

Abstract
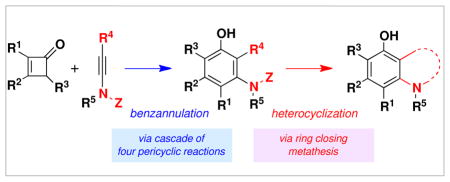
A two-stage “tandem strategy” for the synthesis of benzofused nitrogen heterocycles is described that is particularly useful for the construction of systems with a high level of substitution on the benzenoid ring. The first stage in the strategy involves a benzannulation based on the reaction of cyclobutenones with ynamides. This cascade process proceeds via a sequence of four pericyclic reactions and furnishes a multiply substituted aniline derivative which can bear a variety of functionalized substituents at the position ortho to the nitrogen. In the second stage of the tandem strategy, ring closing metathesis generates the nitrogen heterocyclic ring. This two-step sequence provides efficient access to highly substituted dihydroquinolines, benzazepines, benzazocines, and related benzofused nitrogen heterocyclic systems. The application of this chemistry in a concise formal total synthesis of the anticancer agents (+)- FR900482 and (+)-FR66979 is described.
Introduction
Benzofused nitrogen heterocycles are important synthetic targets, incorporated in the structures of numerous natural products and pharmaceutical compounds. A variety of hetereocyclization methods are available for the formation of benzofused nitrogen heterocycles from ortho-substituted anilines, but the regiocontrolled synthesis of aniline substrates with a high level of substitution on the aromatic ring remains a challenging problem in organic synthesis.
Classically, syntheses of highly substituted benzenoid compounds have most often been achieved by employing linear substitution strategies involving sequential electrophilic substitution and metalationalkylation reactions. A more effective approach, however, involves the application of benzannulation methods: convergent strategies in which the aromatic ring system is assembled from two or more precursors in a single step, with all (or most) substituents in place.i Two complementary benzannulation strategies based on the reactions of alkynes and vinylketenes have been developed in our laboratory,ii,iii,iv and these methods have been successfully applied in total syntheses of a number of bioactive natural products.v,vi In this paper we report the extension of this benzannulation strategy to include ynamine derivatives as the alkyne annulation partner. As outlined in Scheme 1, we envisioned that this variant of the benzannulation would provide exceptionally efficient access to diverse highly substituted aniline derivatives strategically designed for immediate participation in a subsequent “heterocyclization” reaction.vii If feasible, this combination of ring-forming processes would constitute a powerful two-stage “tandem strategy” for the synthesis of several classes of benzofused nitrogen heterocyclic compounds including systems with multiple substituents on the aromatic ring.
Scheme 1.

Tandem Strategy for Synthesis of Benzofused Nitrogen Heterocycles
In its original version, the vinylketene-based benzannulation strategy developed in our laboratory proceeds via a “cascade” process involving four successive pericyclic reactions. Scheme 2 outlines the mechanism of the benzannulation, illustrated here with an ynamine derivative serving as the 2-π annulation partner. Thermolysis (typically at 80–150 °C) or irradiation of a cyclobutenone serves as the triggering step for the cascade, effecting reversible electrocyclic ring opening to generate the transient vinylketene intermediate 4. Vinylketene 4 is immediately intercepted by the alkyne annulation partner in a regioselective [2 + 2] cycloaddition to afford a new cyclobutenone 5, which under the reaction conditions undergoes reversible electrocyclic cleavage (vide infra) to generate dienylketene 6. Rapid 6-π electrocyclic closure and then tautomerization furnishes the desired aromatic product 3.
Scheme 2.

Pericyclic Cascade Mechanism of the Vinylketene-Based Benzannulation
Results and Discussion
Ynamine-Based Benzannulations
Alkynyl ethers have served as the ketenophilic annulation partners in most prior applications of this benzannulation strategy, although alkynyl thioethers and aryl acetylenes can also participate in the reaction. At the outset of this investigation, the feasibility of employing ynamine derivatives in efficient benzannulations of this type was far from certain. In early studies employing ynamines,iia we isolated allenes such as 10 as minor byproducts in benzannulations with cyclobutenones. Based on the work of Ghosez,viii the formation of this allene byproduct is believed to result from initial addition of the ynamine across the ketene carbonyl group via a concertedix or stepwise pathway to form an alkylideneoxete 13. Electrocyclic ring opening then transforms this strained intermediate to the allenyl carboxamide. The aromatic product 9 could result from either direct cyclization of the zwitterionic intermediate 12, or via the usual pericyclic cascade pathway (Scheme 2).
Although zwitterions of type 14 are capable of cyclizing to form benzenoid products, in the case of certain substituted vinylketenes the “zwitterionic cyclization” pathway would lead to an aromatic product of type 15 which is regioisomeric with respect to the phenol 3 produced via the pericyclic cascade mechanism (Scheme 4). A second potential complication associated with the ynamine-based variant of the benzannulation emerges from consideration of an unusual rearrangement observed by Ficini in a study of the reactions of cyclobutenones with ynamines.x Should the Ficini rearrangement be operative, then the reaction of vinylketenes with ynamines could lead to a third regioisomeric benzannulation product of type 16. Needless to say, the potential formation of mixtures of regioisomeric products was alarming as the separation of closely related annulation products such as these was expected to be a daunting task.
Scheme 4.

Alternative Regioisomeric Products in Benzannulations of Vinylketenes and Ynamines
In order to suppress these alternative pathways leading to regioisomeric benzannulation products, oxetes, and allenes, we have focused our attention on reactions of ynamides in which the nucleophilicity of the amino alkyne is attenuated by an electron-withdrawing substituent on the nitrogen atom.xi Ynamides are considerably less nucleophilic than ynamines, and it was our expectation that they would react with vinylketenes analogously to alkynyl ethers, i.e., via the desired concerted [2 + 2] cycloaddition rather than via alternative stepwise pathways. This prediction was confirmed in a preliminary study in which ynamides were found to react smoothly with simple non-conjugated ketenes to afford the desired 3- aminocyclobutenones in excellent yield without detectable formation of allenyl carboxamide byproducts.xii
Synthesis of Ynamides
A number of alternative cyclization protocols can be envisioned for the formation of the heterocyclic ring in the second step of the proposed tandem strategy (Scheme 1). For this initial study we focused our attention on the application of ring closing metathesis (RCM) which is now well established as a powerful method for the construction of nitrogen heterocycles.xiii It was our expectation that the vinylketene-based benzannulation reaction could provide an exceptionally expeditious method for the assembly of RCM substrates bearing a high level of substitution on the aniline ring. Ynamides bearing unsaturated substituents on both the nitrogen atom and at C-2 of the alkyne would be required as benzannulation partners in order to generate the desired intermediates for ring closing metathesis. The success of these reactions would of course require that the vinylketene [2 + 2] cycloaddition proceed with high chemoselectivity at the triple bond of the ynamide in the presence of the other unsaturated moieties.
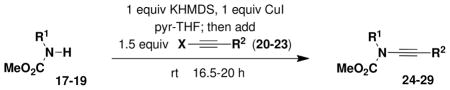
Recent advances in copper-promoted amide coupling reactions have provided the basis for the efficient and convenient synthesis of a variety of ynamides. Most of the ynamides employed in the present study were prepared using the N-alkynylation method recently developed in our laboratory.xiv Although this protocol requires the use of one equivalent of CuI, coupling proceeds smoothly at room temperature and this method thus accommodates the synthesis of a wide range of alkyne derivatives including thermally unstable systems. For many of these ynamides, similar results can be obtained by using the method of Hsung which employs catalytic CuCN or CuSO4 in conjunction with diamine ligands with coupling taking place at elevated temperatures.xv We have found both methods to be reliable and reproducible for reactions on both small and large (i.e., multigram) scale.xvi
For the preparation of alkynyl carbamates, we have found that the protocol developed in our laboratory generally affords the best results (Table 1). The carbamates and alkynyl halides required for the coupling reactions presented in Table 1 are all commercially available or can be prepared in one step as detailed in the Supporting Information. In the case of alkynyl sulfonamides, the Hsung procedure using catalytic CuSO4 was found to be most convenient. Several ynamides were best prepared by alkylation of terminal ynamides obtained by desilylation of C-silyl derivatives (Schemes 5–6). For the synthesis of the “skipped” diynamide 34, the procedure of Caruso and Spinellaxvii was found to be most effective for the propargylation of ynamide 33.
TABLE 1.
Synthesis of Ynamides via N-Alkynylation of Carbamates
 | ||||
|---|---|---|---|---|
| entry | carbamate | halo alkyne | ynamide | yield (%)a |
| 1 |
17 R1 = Me |
20 X = Br, R2 = Hex |
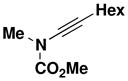 24 |
61b |
| 2 |
18 R1 = CH2CH=CH2 |
21 X = Br, R2 = CH2CH=CH2 |
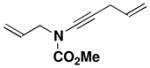 25 |
44 |
| 3 | 18 |
22 X = Br, R2 = C(CH3)=CH2 |
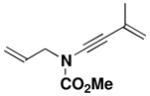 26 |
49 |
| 4 |
19 R1 = (CH2)2CH=CH2 |
21 |
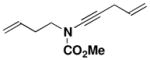 27 |
52 |
| 5 | 19 | 22 |
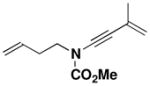 28 |
68b |
| 6 | 18 |
23 X = I, R2 = SiMe3 |
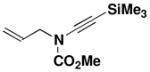 29 |
58 |
Isolated yield of products purified by column chromatography on silica gel.
Ref. 12.
Scheme 5.

Synthesis of Alkynyl Sulfonamide 32
Scheme 6.

Synthesis of Ynamide 34
Feasibility and Scope of the Benzannulation
The feasibility of applying ynamides as alkyne partners in the benzannulation was initially examined using 3-butylcyclobutenone (35)xviii and ynamides 24 and 32 as test substrates. As summarized in Scheme 7, we found that successful benzannulation could be achieved by employing either thermal or photochemical activation, and by microwave irradiation. Benzannulation was most conveniently accomplished in both cases by heating the ynamide and a slight excess of cyclobutenone in toluene at reflux for 1 h. In the case of sulfonamide 32, the yield of phenol could be improved by treating the crude product with KOH in methanol. This treatment serves to hydrolyze the small amount of phenolic esters formed by trapping of the benzannulation product with excess vinylketene during the reaction. Identification of the benzannulation product as 36, the “pericyclic cascade product” of type 3 (Scheme 4) rather than the regioisomeric “Ficini rearrangment product” of type 16, was confirmed by an NOE study (see Supporting Information). Note that in this benzannulation there would be no difference between the product formed via the pericyclic cascade mechanism and that produced by the zwitterionic cyclization pathway.
Scheme 7.

Feasibility of the Benzannulation
Interestingly, reaction of the cyclobutenone and ynamide 24 at a lower temperature (80 °C) for 90 min led to the isolation of a mixture of phenol 36 and a 4-vinylcyclobutenone (38) corresponding to intermediate 5 (Scheme 2) in the pericyclic cascade. Upon further heating in toluene, this vinylcyclobutenone was converted to phenol 36 in 97% yield. The near quantitative yield observed for this transformation suggests that either electrocyclic ring opening of 38 affords exclusively a (Z)- rather than (E)-dienylketene intermediate, or that formation of the dienylketene (intermediate 6 in Scheme 2) is reversible under the reaction conditions. We favor the latter explanation since computational studies by Houk and coworkers on the torquoselectivity of the electrocyclic opening of cyclobutenones suggests that ring opening to the E isomer should be favored.xix
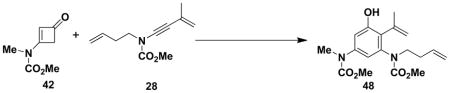
Benzannulations involving ynamides that incorporate unsaturated substituents can generate products that are potential substrates for ring closing metathesis reactions, and systems of this type were the main focus of this study. As demonstrated with the examples in Table 2, ynamides of this type participate in the benzannulation with a variety of cyclobutenonesxx to produce the desired highly substituted aniline derivatives in good to excellent yield. In no case were we able to detect the formation of byproducts resulting from competitive addition of the vinylketene intermediates to alkene double bonds. This important observation supports our expectation that ynamides should exhibit greater ketenophilicity as compared to alkenes, including conjugated double bonds such as those present in ynamides 26 and 28.
TABLE 2.
Benzannulations with Ynamides and Cyclobutenones
| entry | cyclobutenone | ynamide | conditions | benzannulation product | yield (%)a |
|---|---|---|---|---|---|
| 1 |
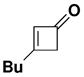 35 |
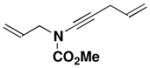 25 |
toluene 80 °C, 1.5 h then 110 °C, 1.5 h |
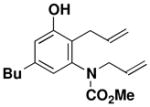 43 |
84 |
| 2 | 35 |
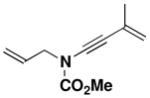 26 |
benzene 80 °C, 3 hb |
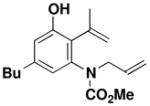 44 |
67–69 |
| 3 |
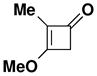 39 |
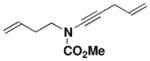 27 |
CHCl3 150 °C, 16.5 hb |
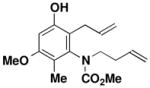 45 |
62 |
| 4 |
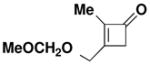 40 |
27 | CHCl3 150 °C, 16.5 hb |
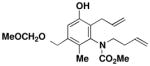 46 |
60 |
| 5 |
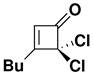 41 |
27 | toluene 135 °C, 4 hc |
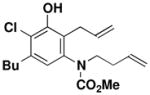 47 |
45 |
| 6 |
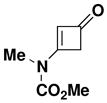 42 |
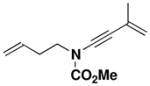 28 |
toluene 110 °C, 10 hb |
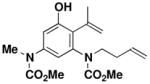 48 |
53 |
| 7 | 35 |
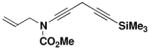 34 |
toluene 80 °C, 1.5 h then 110 °C, 1.5 h |
 49 |
79 |
Isolated yield of products purified by column chromatography on silica gel. Yields based on ynamide (using 1.2–2.0 equiv of cyclobutenone) unless otherwise indicated.
Crude product was heated with 5 M KOH in MeOH (65 °C, 2–3.5 h).
Reaction performed in presence of 2.0 equiv of BHT using 1.0 equiv of cyclobutenone and 1.5 equiv of ynamide.
Optimal conditions for the benzannulation vary from case to case. Cyclobutenones without substituents at the C-2 position generate monosubstituted ketene (“aldoketene”) intermediates upon electrocyclic opening, and these substrates react smoothly at 80–110 °C. In the case of N-allyl ynamides (e.g., 25, 26, and 34), it was found important to perform the reaction at the lowest possible temperature in order to minimize [3,3] sigmatropic rearrangement of the ynamides which begins to occur at temperatures above 125 °C. Reactions with these ynamides are best carried out at 80 °C until all of the starting materials are consumed. In some reactions (entries 1 and 7) this leads to mixtures of the desired phenol and intermediate 4-vinylcyclobutenone, in which case further heating at 110 °C completes conversion to the aromatic product.
Benzannulations that involve disubstituted ketene (“ketoketene”) intermediates (e.g., entries 3 and 4) require higher temperatures due to the slower rate of [2 + 2] cycloaddition in the case of these vinylketenes. Increased reaction rates were observed when these benzannulations were performed in chloroform in place of toluene. Annulation employing the 4,4-dichlorocyclobutenone 41 affords the 2- chlorophenol 47, presumably via an intermediate 6,6-dichlorocyclohexadienone intermediate that undergoes radical-mediated dechlorination at the elevated reaction temperature. Disappointingly, attempted benzannulation with 2,3,4-trisubstituted cyclobutenones furnished the desired aromatic products in low yield. In most cases, optimal yields were obtained under thermal benzannulation conditions rather than via photochemical activation. For example, in the case of the reaction depicted in entry 1, photochemical benzannulation afforded 43 in only 39% yield, presumably because of further reaction of the product by photo-induced cyclization to form a dihydrobenzofuran.xxi
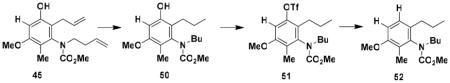
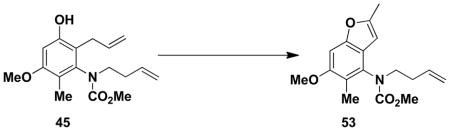
Careful examination of the crude products of these benzannulations indicated that in each case a single phenolic product is generated. As discussed earlier, benzannulations involving disubstituted vinylketenes have the potential to produce regioisomeric products should the alternative zwitterionic cyclization or Ficini rearrangement pathways be operative (see Scheme 4). Confirmation that the benzannulation was proceeding via the desired pericyclic cascade pathway was obtained by the following experiments performed on the product of the benzannulation in entry 3. First, deoxygenation of the phenolxxii was accomplished via the sequence outlined in Scheme 9.xxiii The strong coupling (J = 8.5 Hz) observed for the two aromatic protons in the resulting product 52 indicates that the methyl and allyl groups (corresponding to R1 and R4 in Scheme 4) cannot both be ortho to the hydroxyl group in the benzannulation product, ruling out the zwitterionic cyclization product of type 15. Confirmation that the allyl substituent (R4 in Scheme 4) must be ortho to the hydroxyl was then obtained by treatment of the benzannulation product with N-iodosuccinimide which promoted iodoetherification to form an iodo dihydrobenzofuran that was converted to 53 by elimination of HI with base. This result rules out the alternative regioisomer of type 16, and confirms the identity of the benzannulation product as 45, the regioisomer of type 3 predicted by the pericyclic cascade mechanism.
Scheme 9.

