from sepsis |
| 36 Weeks | Positive | Duodenal atresia, abdominal mass | Combined immunodeficiency, IgG 159, IgM>4, IgA>6.6, CD 3800 | Died at 3 months from sepsis |
Review of HMIA over a period of 30 years.
The details of 24 cases are shown. All the cases have HMIA with immunodeficiency.
ALC, absolute lymphocyte count; GVHD, graft-versus-host disease; HMIA, hereditary multiple intestinal atresia; IgA, IgG and IgM, immunoglobulin A, G and M; ULS, ultrasound.
Genetics
HMIAs involving the gastrointestinal tract from the pylorus to the rectum are the least common intestinal atresias; inheritance was suggested to be autosomal recessive.12 The inheritance of SCID syndrome can be autosomal recessive or X linked.12 Moreno et al explained the inheritance of this association as either autosomal or X-linked recessive with favor to the first possibility. However, an alternative explanation he suggested was single autosomal recessive or X-linked recessive mutation.12
The exact mechanisms of action of the involved genes are, of course, unknown, but Shorter et al4 suggested that the gene responsible for class 3 HMIA plays a critical role in the development of the gastrointestinal tube itself, most likely the mucosal lining, and that the gene responsible for class 4 HMIA is vital for normal development of the superior mesenteric artery and its branches.4
Aetiology and pathogenesis
The two major theories put forward to explain the aetiology of intestinal atresia are: (1) Tandler's concept1 of a lack of recanalisation of the solid cord stage of the intestine during early fetal development and (2) the classic experimental study by Louw and Bernard,6 postulating that a late mesenteric vascular accident is the cause of most jejunoileal and colonic atresias. Tandler presented his theory in 1900.1 The aetiological basis was further expanded by Spriggs in 1912 when he proposed that mechanical accidents including vascular catastrophes may cause atresia.1 In 1955 the theory of vascular accident was further strengthened by the animal model studies of Louw and Barnard.6 Through their experimental studies in puppies, they confirmed the causal role of late mesenteric vascular accidents in most intestinal atresias.6 Later, these findings were further confirmed by animal studies in rabbits by Curtois1 in 1959 and then by Santulli and Blanc1 in 1961. Similar studies were reported in 1968 by Abrams in sheep and in 1975 by Koga in dogs.1
Whereas it is well accepted today that jejunoileal and colonic atresias result from intrauterine vascular accidents occurring in late fetal life, the pathogenesis of HMIA is still speculative. The lack of lanugo hair and squamous cells in the gastrointestinal tract indicate that the process develops very early in intrauterine life. Although some investigators have suggested that HMIA may be a consequence of an intrauterine inflammatory process of unknown origin in genetically susceptible infants, others favour failure of recanalisation during the solid stage of gut development. A third possibility is occlusion of a major part of the gastrointestinal tract caused by abnormal epithelial growth due to a genetic defect.15
Presentation and classification of HMIA
The presenting symptoms of any form of intestinal atresia or stenosis are consistent with bowel obstruction and include bilious vomiting, abdominal distension and failure to pass meconium.1 MIA is a rare and usually fatal disorder. One major complicating factor in the treatment of these patients is the associated combined immunodeficiency. These patients often die from graft-versus-host disease. The donor lymphocytes are supplied by transfused blood products and/or intestinal transplants.16
In 2006, Shorter et al4 proposed a classification of familial intestinal atresia as follows: class 1, pyloric atresia; class 2, duodenal atresia; class 3, HMIA; class 4, apple peel atresia; and class 5, colonic atresia. HMIA affects both the large and small intestines, whereas non-HMIA usually spares the colon.4 This proposed classification would fit with what is known about the embryological development of the gastrointestinal tract, with class 1 being a foregut anomaly, class 2 occurring at the junction between the foregut and midgut, class 3 affecting the entire gastrointestinal tract (ie, derivatives of foregut, midgut and hindgut), class 4 affecting the midgut only and class 5 being an abnormality of hindgut development.4
Antenatal diagnosis
Current advances in fetal medicine and genetics have made it possible to diagnose MIA and immunodeficiency before birth. The decision to terminate depends on the local legal, cultural and social values as well as parental wishes and beliefs. In Muslim countries including the State of Qatar, families are less likely to opt for elective termination and therefore the incidence of and neonatal mortality due to lethal congenital malformations is notably high in these countries.17 18 Prenatal diagnosis and termination of fetuses with HMIA and immunodeficiency may be a better practical option in non-conservative societies.
