

Abstract
Familial multiple lipomatosis is rare. Several modes of inheritance have been proposed but no conclusive evidence shown, although some families have suggested autosomal dominant inheritance. The authors describe a family with multiple lipomatosis showing clear autosomal dominant inheritance, and no mutations within the NF1, SPRED1 or Cowden disease (PTEN) genes. Familial autosomal dominant lipomatosis is a rare but distinct entity.
Background
RAS-MAPK pathway disorders such as neurofibromatosis type 1 and Legius syndrome1 can cause skin lesions including lipomatosis. Their common features are cell proliferation and growth inherited in an autosomal dominant fashion. We report a family with familial autosomal dominant lipomatosis with no evidence of current genes in the RAS-MAPK pathway but is a differential diagnosis in this group.
Case presentation
The proband (individual (III.6), figure 1A) presented, aged 2 years with a possible diagnosis of neurofibromatosis. She had developmental delay and two lumps in the region of her right triceps. Growth parameters were on the 50th centile. She had no dysmorphic features. Her father (II.5), grandfather (I.2) and several family members had multiple subcutaneous lumps.
Figure 1.
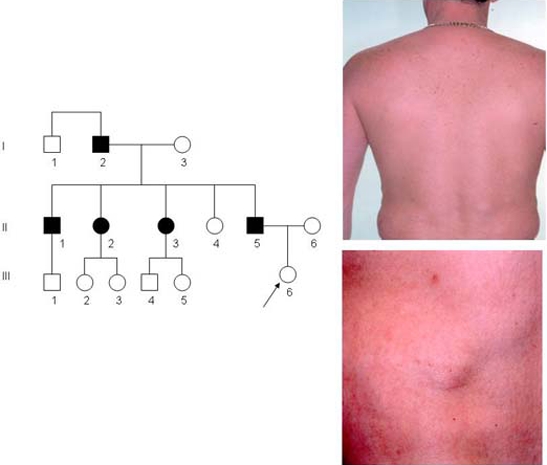
(A) Three generation pedigree affected by familial multiple lipomatosis. Individual shown as affected (shaded symbols) and unaffected (clear symbols). (B) II.5 back, showing multiple lipomas particularly in the lumbar region, and freckles on upper back. (C) II.5 abdomen showing multiple lipomas.
Examination revealed two lumps on her right triceps, measuring 4×4 cm. The clinical findings were consistent with lipomata. Her father (II.5), aged 38 years, had multiple lipomata, distributed over his chest, abdominal wall and back noted from 15 years, and numerous small freckles on his upper back but no café-au-lait spots or axillary freckling. A clinical diagnosis of familial multiple lipomatosis (FML) was made.
Investigations
Molecular genetic testing confirmed a normal chromosomal karyotype, and direct sequencing revealed no mutations within the NF1, SPRED1 or Cowden disease (PTEN) genes. Cardiac ECHO was normal.
Outcome and follow-up
The patient had removal of the lipomata for cosmetic and functional reasons. A large lipoma measuring 6×3 cm over the right pectoralis major muscle and 24 smaller lipomata were excised from his abdominal and chest regions (figure 1B,C) and confirmed as lipomata on histology.
Discussion
FML is rare. Brodie first described FML in 1846,2 and Blaschko noted its familial association.3 Multiple encapsulated painless subcutaneous lipomas over the torso and limbs are characteristics usually noticeable after the third decade.4 Several modes of inheritance have been proposed but no conclusive evidence shown, although some cases of autosomal dominant inheritance have been described and ‘pure’ familial lipomatosis is probably due to an autosomal dominant gene(s).2–8
FML is often confused with neurofibromatosis or Legius syndromes.9 Five out of 15 members of this family were clinically affected and male-to-male transmission on two occasions strongly suggests autosomal dominant inheritance.9
Lipomas usually become apparent after the third decade of life2 but may appear in childhood or adolescence. No gene has yet been identified,9 10 but a recent report in a child with encephalocraniocutaneous lipomatosis (ECCL), with other family members having pure lipomatosis, suggested that the HMGA2 gene could be involved.10 The patient with ECCL had no cutaneous lipomas on her trunk or limbs and the paternal family history of lipomas may be co-incidental as there was an additional maternal history of multiple nevi and melanomas; however, PTEN and NF1 testing were normal in the case, although none of the family members with lipomas appear to have been tested and further investigations are in progress. Isolated lipomas have shown various karyotypic anomalies but no germline mutations have been identified. In this case, no germline cytogenetic abnormalities were identified in the proband or her father, although karyotyping of individual lipomas was not carried out.
Conclusion
We show that one mode of inheritance of multiple lipomatosis is clearly autosomal dominant with an early onset and normal genetic testing for NF1, PTEN and SPRED1. Lipomatosis can be easily confused with neurofibromatosis or Legius syndrome, and all cases of multiple lipomatosis merit taking a detailed family history.
Learning points.
-
▶
This case confirms that familial lipomatosis is a discrete clinical entity.
-
▶
Familial lipomatosis may be autosomal dominant.
-
▶
Age at onset or diagnosis is variable within families and may present in adolescence or in middle age.
Footnotes
Competing interests None.
Patient consent Obtained.
References
- 1.Wright EM, Kerr B. RAS-MAPK pathway disorders: important causes of congenital heart disease, feeding difficulties, developmental delay and short stature. Arch Dis Child 2010;95:724–30 [DOI] [PubMed] [Google Scholar]
- 2.Muller R. Observation sur la transmission héréditaire de la lipomatose circonscrite multiple. Dermatologica 1951;103:258–64 [PubMed] [Google Scholar]
- 3.Toy BR. Familial multiple lipomatosis. Dermatol Online J 2003;9:9. [PubMed] [Google Scholar]
- 4.Stephens FE, Isaacson A. Hereditary multiple lipomatosis. J Hered 1959;50:51–3 [Google Scholar]
- 5.Leffell DJ, Braverman IM. Familial multiple lipomatosis. Report of a case and a review of the literature. J Am Acad Dermatol 1986;15:275–9 [PubMed] [Google Scholar]
- 6.Keskin D, Ezirmik N, Celik H. Familial multiple lipomatosis. Isr Med Assoc J 2002;4:1121–3 [PubMed] [Google Scholar]
- 7.Golsch S, Worret WI. Familial multiple lipomatosis with polyneuropathy. Eur J Dermatol 1995;5:283–5 [Google Scholar]
- 8.Mohar H. Familial multiple lipomatosis. Acta Derm Venereol 1980;60:509–13 [PubMed] [Google Scholar]
- 9.Burman M. Familial multiple circumscribed subcutaneous lipomatosis (neurolipomatosis?), a syndrome which may be mistaken for neurofibromatosis. Bull Hosp Joint Dis 1950;11:192–5 [PubMed] [Google Scholar]
- 10.Prontera P, Stangoni G, Manes I, et al. Encephalocraniocutaneous lipomatosis (ECCL) in a patient with history of familial multiple lipomatosis (FML). Am J Med Genet A 2009;149A:543–5 [DOI] [PubMed] [Google Scholar]