Confirmation of Benzannulation Regiochemistry
Ring Closing Metathesis of Benzannulation Products
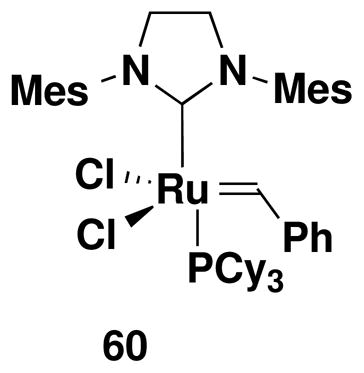
As shown in Table 3, exposure of benzannulation products 43–48 to the action of 5 mol% of the “second generation” Grubbs catalyst 60xxiv in dichloromethane at reflux furnished the desired benzofused nitrogen heterocycles in excellent yield. Limiting the reaction time to under 1 h avoids the formation of byproducts resulting from double bond isomerization.xxv For example, extended reaction (12 h) in the case of entry 3 led to the formation of a 60:40 mixture of 56 and the isomer in which the benzazocine double bond has moved into conjugation with the aromatic ring. No isomerization was observed when the reaction was carried out for 40 min. Disappointingly, attempts to effect enyne ring closing metathesisxxvi with phenol 49 or various derivativesxxvii with any of several metathesis catalysts have proved unsuccessful.
TABLE 3.
Synthesis of Benzofused Nitrogen Heterocycles via RCM of Benzannulation Products
| entry | benzannulation product | cyclizationa product | yield (%)b |
|---|---|---|---|
| 1 | 43 |
 54 |
95 |
| 2 | 44 |
 55 |
98 |
| 3 | 45 |
 56 |
86 |
| 4 | 46 |
 57 |
94 |
| 5 | 47 |
 58 |
87 |
| 6 | 48 |
 59 |
76 |
Conditions: 5 mol% Grubbs-II (60), CH2Cl2 (0.03–0.07 M in diene substrate), reflux, 40 min (2.5 h for entry 6).
Isolated yields of products purified by column chromatography.
Formal Total Synthesis of (+)-FR900482 and (+)-FR66979
We next turned our attention to the application of the tandem benzannulation-RCM strategy in a formal total synthesis of the potent antitumor agents (+)-FR900482 and (+)-FR66979. This exercise was expected to provide an excellent test for our methodology by examining it in the demanding context of a densely functionalized and highly substituted system.
(+)-FR900482 (61) and (+)-FR66979 (62) were isolated in 1987 and 1989 from the fermentation broth of Streptomyces sandaensis by researchers at the Fujisawa Pharmaceutical Co.xxviii These highly potent antitumor agents crosslink DNA following reductive activation and are believed to possess a mode of action closely related to that of mitomycin C.xxix Among the seven total synthesesxxx,xxxi,xxxii,xxxiii,xxxiv and formal total synthesesxxxv,xxxvi,xxxvii of these antitumor antibiotics reported to date, two proceed via related benzazocine intermediates (Scheme 10, 63 and 64), with 63 being prepared by an elegant RCM strategy in 16 steps beginning with 5-nitrovanillin.xxxv As detailed below, we anticipated that our tandem ynamide benzannulation/ring closing metathesis strategy would provide an expeditious enantioselective route to benzazocines of this type requiring as few as 9 steps in the longest linear sequence.
Scheme 10.

Approaches to the Total Synthesis of (+)-FR900482
Scheme 11 outlines our approach. Union of cyclobutenone 67 and ynamide 68 via benzannulation was expected to provide 66, the substrate for ring closing metathesis to generate benzazocine 65. The requisite ynamide would be prepared via N-alkynylation of bromo alkyne 69, which could be derived from an aldehyde of type 70 by means of any of several homologation methods. For the enantioselective synthesis of 70, we decided to focus our attention on a chiral auxiliary-directed syn aldol condensation involving a suitable enolate derivative and acrolein.
Scheme 11.

Formal Total Synthesis of (+)-FR900482: Retrosynthetic Analysis
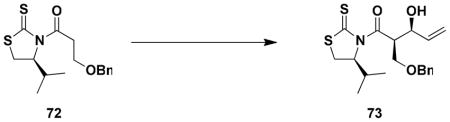
The strategy outlined above in principle can accommodate the preparation of a variety of benzazocines of type 65 in which the four hydroxyl groups are differentially protected, thus enabling maximum flexibility in the later stages of the synthesis. Initially we targeted a system with orthogonal blocking groups; specifically, R1 = TBDMS, R2 = benzyl, and R3 = CH2OMe (MOM ether). For the syn aldol condensation, an issue of some concern was the potential for enolate derivatives of type 71 to undergo β-elimination under the conditions of the reaction. For this reason, we concentrated our attention on Mukaiyama-type aldol chemistry involving tin enolates since these reactions are known to tolerate the use of enolate derivatives with β-alkoxy substituents.xxxviii Best results were obtained by employing the method of Nagaoxxxix,xl and the tin enolate derivative of thiazolidinethione 72, the enantiomer of which has previously been prepared by Nagao.xxxixb Acylation of (S)-4-isopropyl-1,3-thiazolidine-2-thionexli with 3-benzyloxypropionyl chloridexlii furnished convenient access to 72, which was treated with N-ethylpiperidine and freshly prepared tin(II) triflate at −78 °C. Reaction of the resulting tin enolate with acrolein at −78 °C then afforded the expected aldol product with greater than 95% selectivity for the desired isomer.xliii Attempts to employ titanium(IV) enolate derivatives of N-acyl thiazolidinethiones according to the method of Crimminsxliv for this transformation led to significant decomposition of the substrate, and reactions using the corresponding oxazolidinones were not successful under a range of conditions.
Silylation of the secondary alcohol in 73 and reduction of the N-acyl thiazolidinethione with DIBAL-H next furnished aldehyde 74 in excellent yield. The chiral auxiliary, (S)-4-isopropyl-1,3- thiazolidine-2-thione, was recovered in 75% yield simply by trituration of the crude product with hexanes prior to purification of the aldehyde by column chromatography. Conversion of 74 to the bromo alkyne 76 was effected in a convenient one-pot operation by the general method of Michel and Rassat.xlv Thus, reaction of the aldehyde with ylide 75xlvi afforded the expected 1,1-dibromo alkene, which without isolation was treated in the same flask with excess KOt-Bu to effect elimination producing bromo alkyne 76 in excellent yield.
Scheme 13 outlines the preparation of the two cyclobutenones examined as vinylketene precursors for the key benzannulation step. Each of these compounds was prepared from 3-ethoxycyclobutenonexlvii by addition of an alkoxylithium reagent generated by transmetallation from the appropriate organostannane.xlviii In situ hydrolysis of the resulting vinylogous hemiacetal employing the protocol introduced by Liebeskindxlix furnished the desired cyclobutenones, in each case in 65% yield.
Scheme 13.

Synthesis of Cyclobutenone Benzannulation Components
The next step in the synthesis, N-alkynylation of allyl carbamate 82 with bromo acetylene 76, proved unexpectedly challenging. Application of our usual protocolxiv afforded 83 in at best 42% yield accompanied by considerable diyne resulting from homocoupling of the alkynyl bromide. Improved results were obtained by employing the Hsung “second generation” protocolxv as outlined in Scheme 13. In this reaction homodimerization was minimized by using 3 equiv of the carbamate and by adding 0.4 equiv of copper sulfate and 0.8 equiv of 1,10-phenanthroline (total) in two equal portions over 46 h with reaction at 80–85 °C. The desired ynamide 83 was isolated in 68% yield and none of the homodimer byproduct was detected under these conditions.
With both ynamide 83 and the requisite cyclobutenones in hand, we were ready to examine the key tandem benzannulation/RCM stage of the synthetic plan. As shown in Scheme 14, benzannulation employing cyclobutenone 81 proceeded smoothly under our standard conditions to afford 84, and subsequent exposure of the product to the action of the Grubbs “second generation” metathesis catalyst provided the desired benzazocine 85 in excellent yield. Overall this strategy provides access to the benzazocine core of (+)-FR900482 in only 9 steps in the longest linear sequence.
Scheme 14.

Synthesis of the Benzazocine Core of (+)-FR900482
Completion of a formal total synthesis of (+)-FR900482 and (+)-FR66979 required that we link our route to an intermediate previously transformed to the natural products. As outlined in Scheme 15, we targeted Ciufolini’s intermediate 64, which was prepared in racemic form in the course of his synthesis of FR66979.xxxvi Williams has previously reported conversion of FR66979 to FR900482 via Swern oxidation.xxxiii
Scheme 15.