The possibility of antenatal diagnosis by visualising dilatation of the fetal bowel at 17 weeks gestation during routine ultrasound scanning was first proposed by Boyd et al in 1994.19 Other studies have reported that antenatal diagnosis of multiple gastrointestinal atresias is possible by ultrasound examination from 21 weeks gestation onwards.12 In 2002, Chou et al described the antenatal sonographic appearance of a case of HMIA at 30 weeks gestation.20 The fetus had a markedly dilated cystic mass in the right upper quadrant which was non-communicating with the stomach. At laparotomy, a prepyloric septal atresia, multiple-level small intestinal atresias and a rectal diaphragm were discovered. The infant died 52 days after the operation.
Polyhydramnios is the presenting feature in 20–35% of cases and is more frequently associated with proximal intestinal atresias. Antenatal ultrasonography is more reliable in detecting duodenal atresia than more distal lesions. Overall, 42% of antenatally diagnosed gastrointestinal tract malformations are confirmed postnatally and 16% of gastrointestinal tract malformations seen at birth are diagnosed by antenatal ultrasound.1
Antenatal diagnosis of SCID is based on the number of lymphoid cells, total T lymphocytes, T lymphocyte subsets and B lymphocytes present in a micro sample of pure fetal blood taken during the mid-to-late second trimester.12
Outcome
The outcome of MIA depends upon the length and regions of the gut involved. The degree of postoperative short bowel syndrome is a major determinant of survival. The most common cause of death is infection related to pneumonia, peritonitis and sepsis. Other factors contributing to morbidity and mortality are associated anomalies, prematurity and postoperative obstruction.1 The outcome of combined HMIA and immunodeficiency is bleak. Two of our reviewed cases had undergone liver-small bowel transplantation; one developed graft-versus-host disease21 and the second died at the age of 13 months from parainfluenza virus infection.16 BMT is an established successful mode of treatment for SCID.22 Some BMT centres have included MIA with SCID as a standard indication for BMT.23
Recent advances
Ultra short bowel syndrome requiring long term TPN which can be complicated by liver disease is the major cause of morbidity and mortality in these patients. Early use of growth factors to maximise intestinal adaptation, administration of growth hormone and nutritional modification may improve the status of patients with short bowel syndrome.1
Future research
Refinements in small bowel transplantation and advances in the science of immunology may reduce the current high complication rate with improved graft and host survival, thus eventually improving the overall survival rate of these patients.1 Improving chances of antenatal diagnosis using genetic studies is another open area of research in HMIA with SCID.
Learning points.
-
▶
Every patient with hereditary multiple intestinal atresia (HMIA) should be screened for immunodeficiency.
-
▶
Affected babies should be referred to centres with facilities for bone marrow transplantation and bowel lengthening procedures.
-
▶
Genetic counselling should be offered to parents of infants with HMIA with severe combined immunodeficiency.
-
▶
Antenatal and postnatal ultrasonographic follow-up should be carried out during all future pregnancies of parents of affected babies.
Footnotes
Competing interests None.
Patient consent Obtained.
References
- 1.Prasad TR, Bajpai M. Intestinal atresia. Indian J Pediatr 2000;67:671–8 [DOI] [PubMed] [Google Scholar]
- 2.Dalla Vecchia LK, Grosfeld JL, West KW, et al. Intestinal atresia and stenosis: A 25-year experience with 277 cases. Arch Surg 1998;133:490–6; discussion 496–7 [DOI] [PubMed] [Google Scholar]
- 3.Bilodeau A, Prasil P, Cloutier R, et al. Hereditary multiple intestinal atresia: thirty years later. J Pediatr Surg 2004;39:726–30 [DOI] [PubMed] [Google Scholar]
- 4.Shorter NA, Georges A, Perenyi A, et al. A proposed classification system for familial intestinal atresia and its relevance to the understanding of the etiology of jejunoileal atresia. J Pediatr Surg 2006;41:1822–5 [DOI] [PubMed] [Google Scholar]