Completion of the Formal Total Synthesis
For the synthesis of 64, we elected to employ allyl carbamate 86 in which the amino group is protected as a Teoc (2-(trimethylsilyl)ethoxycarbonyl) derivative rather than as a BOC derivative. This modification would enable simultaneous deprotection of the amine and allylic hydroxyl groups in the last step in the sequence. As shown in Scheme 15, the benzannulation in this case again proceeded smoothly, and after benzylation of the phenol, ring closing metathesis furnished benzazocine 90 in excellent yield. Exposure of the RCM product to the action of TBAF in THF then provided 64, with spectroscopic characteristics identical to those reported by Ducray and Ciufolini.xxxvi
Conclusion
The combination of the ynamide-based benzannulation with ring closing metathesis provides an expeditious strategy for the assembly of benzofused nitrogen heterocycles in which the aromatic ring can bear a high level of substitution. The synthetic utility of this tandem strategy is illustrated in its application in an exceptionally concise enantioselective route to the benzazocine core of the antitumor agents (+)-FR900482 and (+)-FR66979. Further elaboration of the benzannulation products via transition metal-mediated coupling reactions of derivatives of the phenolic hydroxyl group and via oxidation to benzoquinones should also be possible based on our previous studies.ii,v Tandem strategies based on the combination of the ynamide-based benzannulation with other heterocyclization methods are under investigation in our laboratory and will be reported in due course.
Experimental Section
General Procedures
All reactions were performed in flame-dried or oven-dried glassware under a positive pressure of argon. Reaction mixtures were stirred magnetically unless otherwise indicated. Airand moisture-sensitive liquids and solutions were transferred by syringe or cannula and introduced into reaction vessels through rubber septa. Reaction product solutions and chromatography fractions were concentrated by rotary evaporation at ca. 20 mmHg and then at ca. 0.1 mmHg (vacuum pump) unless otherwise indicated. Thin layer chromatography was performed on precoated glass-backed silica gel 60 F- 254 0.25 mm plates. Column chromatography was performed on silica gel 60 (230–400 mesh).
Materials
Commercial grade reagents and solvents were used without further purification except as indicated below. Dichloromethane, diethyl ether, and tetrahydrofuran were purified by pressure filtration through activated alumina. Toluene was purified by pressure filtration through activated alumina and Cu(II) oxide. Chloroform, pivaloyl chloride, oxalyl chloride, and trifluoroacetic anhydride were distilled at atmospheric pressure under argon. Triethylamine, pyridine, N-ethylpiperidine, 2,6-lutidine, and benzene were distilled under argon from calcium hydride prior to use. Acrolein was distilled under vacuum at 150 mmHg immediately prior to use. Copper (I) iodide was extracted with THF for 24 h in a Soxhlet extractor and then dried under vacuum (0.1 mmHg). N-Bromosuccinimide was recrystallized from boiling water. Triflic anhydride was distilled under argon from P2O5. n-Butyllithium was titrated according to the Watson- Eastham method using BHT in THF with 1,10-phenanthroline as an indicator.l Potassium tert-butoxide was titrated using p-toluenesulfonic acid in MeOH with phenolphthalein as an indicator.
Instrumentation
Melting points are uncorrected. 1H NMR chemical shifts are expressed in parts per million (δ) downfield from tetramethylsilane (with the CHCl3 peak at 7.27 ppm used as a standard). 13C NMR chemical shifts are expressed in parts per million (δ) downfield from tetramethylsilane (with the central peak of CHCl3 at 77.23 ppm used as a standard).
Experimental Procedures for the Preparation of Ynamides and Cyclobutenones
![]()
Pent-4-en-1-ynyl bromide (21)
A 500-mL, one-necked, round-bottomed flask equipped with a rubber septum and argon inlet needle was charged with 1-penten-4-yne (4.03 g, 60.5 mmol, 1.0 equiv), Nbromosuccinimide (11.8 g, 66.6 mmol, 1,1 equiv), AgNO3 (1.00 g, 6.05 mmol, 0.1 equiv), and 300 mL of acetone, and the resulting mixture was stirred in the dark at rt for 12.5 h. The cloudy yellow reaction mixture was then diluted with 300 mL of pentane and washed with three 125-mL portions of ice-cold water. The combined aqueous layers were extracted with 100 mL of pentane, and the combined organic layers were washed with three 80-mL portions of saturated aq Na2S2O3 solution and two 80-mL portions of brine, dried over MgSO4, filtered, and concentrated at atmospheric pressure through a 9-cm Vigreux column to give 10.27 g of a pale yellow liquid. Bulb to bulb distillation (100 mmHg, 80 °C oven temperature) afforded 6.55 g (75%, ca. 95% pure) of pent-4-en-1-ynyl bromide 21 as a colorless liquid: IR (thin film) 2254, 1077, 1006, and 895 cm−1; 1H NMR (300 MHz, CDCl3) δ 5.74–5.86 (m, 1H), 5.53 (app dq, J = 1.7 Hz, 17.0 Hz, 1H), 5.15 (app dq, J = 1.6, 10.0 Hz, 1H), and 3.00 (app dt, J = 1.8, 5.3 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 131.8, 116.8, 77.1, 40.6, and 24.2; HRMS-ESI (m/z) calcd for C5H4Br, 142.9491; found: 142.9493.
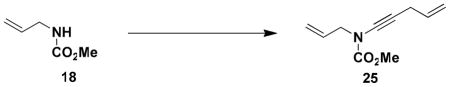
N-(Methoxycarbonyl)-N-[4-penten-1-ynyl]-2-propenylamine (25)
Reaction of a solution of carbamate 18 (1.06 g, 9.20 mmol, 1.0 equiv) in 35 mL of THF with KHMDS (10.1 mL, 9.20 mmol, 0.91 M in THF, 1.0 equiv), pyridine (18.0 mL, 230 mmol, 25 equiv), CuI (1.75 g, 9.20 mmol, 1.0 equiv), and alkynyl bromide 21 (2.0 g, 13.8 mmol, 1.5 equiv) in 14 mL of THF according to the general procedure gave 3.83 g of brown oil. Column chromatography on 60 g of silica gel (gradient elution with 0–5% EtOAc-hexanes) afforded 0.951 g of an orange oil which was further purified on 20 g of silica gel (gradient elution with 0–5% EtOAc-hexanes) to afford 0.722 g (44%) of ynamide 25 as a yellow oil: IR (thin film) 3395, 2957, 2239, 1729, 1446, 1388, 1280, 1235, and 1152 cm−1; 1H NMR (300 MHz, CDCl3) δ 5.79–5.95 (m, 2H), 5.22–5.38 (m, 3H), 5.09–5.15 (m, 1H), 4.07 (d, J = 5.8 Hz, 2H), 3.82 (s, 3H), and 3.09 (td, J = 1.9, 5.2 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 156.1, 133.0, 131.9, 118.5, 116.0, 76.1, 66.7, 54.1, 52.7, and 22.9; HRMS-ESI (m/z) [M+H] calcd for C10H13NO2, 180.1019; found: 180.1019.
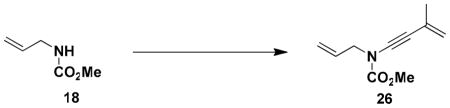
General Procedure for the Synthesis of Ynamides 25–28. N-(Methoxycarbonyl)-N-[3-methyl-3-buten-1-ynyl]-2-propenylamine (26)
A 200-mL, round-bottomed flask fitted with a rubber septum and argon inlet needle was charged with a solution of allyl carbamate 18li (1.15 g, 9.98 mmol, 1.0 equiv) in 40 mL of THF. The solution was cooled at 0 °C while a solution of KHMDS (11.0 mL, 9.98 mmol, 0.91 M in THF, 1.0 equiv) was added dropwise via syringe over 8 min and the resulting yellow slurry was stirred at 0 °C for 15 min. Pyridine (20.0 mL, 250 mmol, 25 equiv) was then added in one portion via syringe, followed by CuI (1.90 g, 9.98 mmol, 1.0 equiv), also in a single portion. The resulting green reaction mixture was allowed to warm to rt over 2 h. A solution of bromoalkyne 22xiva (2.17 g, 15.0 mmol, 1.5 equiv) in 15 mL of THF was added dropwise over 30 min via cannula and the reaction mixture was stirred at rt for 20 h. The reaction mixture was diluted with 160 mL of Et2O and washed with three 100-mL portions of a 2:1 mixture of brine and concentrated aq NH4OH solution. The combined aqueous phases were extracted with two 60-mL portions of Et2O, and the combined organic phases were dried over MgSO4, filtered, and concentrated to give ca. 9.5 g of dark brown oil. Purification by column chromatography on 53 g of silica gel (gradient elution with 0–5% EtOAc-hexanes) afforded 0.880 g (49%) of ynamide 26 as an orange oil: IR (thin film) 3456, 2958, 2257, 1732, 1445, 1368, 1303, 1245, and 1150 cm−1; 1H NMR (300 MHz, CDCl3) δ 5.82–5.95 (m, 1H), 5.12–5.34 (m, 4H), 4.10 (td, J = 1.3, 6.1 Hz, 2H), 3.83 (s, 3H), and 1.91 (dd, J = 1.0, 1.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 155.5, 131.7, 126.5, 119.6, 118.9, 82.2, 72.3, 54.3, 52.9, and 23.9; HRMS-ESI (m/z) [M+Na] calcd for C10H13NO2, 202.0838; found: 202.0844.

N-(Methoxycarbonyl)-N-[5-hexen-1-ynyl]-2-propenylamine (27)
Reaction of a solution of carbamate 19lii (0.765 g, 5.92 mmol, 1.0 equiv) in 24 mL of THF with KHMDS (6.50 mL, 5.92 mmol, 0.91 M in THF, 1.0 equiv), pyridine (12.0 mL, 148 mmol, 25 equiv), CuI (1.20 g, 6.30 mmol, 1.05 equiv), and alkynyl bromide 21 (15.0 ml, 8.88 mmol, 1.5 equiv, 0.6 M in benzene) according to the general procedure gave 3.5 g of brown oil. Column chromatography on 80 g of silica gel (gradient elution with 5–10% EtOAc-hexanes) afforded 0.539 g (52%) of ynamide 27 as a yellow oil: IR (thin film) 3394, 2956, 1726, 1642, 1446, 1391, 1291, 1249, 1154, and 915 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.77–5.89 (m, 2H), 5.35 (qd, J = 1.8, 16.8 Hz, 1H), 5.06–5.16 (m, 2H), 3.80 (s, 3H), 3.54 (t, J = 7.0 Hz, 2H), 3.11 (td, J = 1.8, 5.2 Hz, 2H), and 2.43 (q, J = 7.1 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 156.2, 134.3, 132.9, 117.3, 115.8, 76.7, 74.4, 53.9, 49.2, 32.0, and 22.8; HRMS-ESI (m/z) [M+H] calcd for C11H15NO2, 194.1176; found: 194.1170.
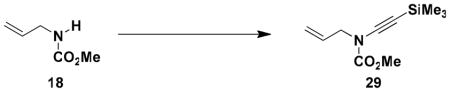
N-(Methoxycarbonyl)-N-(2-propenyl)-2-(trimethylsilyl)ethynylamine (29)
Reaction of a solution of carbamate 18 (0.283 g, 2.45 mmol, 1.0 equiv) in 10 mL of pyridine with KHMDS (2.70 mL, 2.45 mmol, 0.91 M in THF, 1.0 equiv), CuI (0.520 g, 2.71 mmol, 1.0 equiv) and iodoalkyne 23 (6.1 mL, 0.6M in benzene, 3.67 mmol, 1.5 equiv) in 4 mL of pyridine according to the general procedure gave 0.754 g of black oil, which was purified by column chromatography on 20 g of silica gel (gradient elution with 0–10% EtOAc-hexanes) to afford 0.300 g (58%) of ynamide 29 as a dark orange oil: IR (thin film) 2958, 2179, 1737, 1444, 1274, and 866 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.89 (ddt, J = 17.0, 10.3, 6.2 Hz, 1H), 5.25–5.32 (app t, J = 11.2, 17.0 Hz, 2H), 4.07 (d, J = 6.1 Hz, 2H), 3.83, (s, 3H), and 0.21 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 155.6, 131.4, 118.9, 95.5, 72.6, 54.2, 52.7, and 0.4, (peaks corresponding to minor rotamer: 130.9, 119.5, 87.5, 54.6); HRMS-ESI (m/z) [M+Na] calcd for C10H17NO2Si; 234.0921, found 234.0925.
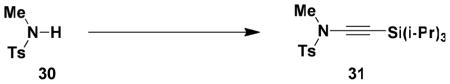
N-(Methyl)-N-(p-toluenesulfonyl)-2-(triisopropylsilyl)ethynylamine (31)
A 7-mL capacity resealable tube fitted with a rubber septum was charged with N-methyltoluenesulfonamide 30 (0.250 g, 1.35 mmol, 1.0 equiv), 1,10-phenanthroline (0.049 g, 0.27 mmol, 0.2 equiv), K2CO3 (0.373 g, 2.70 mmol, 2.0 equiv), CuSO4.5H2O (0.034 g, 0.135 mmol, 0.1 equiv), 1-bromo-2-(triisopropylsilyl)acetylene (0.423 g, 1.62 mmol, 1.2 equiv), and 1.1 mL of toluene. The reaction mixture was heated at 65 °C for 24 h. The resulting yellow-green reaction mixture was cooled to rt and filtered, and the filtrate was concentrated to afford 1.1 g of yellow oil which was purified by column chromatography on 15 g of silica gel (gradient elution with 0–10% EtOAc-hexanes) to afford 0.326 g (66%) of ynamide 31 as a white solid: mp 50–51 °C; IR (thin film) 2943, 2865, 2166, 1598, 1463, 1371, 1173, 987 and 883 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 7.9 Hz, 2H), 3.06 (s, 3H), 2.44 (s, 3H), and 1.04 (app br s, 21H); 13C NMR (100 MHz, CDCl3) δ 144.9, 133.3, 129.8, 127.9, 98.2, 67.5, 39.4, 21.7, 18.7, and 11.4. Anal. Calcd for C19H31NO2SSi: C, 62.42; H, 8.55; N, 3.83. Found: C, 62.13; H, 8.67; N, 3.82.

N-(Methyl)-N-(p-toluenesulfonyl)ethynylamine (S1)
A two-necked, 50-mL pear flask fitted with a rubber septum and an argon inlet adapter was charged with ynamide 31 (1.22 g, 3.34 mmol, 1.0 equiv) and 20 mL of THF. The solution was cooled to −40 °C and tetrabutylammonium fluoride (3.60 mL, 3.67 mmol, 1.0 M solution in THF, 1.1 equiv) was added dropwise over 2 min via syringe. The resulting cloudy yellow mixture was allowed to warm to rt over 1 h. The resulting reaction mixture was diluted with 25 mL of satd NH4Cl solution and 20 mL of Et2O and the aqueous phase was extracted with three 10-mL portions of Et2O. The combined organic phases were washed with 30 mL of brine, dried over MgSO4, filtered, and concentrated to give 1.41 g of pale yellow solid. This material was recrystallized from ca. 3 mL of hot hexanes to afford 0.601 g (86%) of ynamide S1 as white crystals: mp 74–76 °C; IR (thin film) 3280, 2936, 2137, 1597, 1450, 1361, 1174, and 960 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.3 Hz, 2H), 7.36 (d, J = 8.1 Hz, 2H), 3.04 (s, 3H), 2.69 (s, 1H), and 2.44 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 145.1, 133.1, 130.0, 127.9, 77.6, 57.6, 39.0, and 21.8. Anal. Calcd for C10H11NO2S: C, 57.39; H, 5.30; N, 6.69. Found: C, 57.31; H, 5.28; N, 6.56.

N-(Methyl)-N-(p-toluenesulfonyl)-2-methylethynylamine (32)
A 50-mL, two-necked, pear flask fitted with a rubber septum and argon inlet adapter was charged with N-(Methyl)-N-(p-toluenesulfonyl) ethynylamine (0.466 g, 2.23 mmol, 1.0 equiv) and 20 mL of THF. The solution was cooled to −78 °C and KHMDS (3.2 mL, 2.89 mmol, 0.91 M solution in THF, 1.3 equiv) was added dropwise via syringe over 3 min. The reaction mixture was stirred at −78 °C for 1.5 h. MeI (0.25 mL, 0.569 g, 4.01 mmol, 1.8 equiv) was then added to the cloudy yellow reaction mixture in one portion by syringe. The resulting mixture was allowed to warm to rt over 1.5 h and then diluted with 15 mL of satd NH4Cl solution and 15 mL of Et2O. The aqueous layer was extracted with two 10-mL portions of Et2O and the combined organic layers were washed with 20-mL of brine, dried over MgSO4, filtered, and concentrated to afford 0.520 g of orange-yellow solid. Recrystallization from ca. 5 mL of hot hexanes (with cooling to −18°C) provided 0.446 g of ynamide 32 (90%), as pale yellow flakes: mp 93–95 °C; IR (thin film) 2931, 2262, 1595, 1451, 1357, 1169, and 1038 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 8.3 Hz, 2H), 7.35 (d, J = 8.1 Hz, 2H), 3.00 (s, 3H), 2.45 (s, 3H), and 1.86 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 144.7, 133.2, 129.8, 127.9, 73.8, 64.2, 39.4, 21.8 and 3.3. Anal. Calcd for C11H13NO2S: C, 59.17; H, 5.87; N, 6.27. Found, C, 58.97; H, 5.75; N, 6.27.

N-(Methoxycarbonyl)-N-(2-propenyl)ethynylamine (33)
A 100-mL recovery flask fitted with a rubber septum and argon inlet needle was charged with ynamide 29 (1.11 g, 5.25 mmol, 1.0 equiv) and 52 mL of THF. The yellow solution was cooled to −78 °C, and TBAF (5.80 mL, 1.0 M in THF, 5.78 mmol, 1.1 equiv) was added dropwise via syringe over 8 min. The resulting dark green solution was stirred at −78 °C for 20 min and then allowed to warm to rt over 20 min. The reaction mixture was quenched by addition of 40 mL of satd NH4Cl solution and the aqueous layer was separated and extracted with three 30-mL portions of Et2O. The combined organic layers were washed with 50 mL of brine, dried over MgSO4, filtered, and concentrated to give 1.0 g of dark brown oil. Column chromatography on 30 g of silica gel (elution with 10% EtOAc-hexanes) provided 0.617 g (84%) of ynamide 33 as a yellow oil: IR (thin film) 3298, 2958, 2241 (weak), 2146, 1734, 1445, 1362, and 1277 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.88 (ddt, J = 6.0, 10.5, 16.9 Hz, 1H), 5.26–5.32 (app t, J = 10.3, 17.2 Hz, 2H), 4.08 (d, J = 6.0 Hz, 2H), 3.84 (s, 3H), and 2.83 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 155.8, 131.4, 119.0, 77.5, 59.0, 54.4, and 52.5; HRMS-ESI (m/z) [M+Na] calcd for C7H9NO2, 162.0525; found, 162.0531.
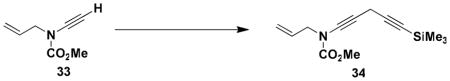
N-(Methoxycarbonyl)-N-(2-propenyl)-5-(trimethylsilyl)-penta-1, 4-diynylamine (34)
A 25- mL, pear flask fitted with a rubber septum and argon inlet needle was charged with ynamide 33 (0.127 g, 0.913 mmol, 1.0 equiv) and 2 mL of DMF. Sodium iodide (0.137 g, 0.913 mmol, 1.0 equiv), Cs2CO3 (0.297 g, 0.913 mmol, 1.0 equiv), and CuI (0.174 g, 0.913 mmol, 1.0 equiv) were added and the resulting bright yellow heterogenous mixture was stirred at rt for 25 min. 3-Trimethylsilylpropargyl bromide (0.155 mL, 0.209 g, 1.10 mmol, 1.2 equiv) was then added dropwise over 4 min by syringe, and the reaction mixture was stirred at rt for 16 h. The resulting dark red-brown mixture was diluted with 20 mL of Et2O and 10 mL of satd NH4Cl solution. The organic layer was washed with two 10-mL portions of H2O, and the combined aqueous layers were extracted with two 15-mL portions of Et2O. The combined organic layers were washed with 20 mL of brine, dried over MgSO4, filtered, and concentrated to give 0.282 g of dark brown oil. Column chromatography on 20 g of silica gel (elution with 5% EtOAc-hexanes) afforded 0.142 g (62%) of diynamide 34 as a dark yellow oil: IR (thin film) 2959, 2269, 2182, 1732, 1446, 1251, and 845 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.87 (ddt, J = 6.0, 10.1, 15.8 Hz, 1H), 5.23–5.31 (app t, J = 11.1, 19.4 Hz, 2H), 4.06 (d, J = 5.8 Hz, 2H), 3.81 (s, 3H), 3.34 (s, 2H), and 0.17 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 156.1, 131.9, 118.9, 100.7, 85.2, 74.3, 64.4, 54.5, 54.3, 52.8, 11.1, and 0.2; HRMS-ESI (m/z) [M+Na] calcd for C13H19NO2Si, 272.1077; found 272.1080.

3-Methoxy-2-methylcyclobutenone (39)
Ketene was generated by pyrolysis of acetone over an electrically heated metal filament using the apparatus described by Williams and Hurd.liii A 250-mL, three-necked flask fitted with a rubber septum, argon inlet adapter and glass stopper was charged with methoxypropyneliv (7.60 g, 108 mmol, 1.0 equiv) in 75 mL of CH3CN and cooled to 0 °C. The argon inlet adapter was replaced with an adapter fitted with a glass pipette connected via Tygon tubing to the ketene generator. The septum was fitted with an outlet needle connected via tubing to a column of CaSO4 leading to a trap of H2O. Ketene was bubbled into the reaction mixture at 0 °C for 8 h and then concentrated to 15.6 g of red oil. This oil was concentrated at 5 mmHg for 1.5 h to remove excess diketene and then distilled through a 10-cm Vigreux column at 0.4 mmHg (bp 54–55 °C) to give 5.90 g (49%) of cyclobutenone 39 as a colorless oil: IR (thin film) 2925, 1757, 1627, 1461, 1385, 1343, 1125, and 970 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.07 (s, 3H), 3.13 (q, J = 2.5 Hz, 2H), and 1.63 (t, J = 2.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 186.0, 177.0, 117.4, 59.9, 46.8, and 6.8; HRMS-ESI (m/z) [M+H] calcd for C6H8O2, 113.0597; found 113.0600.
Experimental Procedures for Benzannulations
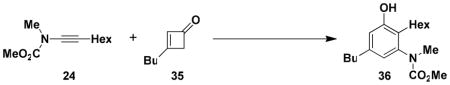
N-(Methoxycarbonyl)-N-methyl[5-butyl-2-hexyl-3-hydroxyphenyl]-amine (36.) Thermal Benzannulation
A 8-mL resealable tube fitted with a rubber septum and argon inlet needle was charged with ynamide 24xii (0.089 g, 0.50 mmol, 1.0 equiv), cyclobutenone 35xviii (0.067 g, 0.54 mmol, 1.2 equiv) and 0.5 mL of toluene. The reaction mixture was heated at reflux for 1 h, then cooled to rt and concentrated to 0.254 g yellow oil. Purification by column chromatography on 10 g of silica gel (elution with 10% EtOAc-hexanes) afforded 0.129 g (80%) of aniline 36 as a white solid: mp 66–68 °C; IR (thin film) 3342, 2956, 2929, 2858, 1679, 1618, 1583, 1459, 1378, 1198, and 1163 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.60 (s, 1H), 6.55 (s, minor rotamer), 6.53 (s, 1H), 6.50 (s, minor rotamer), 5.47 (br s, minor rotamer), 5.22 (br s, 1H), 3.80 (s, minor rotamer), 3.64 (s, 3H), 3.20 (s, 3H), 2.44–2.53 (m, 4H), and 1.47–1.60 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 157.0, 154.7, 142.4, 142.3, 124.4, 120.0, 115.0, 53.1, 38.7, 35.2, 33.4, 32.0, 30.0, 29.3, 25.7, 22.8, 22.6, and 14.2; HRMS-ESI (m/z) [M+Na] calcd for C19H31NO3, 344.2196; found: 344.2186. The regiochemistry of the benzannulation product was confirmed by an NOE study. Irradiation of the benzylic protons on the butyl substituent resulted in enhancement of both aromatic ring protons, ruling out the “Ficini rearrangement” type product (Scheme 4, 16) in which one of these protons would be para rather than ortho to the butyl substituent.
Photochemical Benzannulation
A 5-mL Vycor tube equipped with a rubber septum was charged with cyclobutenone 35 (0.025 g, 0.20 mmol, 1.0 equiv) and ynamide 24 (0.059 g, 0.30 mmol, 1.5 equiv), and 0.4 mL of toluene. The solution was then degassed by three freeze-pump-thaw cycles (-196 °C, 0.02 mmHg). The reaction mixture was irradiated at 254 nm at rt in a Rayonet photochemical reactor. The reaction mixture was then concentrated to give 0.148 g of a yellow oil. Purification by column chromatography on 5 g of silica gel (elution with 10% EtOAc-hexanes) provided 0.042 g (66%) of aniline 36 as a white solid.
Microwave-Promoted Benzannulation
A 5-mL microwavable glass tube equipped with a rubber septum was charged with cyclobutenone 35 (0.037 g, 0.29 mmol, 1.0 equiv) and ynamide 24 (0.057 g, 0.29 mmol, 1.0 equiv), and 0.3 mL of benzene. The tube was sealed and the reaction mixture was heated in a CEM “Discover” microwave apparatus at 130 °C for 30 min (70–90W, 40 psi). The reaction mixture was cooled to rt and concentrated to give 0.107 g of a yellow oil. Column chromatography on 5 g of silica gel (elution of 10% EtOAc-hexanes) provided 0.069 g (74%) of aniline 36 as a white solid.
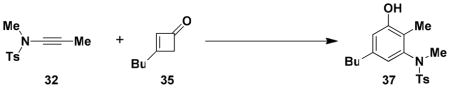
N-(Methyl)-N-(p-toluenesulfonyl)-[5-butyl-3-hydroxy-2-methyl]amine (37)
A 25-mL, roundbottomed flask equipped with a rubber septum and argon inlet needle was charged with ynamide 32 (0.100 g, 0.448 mmol, 1.0 equiv), cyclobutenone 35 (0.069 g, 0.556 mmol, 1.2 equiv) and 0.8 mL of toluene. The septum and needle were replaced with a cold finger condenser with an argon inlet and the pale yellow reaction mixture was heated at reflux for 1h. The reaction mixture was cooled to rt and concentrated to afford 0.179 g of light brown oil. This material was diluted with 2 mL of MeOH and ca. 1.5 mL of 5 M KOH solution and heated at 60–70 °C for 3h. The resulting mixture was cooled to rt, and then diluted with 2 mL of 10% HCl solution and 8 mL of Et2O. The aqueous layer was separated and extracted with two 2-mL portions of Et2O, and the combined organic layers were washed with 5 mL of satd NaHCO3 solution and 5 mL of brine, dried over MgSO4, filtered, and concentrated to 0.197 g of orange oil. Purification by column chromatography on 15 g of silica gel (elution with 15% EtOAc-hexanes) afforded 0.127 g of aniline 37 (81%) as an off-white solid: mp 96–98 °C; IR (thin film) 3447, 2929, 1618, 1586, 1428, 1342, 1160, 1035 and 917 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 8.3 Hz, 2H), 7.30 (d, J = 8.1 Hz, 2H), 6.59 (s, 1H), 5.97 (s, 1H), 5.27 (br s, 1H), 3.11 (s, 3H), 2.46 (s, 3H), 2.35 (t, J = 7.6 Hz, 2H), 2.24 (s, 3H), 1.40 (tt, J = 7.2, 7.6 Hz, 2H), 1.26 (m, 2H), and 0.89 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 155.0, 143.6, 141.4, 141.1, 135.0, 129.6, 128.3, 122.4, 119.0, 115.4, 39.2, 35.1, 33.4, 22.4, 21.8, 14.2, and 11.2. Anal. Calcd for C19H25NO3S: C, 65.38; H, 7.25; N, 4.03. Found: C, 65.38; H, 7.13; N, 3.92.
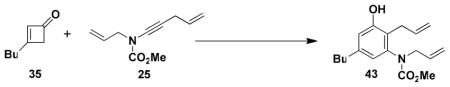
N-(Methoxycarbonyl)-N-allyl-[2-allyl-5-butyl-3-hydroxyphenyl]-amine (43)
A 15-mL resealable tube fitted with a rubber septum and argon inlet needle was charged with ynamide 25 (0.093 g, 0.519 mmol, 1.0 equiv), cyclobutenone 35 (0.077 g, 0.623 mmol, 1.2 equiv), and 0.5 mL of toluene. The reaction mixture was stirred at 80–85 °C for 1.5 h, and then at 110 °C for 1.5 h. The reaction mixture was cooled to rt and concentrated to afford 0.213 g of orange-red oil. Column chromatography on 11 g of silica gel (gradient elution with 10–15% EtOAc-hexanes) afforded 0.132 g (84%) of aniline 43 as a pale yellow solid: mp 39–40 °C; IR (thin film), 3338, 2957, 2930, 1677, 1584, 1459, 1389, 1273, 1197, 1151, and 991 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.67 s, 1H), 6.55 (s, 1H), 5.86–5.94 (m, 2H), 5.50–5.54 (m, 2H), 5.41 (br s, 1H), 5.08–5.18 (m, 4H), 4.35 (dd, J = 6.1, 14.7 Hz, 1H), 3.88–3.93 (m, 1H), 3.80 (s, minor rotamer), 3.62 (s, 3H), 3.30 (m, 2H), 2.43 (t, J = 7.6 Hz, 2H), 1.57 (quintet, J = 7.6 Hz, 2H), 1.34 (q, J = 7.4 Hz, 2H), and 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 156.5, 155.5, 143.1, 140.8, 135.8, 135.4, 121.4, 120.7, 116.9, 116.7, 115.8, 53.1, 50.1, 35.3, 33.3, 30.4, 22.5, and 14.1; HRMS-ESI (m/z) [M+Na] calcd for C18H25NO3, 326.1727; found, 326.1741. Anal. Calcd for C18H25NO3: C, 71.26; H, 8.31; N, 4.62. Found: C, 71.05; H, 8.55; N, 4.34.
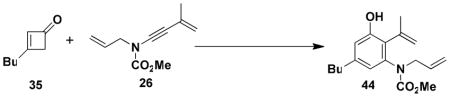
N-(Methoxycarbonyl)-N-allyl-[5-butyl-3-hydroxy-2-isopropenyl phenyl]amine (44)
A 10-mL pear flask equipped with a rubber septum and argon inlet needle was charged with ynamide 26 (0.058 g, 0.324 mmol, 1.0 equiv), cyclobutenone 35 (0.080 g, 0.647 mmol, 2.0 equiv), and 0.4 mL of benzene. The septum was replaced with a cold finger reflux condenser and the yellow reaction mixture was heated at reflux for 3 h. The resulting dark orange mixture was cooled to rt and concentrated to afford 0.163 g of orange oil. Column chromatography on 14 g of silica gel (elution with 15% EtOAc-hexanes) provided 0.066 g (67%) of aniline 44 as a yellow tinted solid: mp 74–76 °C; IR (thin film), 3340, 2957, 1679, 1614, 1575, 1457, 1389, 1315, 1269, 1195, and 1152 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.75 s, 1H), 6.52 (s, 1H), 5.87–5.95 (m, 1H), 5.50–5.54 (m, 2H), 5.06–5.14 (m, 3H), 4.54–4.58 (m, 1H), 3.80 (br s, minor rotamer), 3.63 (s, 3H), 2.54 (t, J = 7.4 Hz, 2H), 1.97 (s, 3H), 1.55–1.61 (m, 2H), 1.26–1.40 (m, 2H), and 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 156.3, 152.6, 143.6, 140.1, 139.3, 133.7, 125.1, 121.5, 119.0, 117.9, 114.6, 53.6, 52.9, 35.4, 33.3, 23.4, 22.5, and 14.1; HRMS-ESI (m/z) [M+Na] calcd for C18H25NO3, 326.1727; found, 326.1734. Anal. Calcd for C18H25NO3: C, 71.26; H, 8.31; N, 4.62. Found: C, 71.17; H, 8.55; N, 4.34.
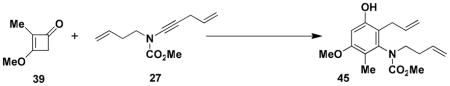
N-(Methoxycarbonyl)-N-(but-3-enyl)-[2-allyl-3-hydroxy-5-methoxy-5-methylphenyl]amine (45)
A threaded Pyrex tube (ca. 5 mL capacity) fitted with a rubber septum and argon inlet needle was charged with ynamide 27 (0.180 g, 0.931 mmol, 1.0 equiv), cyclobutenone 39 (0.208 g, 1.85 mmol, 2.0 equiv), and 1 mL of CHCl3. The resulting yellow solution was degassed via three cycles of freeze-pumpthaw (−196 °C, 0.05 mmHg), and sealed under argon with a threaded Teflon screw cap. The tube was submerged in an oil bath and heated at 150 °C for 16.5 h. After cooling to rt, the reaction mixture was transferred to a 25-mL, round-bottomed flask and concentrated to ca. 0.45 g of dark brown oil, which was diluted with 3 mL of MeOH and 2 mL of 5 M KOH solution. The flask was fitted with a reflux condenser and the reaction mixture was heated at 65 °C for 2 h and then cooled to rt and diluted with 2 mL of 10% HCl solution and 10 mL of Et2O. The aqueous layer was extracted with three 4-mL portions of Et2O, and the combined organic layers were washed with 8 mL of brine, dried over MgSO4, filtered, and concentrated to afford 0.355 g of red-brown oil. Column chromatography on 23 g of silica gel (gradient elution with 20–25% EtOAc-hexanes) afforded 0.176 g (62%) of aniline 45 as a yellow tinted solid: mp 167–170 °C; IR (thin film) 3331, 2954, 1677, 1605, 1461, 1393, 1327, 1309, 1196, 1172, 1124, 1020, and 917 cm−1; 1H NMR (500 MHz, CDCl3, 50 °C) δ 6.46 (s, 1H), 6.32 (s, minor rotamer), 5.88–5.96 (m, 1H), 5.68–5.76 (m, 1H), 5.48 (s, 1H), 5.12–5.17 (m, 2H), 4.99–5.11 (m, 2H), 3.81 (s, minor rotamer), 3.80 (s, 3H), 3.74 (s, minor rotamer), 3.61 (s, 3H), 3.53–3.57 (m, 2H), 3.48–3.51 (m, minor rotamer), 3.26–3.33 (m, 2H), 2.33 (m, 2H), 2.01 (s, minor rotamer) and 2.00 (s, 3H);13C NMR (125 MHz, CDCl3, 50 °C) δ 157.7, 156.7, 154.1, 140.5, 136.4, 135.3, 117.5, 116.8, 116.4, 115.1, 99.5, 55.7, 53.0, 50.6, 32.3, 30.8, and 11.3, (Peaks corresponding to minor rotamer: 137.1, 116.1, 99.7, 33.0, and 31.0); HRMS-ESI (m/z) [M+H] calcd for C17H23NO4, 306.1700; found 306.1704. Anal. Calcd for C17H23NO4: C, 66.86; H, 7.59; N, 4.59. Found: C, 66.59; H, 7.55; N, 4.54.
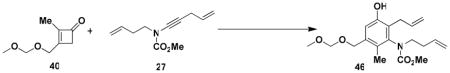
N-(Methoxycarbonyl)-N-(but-3-enyl)-[2-allyl-3-hydroxy-5-(methoxymethoxymethyl)-5-methylphenyl]amine (46)
A 5-mL threaded Pyrex tube fitted with a rubber septum and argon inlet needle was charged with ynamide 27 (0.098 g, 0.507 mmol, 1.0 equiv), cyclobutenone 40 (0.158 g, 1.01 mmol, 2.0 equiv) and 0.7 mL of CHCl3. The pale yellow solution was degassed via three freeze-pumpthaw cycles (−196 °C, 0.05 mmHg), sealed under argon with a Teflon screw cap, and then heated at 150 °C for 16.5 h. The resulting mixture was cooled to rt, transferred to a 25-mL round-bottomed flask, and concentrated to give 0.410 g of dark brown oil. The flask was equipped with a reflux condenser fitted with an argon inlet adapter, 3 mL of MeOH and 2 mL of 5M KOH solution were added, and the resulting mixture was heated at 60–70 °C for 2.5 h. The reaction mixture was cooled to rt and diluted with 2 mL of 10% HCl solution and 4 mL of Et2O. The aqueous layer was extracted with three 2-mL portions of Et2O, and the combined organic layers were washed with 4 mL of satd NaHCO3 solution and 4 mL of brine, dried over MgSO4, filtered, and concentrated to yield 0.222 g of a dark orange oil. Column chromatography on 20 g of silica gel (elution with 30% EtOAc-hexanes) afforded 0.106 g (60%) of aniline 46 as an off-white solid: mp 81–83 °C; IR (thin film) 3336, 2952, 1706, 1676, 1453, 1390, 1326, 1150, 1042 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.93 (s, 1H), 6.83 (s, minor rotamer), 6.24 (s, minor rotamer), 6.21 (s, 1H), 5.85–5.97 (m, 1H), 5.64–5.75 (m, 1H), 4.98–5.09 (m, 4H), 4.74 (s, 2H), 4.70 (s, minor rotamer), 4.50–4.57 (m, 2H), 3.81 (s, minor rotamer), 3.59 (s, 3H), 3.47–3.58 (m, 2H), 3.43 (s, 3H), 3.41 (s, minor rotamer), 3.25–3.39 (m, 2H), 2.28–238 (br dt, 2H), and 2.08 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.7, 153.4, 140.2, 136.1, 136.0, 135.1, 126.3, 123.2, 116.9, 116.0, 115.8, 96.1, 67.7, 55.6, 53.1, 32.3, 31.3, and 13.6. (Peaks corresponding to minor rotamer: 156.4, 153.6, 140.5, 136.2, 126.4, 123.5, 95.9, 53.3, 32.9, and 31.2). Anal. Calcd for C19H27NO5: C, 65.31; H, 7.79; N, 4.01. Found: C, 65.07; H 7.65; N, 3.98.
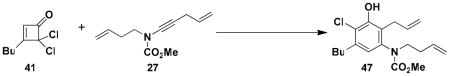
N-(Methoxycarbonyl)-N-(but-3-enyl)-[2-allyl-5-butyl-4-chloro-3-hydroxyphenyl]amine (47)
A 5-mL threaded Pyrex tube fitted with a rubber septum and argon inlet needle was charged with cyclobutenone 41 (0.100 g, 0.518 mmol, 1.0 equiv), ynamide 27 (0.150 g, 0.777 mmol, 1.5 equiv), BHT (0.342 g, 1.55 mmol, 3.0 equiv) and 0.7 mL of toluene. The orange-yellow solution was degassed via three freeze-pump-thaw cycles (−196 °C, 0.05 mmHg) and the tube was sealed under argon with a threaded Teflon screwcap. The tube was heated in an oil bath at 135 °C for 4 h. The resulting solution was cooled to rt, diluted with 10 mL of Et2O, and washed with two 8-mL portions of H2O and 8 mL of satd NaHCO3 solution. The combined aqueous layers were extracted with two 8-mL portions of Et2O and the combined organic layers were washed with 10 mL of brine, dried over MgSO4, filtered, and concentrated to give ca. 0.5 g of dark red-orange oil. Purification by column chromatography on 24 g of silica gel (eluted with 10% EtOAc-hexanes) and then on 15 g of silica gel (elution with 7% EtOAchexanes) provided 0.082 g (45%) of aniline 47 as a viscous orange oil: IR (thin film) 3367, 3078, 2956, 2862, 1694, 1455, 1305, 1152, and 915 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.62, (s, 1H), 5.73–5.93 (m, 2H), 5.82 (s, 1H), 5.02–5.10 (m, 4H), 2.90–3.95 (m, 1H), 3.77 (br s, minor rotamer), 3.59, (s, 3H), 3.21–3.32 (m, 3H), 2.64–2.73 (m, 2H), 2.30–2.35 (m, 2H), 1.56–1.64 (app quintet, J = 7.5 Hz, 2H), 1.35–1.45 (app sextet, J = 7.5 Hz,2H), and 0.96 (t, J = 7.3 Hz); 13C NMR (100 MHz, CDCl3) δ 156.3, 150.5, 139.2, 139.0, 135.3, 135.2, 123.1, 122.3, 119.7, 117.0, 115.7, 53.0, 50.1, 33.4, 31.7, 30.9, 22.6, and 14.1; HRMS-ESI (m/z) [M+Na] calcd for C19H26ClNO3, 374.1493, found 374.1506.
N-(Methoxycarbonyl)-N-(but-3-enyl)-[3-hydroxy-2-isopropenyl-5-(N-methoxycarbonyl-Nmethylamine) phenyl]-amine (48)
A 10-mL, pear flask equipped with a rubber septum and argon inlet needle was charged with ynamide 28 (0.118 g, 0.611 mmol, 1.0 equiv), cyclobutenone 428 (0.168 g, 1.08 mmol, 1.8 equiv) and 0.8 mL of toluene. The septum was replaced with a cold finger reflux condenser and the mixture was stirred at 110 °C for 10 h. The reaction mixture was cooled to rt and concentrated to furnish ca. 0.450 g of a red-brown oil. Unreacted ynamide 28 was removed from this material via column chromatography on 13 g of silica gel (elution with 50% EtOAc-hexanes) to provide 0.222 g of a viscous yellow oil which was transferred into a 25-mL, round-bottomed flask and diluted with 3 mL of MeOH and 2 mL of 5M KOH. The flask was fitted with a reflux condenser and the mixture was stirred at 65 °C for 3.5 h. After cooling to rt, the reaction was quenched with 2 mL of 10% HCl solution and diluted with 10 mL of Et2O. The aqueous layer was separated and the organic layer was washed with 5 mL of H2O and 5 mL of brine, dried over MgSO4, filtered, and concentrated to 0.169 g yellow oil. Column chromatography on 13 g of silica gel (gradient elution with 40–50% EtOAc-hexanes) afforded 0.112 g (53%) of aniline 48 as a pale yellow waxy solid: IR (thin film) 3311, 2956, 1709, 1680, 1609, 1452, 1359, 1264, and 1157 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.83 (s, 1H), 5.51 (s, 1H), 5.68–5.78 (m, 1H), 5.61 (s, minor rotamer), 5.44 (s, 1H), 5.00–5.06 (m, 3H), 3.90–4.04 (m, 1H), 3.74 (s, minor rotamer), 3.72 (s, 3H), 3.28 (s, 3H), 3.17 (s, minor rotamer), 3.03–3.08 (m, 1H), 2.77 (d, minor rotamer), 2.44 (s, minor rotamer), 2.29–2.4 (m, 2H), and 1.95 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.4, 159.4, 156.1, 153.4, 142.8, 139.4, 135.3, 125.7, 118.9, 117.0, 111.6, 109.8, 53.2, 49.7, 37.6, 36.9 (minor rotamer), 32.6, 23.0, and 19.0. Anal. Calcd for C18H24N2O5: C, 62.05; H, 6.94; N, 8.04; Found: C, 61.97; H, 6.73; N, 7.83.
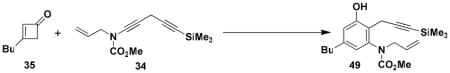
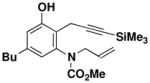
N-(Methoxycarbonyl)-N-allyl-[5-butyl-2-(3-trimethylsilylprop-2-ynyl)-3-hydroxyphenyl] amine (49)
A 50-mL pear flask fitted with a rubber septum and argon inlet needle was charged with the diynamide 34 (0.543 g, 2.18 mmol, 1.0 equiv), cyclobutenone 35 (0.351 g, 2.61 mmol, 1.2 equiv), and 2.1 mL of toluene. The septum and needle were replaced with a cold finger condenser with an argon inlet and the reaction mixture was heated at 80–85 °C for 1.5 h and then at reflux for 1 h. The resulting dark red reaction mixture was cooled to rt and concentrated to afford ca. 1.5 g of red liquid. Column chromatography on 45 g of silica gel (elution with 15% EtOAc-hexanes) provided 0.644 g (79%) of 49 as a yellow solid: mp 106–108 °C, IR (CH2Cl2) 3328, 2957, 2174, 1678, 1588, 1458, 1389, 1249, and 842 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.72 (s, 1H), 6.56 (s, 1H), 6.11 (s, 1H), 5.92 (ddt, J = 6.4, 10.4, 17.1 Hz, 1H), 5.10–5.14 (m, 2H), 4.52 (app dd, J = 5.8, 6.7, and 14.6, 15.6 Hz, 1H), 4.07–4.10 (m, 1H), 3.65 (s, 3H), 3.45 (s, 2H), 2.53 (t, J = 7.6 Hz, 2H), 1.57 (app quintet, J = 7.6 Hz, 2H), 1.35 (app sextet, J = 7.5 Hz, 2H), 0.93 (t, J = 7.3 Hz, 3H) and 0.16 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.6, 155.7, 143.7, 140.2, 133.4, 121.0, 118.9, 118.4, 116.6, 103.1, 87.6, 54.1, 53.5, 35.3, 33.5, 22.7, 17.2, 14.3, and 0.3; HRMSESI (m/z) [M+Na]+ calcd for C21H31NO3Si: 396.1965, found: 396.1955.
N-(Methoxycarbonyl)-N-(butyl)-[4-methoxy-5-methyl-2-propyl phenyl]amine (52)
A 25-mL round-bottomed flask fitted with a rubber septum and argon inlet needle was charged with aniline 45 (0.088 g, 0.288 mmol, 1.0 equiv), 12 mL of EtOH, and 5% Pd/C (0.061 g, 0.029 mmol, 0.1 equiv). The rubber septum and needle were replaced with a Claisen adapter equipped with an argon stopcock adapter and an adapter fitted with a glass pipette. Argon was bubbled through the reaction mixture for 5 min and then a balloon of H2 was fitted over the pipette. H2 was bubbled into the solution at a steady rate at rt for 4 h (the balloon was refilled with H2 at ca. 30–45 min intervals) after which the reaction mixture was purged with argon for 5 min. The resulting mixture was then carefully filtered through a pad of Celite (rinsed with four 10-mL portions of EtOH) and the filtrate was concentrated to provide 0.077 g (82%) of N- (methoxycarbonyl)-N-(butyl)-[3-hydroxy-4-methoxy-5-methyl-2-propylphenyl]amine (50) as a pale yellow oil which was used in the next step without further purification: IR (thin film); 3343, 2959, 1675, 1604, 1464, 1309, and 1125 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.39 (s, 1H), 6.21 (s, minor rotamer), 5.43 (s, minor rotamer), 4.91 (s, 1H), 3.82 (s, minor rotamer), 3.79 (s, 3H), 3.72 (s, minor rotamer), 3.62 (s, 3H), 3.57–3.61 (m, 1H), 3.32 (app ddd, J = 5.5, 11.3, 13.1 Hz), 2.37–2.48 (m, 2H), 1.99 (s, minor rotamer), 1.97 (s, 3H), 1.46–1.64 (m, 4H), 1.22–1.31 (m, 2H), 0.97 (t, J = 7.3 Hz, 3H), and 0.89 (t, J = 7.3 Hz, 3H).
A 25-mL recovery flask fitted with a rubber septum and argon inlet needle was charged with phenol 50 (0.074 g, 0.239 mmol, 1.0 equiv), DMAP (0.066 g, 0.540 mmol, 2.3 equiv), and 1.2 mL of CH2Cl2. The resulting mixture was cooled to −20 °C and triflic anhydride (0.100 mL, 0.162 g, 0.574 mmol, 2.4 equiv) was added dropwise via syringe over ca. 1 min, resulting in a white precipitate. The cooling bath was removed and the reaction mixture was allowed to warm to rt over 6 h. The resulting mixture was diluted with 6 mL of CH2Cl2 and washed with 3 mL of H2O and 3 mL of 1 M HCl solution. The combined aqueous layers were extracted with three 3-mL portions of CH2Cl2 and the combined organic layers were washed with 6 mL of brine, dried over MgSO4, filtered, and concentrated to afford 0.150 g of light brown oil. Purification by column chromatography on 6 g of silica gel (elution with 10% EtOAc-hexanes) provided 0.077 g (73%) of N-(methoxycarbonyl)-N-(butyl)-[4-methoxy-5-methyl-2- propyl-3-(trifluoromethane-sulfonyl) phenyl]amine (51) as a pale yellow oil: IR (thin film) 2963, 1777, 1715, 1611, 1445, 1422, 1214, and 1143 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.74 (s, 1H), 3.84 (s, 3H), 3.83 (s, minor rotamer), 3.82 (s, minor rotamer), 3.65 (s, 3H), 3.58–3.66 (m, 1H), 3.28–3.33 (app ddd, J = 3.0, 10.9, 13.6 Hz), 2.41–2.59 (m, 2H), 2.07 (s, minor rotamer), 2.04 (s, 3H), 1.43–1.59 (m, 4H), 1.22–1.32 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H), and 0.90 (t, J = 7.3 Hz, 3H).
A 10-mL pear flask fitted with a rubber septum and argon inlet needle was charged sequentially with the aryl triflate 51 (0.072 g, 0.163 mmol, 1.0 equiv), 0.4 mL of DMF, Et3N (0.070 mL, 0.489 mmol, 3.0 equiv), formic acid (0.012 mL, 0.326 mmol, 2.0 equiv), Pd(OAc)2 (0.002 g, 0.0098 mmol, 0.06 equiv), and dppf (0.005 g, 0.0098 mmol, 0.06 equiv). The rubber septum was replaced with a cold finger condenser and the reaction mixture was heated at 85–90 °C for 12.5 h. The resulting mixture was cooled to rt, diluted with 6 mL of Et2O, and washed with two 3-mL portions of 1M HCl and then 3 mL of H2O. The combined aqueous layers were extracted with three 3-mL portions of Et2O and the combined organic layers were washed with 6 mL of brine, dried over MgSO4, filtered, and concentrated to provide 0.050 g of dark brown oil. Column chromatography on 10 g of silica gel (elution with 10% EtOAc-hexanes) afforded 0.033 g (69%) of aniline 52 as a colorless oil: IR (thin film) 2958, 1708, 1600, 1486, 1450, 1304, and 1261 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 8.5 Hz), 6.80 (d, J = 8.5 Hz), 3.84 (s, 3H), 3.82 (s, minor rotamer), 3.79 (s, minor rotamer), 3.61 (s, 3H), 3.54–3.60 (m, 1H), 3.38 (app ddd, J = 5.7, 8.8, 10.7 Hz, 1H), 2.35–2.43 (m, 2H), 2.09 (s, minor rotamer), 2.06 (s, 3H), 1.48–1.65 (m, 4H), 1.24–1.33 (app sextet, J = 7.3 Hz), 0.95 (t, J = 7.3 Hz, 3H), and 0.90 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 156.7, 156.3, 140.0, 132.0, 126.8, 125.1, 109.4, 55.7, 52.9, 51.1, 33.0, 30.2, 23.7, 20.6, 14.6, 14.0, and 11.7; HRMS-ESI (m/z) [M+Na] calcd for C17H27NO3; 316.1883, found 316.1893.
4-{N-(3-butenyl)-N-(carbomethoxy)amino}-2, 5-dimethyl-6-methoxy-benzo[b]furan (53)
A 25-mL pear flask fitted with a rubber septum and argon inlet needle was charged with phenol 45 (0.048 g, 0.157 mmol, 1.0 equiv) and 1.6 mL of CH2Cl2. N-iodosuccinimide (0.042 g, 0.189 mmol, 1.2 equiv) was added in one portion, and the resulting red-orange mixture was stirred at rt for 2 h. The reaction mixture was diluted with 10 mL of CH2Cl2 and washed with two 5-mL portions of satd NaS2O3 solution and 5 mL of brine, dried over MgSO4, filtered, and concentrated to afford 0.092 g of pale yellow oil. This material was diluted with 3 mL of MeOH, NaOH (ca. 0.050 g, 1.25 mmol) was added, and the resulting mixture was heated at 65 °C for 2 h. The reaction mixture was cooled to rt and diluted with 2 mL of 1M HCl solution and 3 mL of CH2Cl2. The aqueous layer was extracted with three 2-mL portions of CH2Cl2, and the combined organic layers were washed with 5 mL of satd NaHCO3 solution and 5 mL of brine, dried over MgSO4, filtered, and concentrated to provide 0.062 g of dark yellow oil. Purification by column chromatography on 5 g of silica (gradient elution with 5%-15% EtOAc-hexanes) afforded 0.027 g (56%) of benzofuran 53 as a white solid: mp 79–81 °C; IR (thin film) 2952, 1709, 1592, 1452, 1317 and 1120 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.93 (s, 1H), 6.18 (s, 1H), 5.73 (ddt, J = 6.7, 10.4, 17.2 Hz, 1H), 5.04 (d, J = 17.3 Hz, 1H), 5.00 (d, J = 10.4 Hz, 1H), 3.87 (s, 3H), 3.83 (s, minor rotamer), 3.75–3.81 (m, 2H), 3.62 (s, 3H), 3.57–3.61 (m, minor rotamer), 2.42 (s, 3H), 2.28–2.34 (m, 2H), 2.13 (s, minor rotamer), and 2.10 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.6, 155.7, 154.4, 153.8, 135.3, 131.6, 120.5, 120.1, 116.9, 100.6, 93.5, 56.1, 53.2, 49.7, 32.8, 14.3, and 11.3. Anal. Calcd for C17H21NO4: C, 67.31; H, 6.98; N, 4.62. Found: C, 67.24; H, 6.95, N, 4.59.
Experimental Procedures for Ring Closing Metathesis Reactions