- 5.Feggetter S. Congenital intestinal atresia. Br J Surg 1955;42:378–88 [DOI] [PubMed] [Google Scholar]
- 6.Louw JH, Barnard CN. Congenital intestinal atresia; observations on its origin. Lancet 1955;269:1065–7 [DOI] [PubMed] [Google Scholar]
- 7.Winter ST, Zeltzer M. Congenital atresia of the ileum in two brothers. J Pediat 1956;49:194–6 [DOI] [PubMed] [Google Scholar]
- 8.Mishalany HG, Najjar FB. Familial jejunal atresia: three cases in one family. J Pediatr 1968;73:753–5 [DOI] [PubMed] [Google Scholar]
- 9.Mishalany HG, Der Kaloustian VM. Familial multiple-level intestinal atresias: report of two siblings. J Pediatr 1971;79:124–5 [DOI] [PubMed] [Google Scholar]
- 10.Puri P, Fujimoto T. New observations on the pathogenesis of multiple intestinal atresia. J Pediatric Surg 1988;23:221–5 [DOI] [PubMed] [Google Scholar]
- 11.Nawaz A, Matta H, Jacobsz AW, et al. Neonatal intestinal atresia. Saudi Med J 1999;6:438–43 [PubMed] [Google Scholar]
- 12.Moreno LA, Gottrand F, Turck D, et al. Severe combined immunodeficiency syndrome associated with autosomal recessive familial multiple gastrointestinal atresias: study of a family. Am J Med Genet 1990;37:143–6 [DOI] [PubMed] [Google Scholar]
- 13.Walker MW, Lovell MA, Kelly TE, et al. Multiple areas of intestinal atresia associated with immunodeficiency and posttransfusion graft-versus-host disease. J Pediatr 1993;123:93–5 [DOI] [PubMed] [Google Scholar]
- 14.Rothenberg ME, White FV, Chilmonczyk B, et al. A syndrome involving immunodeficiency and multiple intestinal atresias. Immunodeficiency 1995;5:171–8 [PubMed] [Google Scholar]
- 15.Lambrecht W, Kluth D. Hereditary multiple atresias of the gastrointestinal tract: report of a case and review of the literature. J Pediatr Surg 1998;33:794–7 [DOI] [PubMed] [Google Scholar]
- 16.Jung LKL, Bonilla F, Botha L, et al. Immunodeficiency associated with multiple intestinal atresias. Primary Immunodeficiency Diseases consortium conference; 12 May 2005, Boston (MA) [Google Scholar]
- 17.Salameh K, Rahman S, Al Rifai H, et al. An analytic study of trends in perinatal and neonatal mortality rates in the state of qatar over a 30 years period (1977–2007): a comparative study with regional and developed countries. J Perinatol 2009;29:765–70 [DOI] [PubMed] [Google Scholar]
- 18.March of Dimes White Paper on Preterm Birth. The Global and Regional Toll. 2009, 1–20 (http://www.marchofdimes.com/globalprograms (accessed 20 Sep 2010)
- 19.Boyd PA, Chamberlain P, Gould S, et al. Hereditary multiple intestinal atresia-ultrasound findings and outcome of pregnancy in an affected case. Prenat Diagn 1994;14:61–4 [DOI] [PubMed] [Google Scholar]
- 20.Chou MM, Tseng JJ, Ho ES, et al. In utero sonographic findings in a fetus with a hereditary multiple intestinal atresia. Zhonghua Yi Xue Za Zhi (Taipei) 2002;65:131–4 [PubMed] [Google Scholar]
- 21.Gilroy RK, Coccia PF, Talmadge JE, et al. Donor immune reconstitution after liver-small bowel transplantation for multiple intestinal atresia with immunodeficiency. Blood 2004;103:1171–4 [DOI] [PubMed] [Google Scholar]
- 22.Bonilla FA. Hematopoietic cell transplantation for primary immunodeficiency. http://www.uptodate.com/patients/content/topic.do?topicKey=~jyAAspzNzzJTTRx (accessed 22 Sep 2010)
- 23.University of California San Francisco, Benioff Children's Hospital Severe combined immunodeficiency disease treatment. http://www.ucsfbenioffchildrens.org/treatments/severe_combined_immunodeficiency_disease_treatment/ (accessed 22 Sep 2010)
- 24.Moore SW, de Jongh G, Bouic P, et al. Immune deficiency in familial duodenal artesia. Pediatr Surg 1996;31:1733–5 [DOI] [PubMed] [Google Scholar]
- 25.Kim S, Yedlin S, Idowu O. Colonic atresia in monozygotic twins. Am J Med Genet 2000;91:204–6 [PubMed] [Google Scholar]
- 26.Bass J. Pyloric atresia associated with multiple intestinal atresia and immune deficiency. J Pediatr Surg 2000;37:941–2 [DOI] [PubMed] [Google Scholar]
- 27.Al-Salem AH. Congenital pyloric atresia and associated anomalies. Pediatr Surg Int 2007;23:559–63 [DOI] [PubMed] [Google Scholar]
- 28.Chen K. A Familial Case of Hereditary Multiple intestinal Atresia with Immunodeficiency. Primary Immunodeficiency Diseases Consortium Conference; 11 June 2009, San Francisco (CA) [Google Scholar]