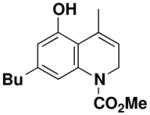
General Procedure for Ring-Closing Metathesis. 7-Butyl-5-hydroxy-4-methyl-2H-quinoline- 1-carboxylic acid methyl ester (55)
A 100-mL, round-bottomed flask equipped with a rubber septum and argon inlet needle was charged with a solution of the Grubbs catalyst 60 (0.015g, 0.018 mmol, 0.05 equiv) in 28 mL of CH2Cl2. The aniline 44 (0.109 g, 0.359 mmol, 1.0 equiv) in 5 mL of CH2Cl2 was added to the reaction flask via cannula in one portion. The rubber septum was replaced with a reflux condenser fitted with an argon inlet adapter. The pale pink-colored reaction mixture was heated at reflux for 40 min, allowed to cool to rt, and then concentrated to afford ca. 0.131 g of green-brown foam. Column chromatography on 10 g of silica gel (elution with 15% EtOAc-hexanes) provided 0.097 g (98%) of dihydroquinoline 55 as a tan waxy solid: IR (thin film) 3360, 2957, 2923, 1680, 1616, 1502, 1440, 1351, 1297, 1248, 1220 and 1146 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.97 (br s, 1H), 6.39 (d, J = 1.5 Hz, 1H), 5.72, (dt, J = 1.5, 4.7 Hz, 1H), 5.64 (br s, 1H), 4.14–4.15 (m, 2H), 3.78 (s, 3H), 2.51 (q, J = 7.6 Hz, 2H), 2.24 (q, J = 1.5 Hz, 3H), 1.54–1.60 (m, 2H), 1.36 (app dt, J = 7.5, 15.0 Hz, 2H), and 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 154.9, 152.8, 142.8, 138.5, 132.6, 121.6, 117.0, 116.0, 113.2, 53.9, 53.3, 42.9, 35.5, 33.4, 22.5, 21.9 and 14.1; HRMS-ESI (m/z) [M+Na] calcd for C16H21NO3, 298.1414; found, 298.1417.

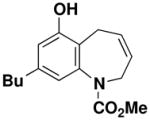
8-Butyl-6-hydroxy-2,5-dihydro-benzo[b]azepine-1-carboxylic acid methyl ester (54)
Reaction of a solution of aniline 43 (0.132 g, 0.435 mmol, 1.0 equiv) in 43 mL of CH2Cl2 with Grubbs catalyst 60 (0.018g, 0.0217 mmol, 0.05 equiv) at reflux for 40 min according to the general procedure afforded ca. 0.195 g of brown foam. Column chromatography on 10 g of silica gel (gradient elution with 15–20% EtOAc-hexanes) provided 0.114 g (95%) of dihydrobenzoazepine 54 as a tan wax: IR (thin film) 3348, 2956, 2930, 1682, 1620, 1587, 1443, 1364, 1267, 1231, and 1020 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.60 (s, 1H), 6.40 (s, 1H), 6.26 (br s, minor rotamer), 5.76–5.83 (m, 2H), 5.49 (d, J = 11.0, 1H), 4.94 (br s, 3H), 4.23 (br s, 1H), 3.83 (s, minor rotamer), 3.68 (s, 3H), 3.38 (br s, 2H), 2.54–2.55 (m, 2H), 1.51–1.62 (m, 2H), 1.25–1.42 (m, 2H), and 0.93 (t, J = 7.3 Hz, 3H);13C NMR (125 MHz, CDCl3) δ 156.5, 152.6, 142.4, 142.4, 142.3, 127.0, 125.3, 123.6, 120.0, 115.1, 53.4, 47.9, 35.3, 33.4, 22.6, 22.5, and 14.1; HRMS-ESI (m/z) [M+Na] calcd for C16H21NO3, 298.1414; found, 298.1424.

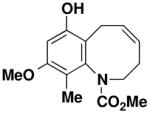
7-Hydroxy-9-methoxy-10-methyl-3,6-dihydro-2H-benzo[b]azocine-1-carboxylic acid methyl ester (56)
Reaction of a solution of aniline 45 (0.103 g, 0.337 mmol, 1.0 equiv) in 33 mL of CH2Cl2 with Grubbs catalyst 60 (0.014 g, 0.0169 mmol, 0.05 equiv) at reflux for 40 min, gave ca. 0.120 g of grey foam. Column chromatography on 9 g of silica gel (elution with 20% EtOAc-hexane) yielded 0.083 g (86%) of benzoazocine 56 as a grey-white solid: mp 165–168 °C; IR (thin film) 3325, 2936, 1704, 1674, 1606, 1464, 1390, 1322, 1192, 1165, 1129, and 1105 cm−1; 1H NMR (500 MHz, CDCl3, 55 °C) δ 6.34 (s, 1H), 6.05 (s, minor rotamer), 5.92 (app q, J = 8.7 Hz, 1H), 5.66 (app q, J = 8.4 Hz, 1H), 5.59 (app q, minor rotamer), 4.86 (s, 1H), 4.38 (dd, J = 6.9, 13.0 Hz, 1 H), 4.21 (dd, minor rotamer), 3.88 (s, minor rotamer), 3.79 (s, 3H), 3.69 (s, minor rotamer), 3.67 (s, 3H) 3.26–3.34 (m, 1 H), 3.09–3.14 (m, 1H), 2.57–2.70 (m, 2H), 2.13–2.19 (m, 1H), 1.97 (s, minor rotamer), 1.95 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 157.2, 156.5, 156.4, 155.9, 152.3, 151.5, 140.8, 139.6, 133.5, 132.2, 128.3, 127.1, 120.7, 120.1, 116.9, 115.0, 98.9, 98.8, 55.7, 55.0, 53.6, 53.3, 49.4, 49.2, 27.5, 26.9, 25.1, 25.0, 10.2, and 10.0, (Peaks corresponding to minor rotamer: δ 156.4, 151.5, 140.8, 132.2, 128.3, 120.1, 116.9, 98.8, 55.7, 53.3, 49.2, 26.9, 25.0, and 10.2.). Anal. Calcd for C15H19NO4: C, 64.97; H, 6.91; N, 5.05. Found: C, 64.88; H, 6.83; N, 4.91.
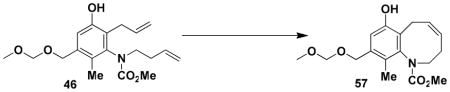
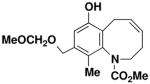
7-Hydroxy-9-(methoxymethoxymethyl)-10-methyl-3,6-dihydro-2H-benzo[b]azocine-1- carboxylic acid methyl ester (57)
Reaction of a solution of aniline 46 (0.111 g, 0.309 mmol, 1.0 equiv) in 30 mL of CH2Cl2 with Grubbs catalyst 60 (0.013g, 0.015 mmol, 0.05 equiv) at reflux for 40 min according to the general procedure afforded ca. 0.125 g of brown foam. Column chromatography on 10 g of silica gel (elution with 40% EtOAc-hexane) yielded 0.093 g (94%) of benzoazocine 57 as a grey-white solid: mp 110–112 °C; IR (thin film) 3338, 2939, 1704, 1676, 1607, 1454, 1389, 1322, 1037, and 919; 1H NMR (500 MHz, CDCl3) δ 6.82 (s, 1H), 6.74 (br s, minor rotamer), 5.95 (app q, J = 8.5 Hz), 5.62–5.70 (m, 1H), 4.73 (s, 3H), 4.70 (s, minor rotamer), 4.52 (AB, J = 12.2 Hz, each 1H), 4.37–4.51 (m, 1H), 3.85 (s, minor rotamer), 3.66 (s, 3H), 3.43 (s, 3H), 3.42 (s, minor rotamer), 3.37 (app dd, J = 7.2, 13.3 Hz, 1H), 3.13 (app dd, J = 8.9, 13.4 Hz, 1H), 2.61–2.76 (m, 1H), 2.54 (app dd, J = 10.0, 13.7 Hz), 2.19 (app quintet, J = 7.3 Hz, 1H), 2.06 (s, minor rotamer), and 2.03 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.5, 151.6, 140.7, 134.6, 132.1, 128.5, 128.2, 116.3, 115.5, 95.9, 67.8, 55.6, 53.3, 49.0, 27.0, 25.4, and 12.6, (Peaks corresponding to minor rotamer: δ 156.6, 151.8, 140.3, 132.8, 127.7, 116.3, 53.4, and 27.6). Anal. Calcd for C17H23NO5: C, 63.54; H, 7.21; N, 4.36. Found: C, 63.23; H, 7.11; N, 4.25.

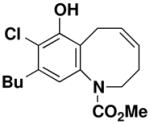
9-Butyl-8-chloro-7-hydroxy-3,6-dihydro-2H-benzo[b]azocine-1-carboxylic acid methyl ester (58)
Reaction of a solution of aniline 47 (0.134 g, 0.381 mmol, 1.0 equiv) in 40 mL of CH2Cl2 with Grubbs catalyst 60 (0.016 g, 0.019 mmol, 0.05 equiv) at reflux for 30 min according to the general procedure gave 0.203 g of dark brown foam. Column chromatography on 11 g of silica gel (elution with 10% EtOAc-hexanes) yielded 0.107 g (87%) of benzoazocine 58 as a light brown solid: mp 112–114 °C, IR (thin film) 3367, 2956, 1693, 1571, 1456, 1323, and 1181 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.67 (s, minor rotamer), 6.60 (s, 1H), 5.93–6.00 (m, 1H), 5.84 (s, 1H), 5.65–5.70 (m, 1H), 4.36–4.40 (m, 1H), 3.82 (s, minor rotamer), 3.66 (s, 3H), 3.42–3.47 (app dd, J = 13.6, 7.1 Hz, 1H), 3.09–3.15 (m, 1H), 2.60–2.68 (m, 4H), 2.16–2.20 (m, 1H), 1.54–1.62 (tt, J = 7.1, 7.5 Hz, 2H), 1.34–1.43 (app sextet, J = 7.4 Hz, 2H), and 0.95 (t, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 156.2, 149.2, 140.4 (minor rotamer), 140.0, 139.1 (minor rotamer), 138.8, 131.7 (minor rotamer), 131.3, 128.9, 128.4 (minor rotamer), 126.9, 121.7, 119.5, 53.1, 49.9, 33.4, 31.7, 27.8 (minor rotamer), 27.1, 25.7, 22.5, and 14.0; HRMS-EI (m/z) [M+] calcd for C17H22ClNO3; 323.1283, found 323.1286. Anal. Calcd for C17H22ClNO3: C, 63.06; H, 6.85; N, 4.33. Found: C, 62.88; H, 6.80; N, 4.35.

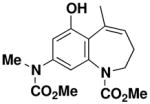
8-(N-Methoxycarbonyl-N-methylamino)-5-methyl-2,3-dihydro-benzo[b]azepine-1-carboxylic acid methyl ester (59)
Reaction of a solution of aniline 48 (0.113 g, 0.324 mmol, 1.0 equiv) in 26 mL of CH2Cl2 with Grubbs catalyst 60 (0.013g, 0.0153 mmol, 0.05 equiv) at reflux for 2 h according to the general procedure gave ca. 0.143 g of brown foam. Column chromatography on 15 g of silica gel (elution with 50% EtOAc-hexanes) provided 0.079 g (76%) of dihydrobenzoazepine 59 as an off-white solid: mp 200–202 °C; IR (thin film) 3297, 2954, 1706, 1676, 1609, 1580, 1450, and 1365 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.94 (br s, 1H), 6.82 (s, 1H), 6.72 (br s, minor rotamer), 6.61 (s, 1H), 5.90 (m, 1H), 4.35–4.42 (m, 1H), 4.19 (br s, minor rotamer), 3.78 (s, 3H), 3.75 (s, minor rotamer), 3.60 (s, 3H), 3.48–3.51 (m, 1H), 3.25 (s, 3H), 3.23 (s, minor rotamer), 2.00 (s, 3H), 1.98–2.01 (m, 2H), and 1.85 (br s, minor rotamer); 13C NMR (100 MHz, CDCl3) δ 157.0, 156.6, 154.6, 142.4, 139.7, 135.9, 126.3, 124.8, 54.9, 53.4, 53.1, 37.7, 22.2, and 21.3. Anal. Calcd for C16H20N2O5: C, 59.95; H, 6.29; N, 8.74. Found: C, 59.85; H, 6.15; N, 8.64.
Experimental Procedures for the Formal Total Synthesis of (+)-FR900482
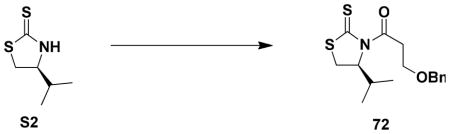
(4S)-3-[3-Benzyloxy]propanoyl]-4-isopropyl-1,3-thiazolidine-2-thione (72)
Procedure A
A 100-mL, round-bottomed flask fitted with a rubber septum and argon inlet needle was charged with 3- (benzyloxy)propionic acidlv (3.66 g, 20.3 mmol, 1.0 equiv) and 40 mL of CH2Cl2. Oxalyl chloride (3.2 mL, 36.6 mmol, 1.8 equiv) was added dropwise over 3 min via syringe, followed by 1–2 drops of DMF, resulting in rapid effervescence. The reaction mixture was stirred at rt for 1.5 h, then concentrated to afford the 3-(benzyloxy)propionyl chloride as pale yellow oil which was used immediately in the next step without further purification.
A 100-mL, two-necked pear flask fitted with a rubber septum and argon inlet adapter was charged with thiazolidine thione S2lvi (2.69 g, 16.67 mmol, 1.0 equiv) and 30 mL of THF. The solution was cooled at −78 °C, while n-BuLi solution (6.5 mL, 2.57 M in hexanes, 1.0 equiv) was added dropwise via syringe over 3 min. The resulting mixture was stirred at −78 °C for 30 min. The solution of 3- (benzyloxy)propionyl chloride (ca 1.2 equiv) in 20 mL of THF was rapidly added to the reaction mixture over 5 min via cannula, and the resulting mixture was stirred at −78 °C for 30 min. The cooling bath was then removed and the reaction mixture was allowed to warm to rt over 1.5 h. To the resulting bright orange mixture was then added 30 mL of H2O and the aqueous layer was separated and extracted with three 20-mL portions of Et2O. The combined organic phases were washed with 80 mL of brine, dried over MgSO4, filtered, and concentrated to afford 6.16 g of a dark orange oil. Purification by column chromatography on 70 g of silica gel (gradient elution with 5–10% EtOAc-hexanes) provided 4.08 g (76%) of the N-acyl thiazolidine-2-thione 72 as a bright yellow oil: [α]21 D +292.0 (c 1.0, CHCl3); IR (thin film) 2963, 1694, 1365, 1260, 1169, and 1094 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.38 (app s, 2H), 7.35 (app s, 2H), 7.27–7.32 (m, 1H), 5.15 (app t, J = 7.0 Hz, 1H), 4.59, 4.53 (AB, J = 11.9 Hz, 1H each), 3.78–3.90 (m, 2H), 3.53–3.60 (m, 2H), 3.49 (dd, J = 8.0, 11.4 Hz, 1H), 3.02 (app d, J = 11.5 Hz), 2.32–2.44 (m, 1H), 1.05 (d, J = 7.2 Hz, 3H), and 0.97 (d, J = 6.8 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 203.0, 171.8, 138.3, 128.6, 128.0, 127.9, 73.4, 71.8, 65.6, 39.1, 31.0, 30.7, 19.3, and 17.9. Anal Calcd for C16H21NO2S2: C, 59.41; H, 6.54; N, 4.33. Found: C, 59.33; H, 6.68; N, 4.26.
Procedure B
A two-necked, 25-mL pear flask fitted with an argon inlet adapter and rubber septum was charged with a solution of 3-(benzyloxy)propionic acid (0.208 g, 1.15 mmol, 1.1 equiv) in 4 mL of THF. The solution was cooled to −78 °C and Et3N (0.15 mL, 1.26 mmol, 1.2 equiv) was added in one portion via syringe, followed by pivaloyl chloride (0.16 mL, 1.32 mmol, 1.2 equiv) rapidly dropwise via syringe. The resulting thick white slurry was stirred at 0 °C for 1 h and then recooled to −78 °C. A solution of thiazolidinone thione S2 (0.169 g, 1.05 mmol, 1.0 equiv), Et3N (0.15 mL, 1.24 mmol, 1.0 equiv), and DMAP (0.012 g, 0.105 mmol, 0.1 equiv) in 3 mL of THF was added dropwise over 5 min via cannula and the resulting mixture was allowed to warm to rt over 20 h. The reaction mixture was then diluted with 25 mL of Et2O and washed with 10 mL of H2O, three 10-mL portions of 1M NaOH solution, and 10 mL of brine. The combined organic phases were dried over MgSO4, filtered, and concentrated to give 0.423 g yellow oil. Purification by column chromatography on 10 g of silica gel (elution with 10% EtOAc-hexanes) provided 0.252 g (74%) of the N-acyl thiazolidinethione 72.
(4S)-3-[(2R,3S-3-Hydroxy-2-[(benzyloxy)methyl]pent-4-en-oyl]-4-isopropyl-1,3-thiazolidine- 2-thione (73)
A 200-mL recovery flask fitted with a rubber septum and argon inlet needle was charged with freshly purified tin (II) trifluoromethanesulfonatelvii (7.77 g, 18.6 mmol, 1.7 equiv) and 54 mL of CH2Cl2 and cooled to −78 °C. Freshly distilled N-ethylpiperidine (2.70 mL, 20.6 mmol, 1.8 equiv) was then added in one portion via syringe. The resulting off-white suspension was stirred at −78 °C for 5 min, and then a solution of the N-acyl thiazolidinethione 72 (3.57 g, 11.0 mmol, 1.0 equiv) in 30 mL of CH2Cl2 was added dropwise via cannula over 20 min. The cloudy yellow reaction mixture was stirred at −78 °C for 3 h, after which freshly distilled acrolein (1.50 mL, 21.9 mmol, 2.0 equiv) was added rapidly dropwise via syringe. After stirring at −78 °C for 1.5 h, the cold reaction mixture was poured into 80 mL of pH 7 phosphate buffer, resulting in the formation of a thick white precipitate which was filtered through Celite with the aid of four 50-mL portions of CH2Cl2. The filtrate was washed with 250 mL of brine, dried over MgSO4, filtered, and concentrated to afford 6.79 g of a viscous orange oil. Purification by column chromatography on 120 g of silica gel (gradient elution with 15–20% EtOAc-hexanes) afforded 1.94 g (73%) of 73 as a viscous bright yellow oil: [α]21 D +354.8 (c 0.95, CHCl3); IR (thin film) 3428, 2964, 1694, 1372, and 1160 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.24–7.34 (m, 5H), 5.90 (ddd, J = 5.7, 10.5, 16.2 Hz, 1H), 5.31 (app dt, J = 1.5, 17.2 Hz, 1H), 5.18–5.23 (m, 1H), 5.17 (app dt, J = 1.5, 10.5 Hz), 5.00 (app t, J = 6.7 Hz, 1H), 4.62–4.67 (m, 1H), 4.41, 4.46 (AB, J = 11.8 Hz, each 1H), 3.86 (app t, J = 8.4 Hz, 1H), 3.71 (dd, J = 4.8, 9.3 Hz, 1H), 3.25 (dd, J = 7.8, 11.4 Hz, 1H), 3.20 (d, J = 3.1 Hz, 1H), 2.91 (dd, J = 0.9, 11.4 Hz, 1H), 2.27–2.39 (m, 1H), 1.01 (d, J = 6.8 Hz, 3H), and 0.94 (d, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 204.2, 174.8, 138.1, 137.2, 128.5, 127.9, 127.7, 116.6, 73.6, 72.6, 72.3, 69.2, 48.3, 31.1, 30.9, 19.3, and 18.1. Anal. Calcd for C19H25NO3S2: C, 60.13; H, 6.64; N, 3.69. Found: C, 59.95; H, 6.53; N, 3.72.
Assignment of the relative stereochemistry of the major and minor aldol products was carried out by sodium borohydride reduction of each compound and comparison of NMR data for the resulting 1,3- diols to that reported for closely related diols by Breit and Zahn.lviii Further confirmation was obtained by conversion of the diols to cyclic acetals (PhCH(OMe)2, cat. TsOH, DCM, 4A sieves, rt) and analysis of the coupling constants for the vicinal protons on the ring. The absolute stereochemistry of the aldol product was determined by cleavage of the auxiliary to form the methyl ester (MeOH, imidazole, DCM, rt, 17 h) and application of Kakisawa’s modified Mosher ester analysislix to the (R) and (S)-MPTA esters prepared by reaction with the appropriate MPTA acids in the presence of EDAC and DMAP (rt, 16 h).
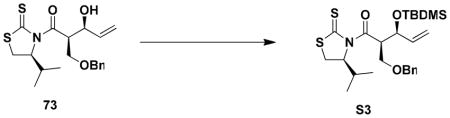
(4S)-3-[(2R,3S-3-tert-Butyldimethylsilyloxy-2-[(benzyloxy)methyl]pent-4-en-oyl]-4-isopropyl- 1,3-thiazolidine-2-thione (S3)
A 100-mL recovery flask fitted with a rubber septum and argon inlet needle was charged with alcohol 73 (2.50 g, 6.59 mmol, 1.0 equiv) and 30 mL of CH2Cl2 and cooled to 0 °C. 2,6-Lutidine (1.7 mL, 1.56 g, 14.6 mmol, 2.2 equiv) and then TBSOTf (1.7 mL, 1.92 g, 7.25 mmol, 1.1 equiv) were added rapidly dropwise via syringe. The yellow reaction mixture was stirred at 0 °C for 50 min, and then 25 mL of H2O was added. The aqueous layer was separated and extracted with two 10- mL portions of CH2Cl2, and the combined organic phases were washed with 35 mL of brine, dried over MgSO4, filtered, and concentrated to give 4.40 g of a dark yellow oil. Purification by column chromatography on 50 g of silica gel (gradient elution with 0–5% EtOAc-hexanes) provided 3.11 g (96%) of S3 as a viscous bright yellow oil, which solidified to a yellow solid: mp 74–76 °C; [α]22 D +266.3 (c 0.62, CHCl3); IR (thin film) 2929, 1694, 1471, 1363, 1168, 1091, 838 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.25–7.35 (m, 5H), 5.99 (ddd, J = 7.0, 10.3, 17.2 Hz, 1H), 5.18–5.25 (m, 2H), 5.10 (app d, J = 10.4 Hz, 1H), 4.99 (app t, J = 6.9 Hz, 1H), 4.58 (app t, J = 6.9 Hz, 1H), 4.43, 4.49 (AB, J = 11.9 Hz, each 1H), 3.91 (app t, J = 9.1 Hz, 1H), 3.81 (dd, J = 4.5, 9.1 Hz, 1H), 3.20 (dd, J = 7.8, 11.3 Hz, 1H), 2.88 (app d, J = 11.4 Hz, 1H), 2.30–2.38 (m, 1H), 1.03 (d, J = 6.8 Hz, 3H), 0.95 (d, J = 6.9 Hz, 3H), 0.89 (s, 9H), 0.03 (s, 3H) and 0.02 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 203.8, 173.7. 139.3, 138.5, 128.4, 127.7, 127.6, 116.3, 73.9, 73.3, 72.6, 70.7, 49.8, 30.9, 30.8, 26.0, 19.4, 18.3, 18.0, −3.9, and −4.8. Anal. Calcd for C25H39NO3S2Si: C, 60.81; H, 7.96; N, 2.84. Found: C, 60.69; H, 8.04; N, 2.87.

(2R,3S)-2-(Benzyloxy)methyl-3-(tert-butyldimethylsilyloxy)pent-4-en-al (74)
A 25-mL recovery flask equipped with a rubber septum and argon inlet needle was charged with S3 (1.01 g, 2.06 mmol, 1.0 equiv) and 10 mL of CH2Cl2 and cooled to −78 °C. DIBAL-H (1.0 M in hexanes, 3.7 mL, 3.71 mmol, 1.8 equiv) was added dropwise via syringe over 3 min. The bright yellow solution was stirred for ca. 10 min (until the reaction mixture became colorless), and 5 mL of satd Na/K tartrate solution was added. The reaction mixture was allowed to warm to rt over 1.5 h and the aqueous layer was then extracted with two 5-mL portions of CH2Cl2. The combined organic layers were washed with 15 mL of brine, dried over MgSO4, filtered, and concentrated to provide 1.05 g of an off-white semi-solid consisting of the aldehyde and white crystals of (4S)-4-isopropyl-1,3-thiazolidine-2-thione. This material was treated with ca. 15 mL of hexanes, and the resulting solution was separated from the chiral auxiliary, concentrated, and purified by column chromatography on 6 g of silica gel (elution with 5% EtOAchexanes), to afford 0.586 g (85%) of the aldehyde 74 as a very pale yellow oil: [α]22 D −2.45 (c 1.35, CHCl3); IR (thin film) 2858, 1727, 1472, 1362, 1254, and 1092 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.78 (d, J = 1.8 Hz, 1H), 7.27–7.40 (m, 5H), 5.82 (ddd, J = 6.3, 10.4, 16.9 Hz, 1H), 5.25 (app d, J = 17.1 Hz, 1H) 5.15 (app d, J = 10.4 Hz, 1H), 4.65 (app t, J = 6.1 Hz, 1H), 4.48, 4.55 (AB, J = 11.9 Hz, 1 H each), 3.91 (dd, J = 7.2, 9.6 Hz, 1H), 3.67 (dd, J = 5.9, 9.6 Hz, 1H), 2.72–2.77 (m, 1H), 0.88 (s, 9H), 0.08 (s, 3H), and 0.03 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 203.7, 138.4, 138.1, 128.6, 127.9, 127.8, 116.4, 73.6, 71.9, 66.0, 58.2, 25.9, 18.3, −4.1, and −5.0. Anal. Calcd for C19H30O3Si: C, 68.22; H, 9.04. Found: C, 68.09; H, 9.08.

(2R,3S)-2-(Benzyloxy)methyl-1-bromo-3-(tert-butyldimethylsilyloxy)hex-5-en-1-yne (76)
A two-necked, 25-mL pear flask equipped with a rubber septum and argon inlet adapter was charged with phosphonium bromide 75lx (0.110 g, 0.329 mmol, 2.5 equiv) and 1.6 mL of THF. A solution of KOt-Bu (0.82 mL, 0.790 mmol, 0.96 M in THF, 2.4 equiv) was added rapidly dropwise via syringe over 2 min. The resulting dark brown reaction mixture was stirred for 3 min at rt and then cooled to 0 °C. A solution of the aldehyde 74 (0.110 g, 0.329 mmol, 1.0 equiv) in 1.6 mL of THF was added dropwise over 4 min via cannula. The reaction mixture was stirred at 0 °C for 10 min, and then cooled to −78 °C. A second portion of KOt-Bu solution (1.5 mL, 1.45 mmol) was added rapidly via syringe, resulting in a dark brown reaction mixture which was allowed to stirred at −78 °C for 20 min. The reaction was diluted with 6 mL of brine and the aqueous layer was separated and extracted with three 6-mL portions of Et2O. The combined organic phases were washed with 10 mL of brine, dried over MgSO4, filtered, and concentrated to afford 0.562 g of a brown semi-solid which was dissolved with 2 mL of CH2Cl2 and concentrated onto ca. 0.8 g of silica. The resulting free-flowing powder was added to the top of a column of 10 g of silica gel and eluted with 2% EtOAc-hexanes to provide 0.127 g (92%) of bromo alkyne 76 as a yellow oil: [α]22 D −1.54 (c 0.32, CHCl3); IR (thin film) 2928, 1472, 1361, 1252, 1082, 927, and 836 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.36 (app s, 2H), 7.35 (app s, 2H), 7.28–7.34 (m, 1H), 5.92 (ddd, J = 6.6, 10.4, 17.0 Hz, 1H), 5.23 (app d, J = 17.2 Hz, 1H), 5.17 (app d, J = 10.4 Hz, 1H), 4.53, 4.62 (AB, J = 12.2 Hz, each 1H), 4.32 (app t, J = 6.4 Hz, 1H), 3.59 (d, J = 9.3 Hz, 2H), 2.78 (app q, J = 6.0 Hz, 1H), 0.90 (s, 9H), 0.08 (s, 3H), and 0.04 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 139.0, 138.3, 128.5, 127.8, 127.7, 116.3, 79.3, 73.3, 73.2, 69.0, 41.5, 41.2, 25.9, 18.3, −4.2, and −4.8. Anal. Calcd for C20H29BrO2Si: C, 58.67; H, 7.14. Found: C, 58.72; H, 7.09.
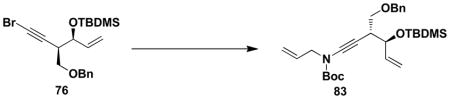
(3R,4S)-N-(tert-butoxycarbonyl)-N-(2-propenyl)-hex-5-en-3-(benzyloxymethyl)-4-(tertbutyldimethylsilanyloxy)- 1-ynylamine (83)
A 15-mL resealable Pyrex tube fitted with a rubber septum and argon inlet adapter was charged with CuSO4.5H2O (0.032 g, 0.128 mmol, 0.2 equiv), 1,10- phenanthroline (0.046 g, 0.257 mmol, 0.4 equiv), K3PO4 (0.273 g, 1.28 mmol, 2.0 equiv), and a solution of the bromo alkyne 76 (0.263 g, 0.642 mmol, 1.0 equiv) and tert-butylallyl carbamate 82lxi (0.303 g, 1.93 mmol, 3.0 equiv) in 2 mL of toluene. The resulting heterogenous reaction mixture was heated at 80–85 °C for 24 h, and then a second portion of CuSO4.5H2O (0.032 g, 0.128 mmol, 0.2 equiv) and 1,10- phenanthroline (0.036 g, 0.257 mmol, 0.4 equiv) was added. The resulting mixture was stirred at 80–85 °C for 22 h, cooled to rt, and then filtered through a plug of ca. 2 g of silica gel with the aid of ca. 15 mL of Et2O and concentrated to give 0.532 g of orange oil. Purification by column chromatography on 30 g of silica gel (gradient elution with 2–5% EtOAc-hexanes) afforded 0.039 g (15%) of unreacted bromo alkyne 76 and 0.213 g (68%) of ynamide 83 as a pale yellow oil: [α]22 D −8.4 (c 0.75, CHCl3); IR (thin film) 3087, 2928, 2263, 1717, 1646, 1473, 1392, 1153, and 924 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.28–7.35 (m, 5H), 5.96 (ddd, J = 6.6, 10.4, 17.1 Hz, 1H), 5.85 (ddt, J = 6.6, 10.3, 17.0 Hz, 1H), 5.11–5.25 (m, 4H), 4.55 (AB, J = 12.1 Hz, each 1H), 4.27 (app t, J = 6.3 Hz, 1H), 3.97 (app bd, J = 5.4 Hz, 2H), 3.57 (app bd, J = 5.5 Hz, 2H), 2.87 (app q, J = 6.1 Hz, 1H), 1.48 (s, 9H), 0.89 (s, 9H), 0.06 (s, 3H), and 0.03 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 154.4, 139.5, 138.6, 132.4, 128.5, 127.8, 127.6, 118.0, 115.7, 82.1, 73.6, 73.2, 70.1, 68.0, 51.9, 40.4, 28.2, 26.0, 18.3, −4.1, and −4.7. Anal Calcd for C28H43NO3Si: C, 69.24; H, 8.92; N, 2.88. Found: C, 69.31; H, 8.88; N, 2.67.
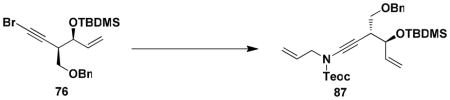
(3R,4S)-N-(2-Trimethylsilylethoxycarbonyl)-N-(2-propenyl)-hex-5-en-3-benzyloxymethyl-4- (tert-butyldimethylsilanyloxy)-1-ynylamine (87)
A 25-mL pear flask fitted with a cold finger condenser with argon inlet was charged with the bromo alkyne 76 (0.161 g, 0.393 mmol, 1.0 equiv), carbamate 86lxii (0.238 g, 0.1.18 mmol, 3.0 equiv), 1,10-phenanthroline (0.028 g, 0.157 mmol, 0.4 equiv), K3PO4 (0.167 g, 0.0.786 mmol, 2.0 equiv), CuSO45•H2O (0.020 g, 0.0786 mmol, 0.2 equiv), and 1 mL of toluene. The heterogenous reaction mixture was heated at 85–90 °C for 24 h and then a second portion of CuSO4.5H2O (0.020 g, 0.0786 mmol, 0.2 equiv) and 1,10-phenanthroline (0.028 g, 0.157 mmol, 0.4 equiv) was added. The reaction mixture was heated at 85–95 °C for 24 h, cooled to rt, and then filtered through a plug of ca. 3 g of celite with the aid of ca. 15 mL of Et2O. Concentration gave ca. 0.4 g of a orange-brown oil. Purification by column chromatography on 24 g of silica gel (gradient elution with 2–5% EtOAc-hexanes)lxiii afforded 0.162 g (78%) of the ynamide 87 as a pale yellow oil: [α]21 D −5.1 (c 0.28, CHCl3); IR (thin film) 2955, 2857, 2265, 1724, 1402, 1251, 1075, 925, and 837 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.27–7.36 (m, 5H), 5.81–5.99 (m, 2H), 5.11–5.27 (m, 4H), 4.56 (AB, J = 12.1 Hz, 1H each); 4.23–4.30 (m, 3H), 4.02 (d, J = 5.7 Hz, 2H), 3.50–3.61 (m, 2H), 2.87 (app q, J = 6.1 Hz, 1H), 1.04 (t, J = 8.5 Hz, 2H), 0.89 (s, 9H), 0.06 (s, 3H), 0.04 (s, 9H), and 0.03 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 155.8, 139.4, 138.6, 132.1, 128.5, 127.8, 127.7, 118.3, 115.9, 76.7, 73.6, 73.2, 70.0, 68.5, 65.5, 52.6, 40.4, 26.0, 18.3, 17.8, −1.3, −4.1, and −4.7; Anal Calcd for C29H47NO4Si2: C, 65.74; H, 8.94; N, 2.64; found: C, 65.69; H, 8.96; N, 2.51.

3-(Methylbenzyloxy)cyclobut-1-enone (80)
A 50-mL, two-necked pear flask fitted with a rubber septum and argon inlet adapter was charged with a solution of tributyl(benzyloxymethyl)stannanelxiv (77) (1.05 g, 2.59 mmol, 1.2 equiv) in 4 mL of THF. The solution was cooled to −78 °C and n-BuLi solution (0.92 mL, 2.59 M in hexanes, 1.1 equiv) was added rapidly dropwise via syringe. The resulting mixture was stirred at −78 °C for 5 min, then a solution of 3- ethoxycyclobutenonexlvii (79) in 3 mL of THF was added rapidly dropwise via cannula. The resulting orange reaction mixture was stirred at −78 °C for 2.5 h, then trifluoroacetic anhydride (0.544 g, 0.36 mL, 2.59 mmol, 1.2 equiv) was added via syringe in a single portion. The reaction mixture was stirred at −78 °C for 3 h and then diluted with 10 mL of satd NaHCO3 solution and allowed to warm to rt over 30 min. The aqueous phase was extracted with three 10-mL portions of Et2O, and the combined organic phases were washed with 15 mL of brine, dried over Na2SO4, filtered, and concentrated to 1.43 g of a biphasic orange oil. Purification by column chromatography on 30 g of acetone-deactivated silica gel (elution with 20% EtOAc-hexanes) afforded 0.265 g (65%) of cyclobutenone 80 as a yellow oil: FT-IR (thin film) 2920, 1769, 1597, 1497, 1454, 1225, 1143, and 1095 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.29–7.47 (m, 5H), 6.14 (s, 1H), 4.65 (s, 2H), 4.48 (s, 2H), and 3.22 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 186.8, 175.4, 137.7, 135.1, 128.8, 128.3, 128.0, 73.5, 68.3, and 49.7; HRMS-ESI (m/z) [M+Na] calcd for C12H12O2: 211.0730, found: 211.0735.

3-(Methoxymethoxymethyl)cyclobut-1-enone (81)
A 25-mL, two-necked pear flask fitted with a rubber septum and argon inlet adapter was charged with a solution of tributyl[(methoxymethoxy)methyl]stannanexlviiib (78) (0.739 g, 2.03 mmol, 1.5 equiv) in 5 mL of THF. The solution was cooled to −78 °C and n-BuLi solution (0.69 mL, 2.36 M in hexane, 1.2 equiv) was added rapidly dropwise via syringe. The reaction mixture was allowed to stir at −78 °C for 2 h and then a solution of 3-ethoxycyclobutenonexlvii 79 (0.151 g, 1.35 mmol, 1.0 equiv) in 2 mL of THF was added rapidly dropwise via cannula. The resulting orange reaction mixture was stirred at −78 °C for 2 h and then trifluoroacetic anhydride (0.250 mL, 1.76 mmol, 1.3 equiv) was added via syringe in a single portion. The reaction mixture was stirred at −78 °C for 75 min and diluted with 3 mL of satd NaHCO3 solution and allowed to warmed to rt over 30 min. The resulting mixture was diluted with 15 mL of CH2Cl2 and the aqueous phase was separated and extracted with two 5-mL portions of CH2Cl2. The combined organic phases were washed with 15 mL of brine, dried over MgSO4, filtered, and concentrated to give 0.901 g of a dark orange, biphasic oil. Purification by column chromatography on 19 g of silica gel (elution with 25% EtOAc-hexanes) afforded 0.125 g (65%) of 81 as an orange oil: FT-IR (neat) 2923, 1772, 1599, 1438, 1152, and 1048 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.10 (s, 1H), 4.74 (s, 2H), 4.54 (s, 2H), 3.40 (s, 3H), and 3.23 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 186.7, 175.2, 135.0, 96.5, 65.6, 55.8, and 49.6; HRMS-ESI (m/z) [M+Na] calcd for C7H10O3: 165.0522, found: 165.0526.
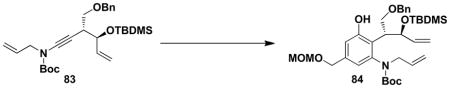
N-Allyl-N-(tert-butoxycarbonyl)-[2-{(1R,2S)-1-(benzyloxy)methyl-2-(tertbutyldimethylsilanyloxy)- but-3-enyl}-3-hydroxy-5-(methoxymethoxy)methylphenyl]amine (84)
A 25-mL pear flask fitted with a rubber septum and argon inlet needle was charged with a solution of ynamide 83 (0.108 g, 0.222 mmol, 1.0 equiv) and cyclobutenone 81 (0.041 g, 0.289 mmol, 1.3 equiv) in 0.7 mL of toluene. The septum was replaced with a cold finger reflux condenser with an argon inlet and the reaction mixture was heated at 80 °C for 1.5 h and then at reflux for 1.5 h. After cooling to rt, the reaction mixture was concentrated to 0.200 g of viscous orange oil. Purification by column chromatography on 14 g of silica gel (elution with 15% EtOAc-hexanes) provided 0.125 g (94%) of 84 as a viscous pale yellow oil: [α]21 D −6.3 (c = 0.125, CHCl3); IR (thin film) 3281, 2927, 1699, 1667, 1624, 1574, 1437, 1254, 1049 and 922 cm−1; 1H NMR (500 MHz, 90 °C, d6-DMSO) δ 9.20 (s, 1H), 9.13 (s, minor rotamer), 7.22–7.30 (m, 3H), 7.15–7.16 (m, 2H), 6.73 (s, minor rotamer), 6.70 (s, 1H), 6.52 (s, minor rotamer), 6.43 (s, 1H), 5.93–5.97 (m, minor rotamer), 5.79–5.83 (m, 1H), 5.62–5.68 (m, 1H), 5.03–5.13 (m, minor rotamer), 4.97–4.99 (m, 2H), 4.87 (app d, J = 18.1 Hz, 1H), 4.73–4.80 (m, 2H), 4.61 (s, 2H), 4.41 (s, 4H), 4.31–4.37 (m, 1H), 4.14 (t, J = 9.0 Hz, 1H), 3.90–3.97 (m, 2H), 3.80 (dd, J = 6.8, 15.3 Hz, 1H), 3.61–3.64 (m, minor rotamer), 3.30 (s, 3H), 3.12 (bs, 1H), 1.38 (s, 9H), 1.32 (s, minor rotamer), 0.88 (s, 9H), 0.05 (s, 3H), and 0.01 (s, 3H); 13C NMR (125 MHz, 90 °C, d6-DMSO) δ 155.9, 153.8, 142.5, 140.1, 138.2, 136.4, 134.0, 127.6, 126.9, 126.8, 126.7, 123.6, 119.9, 116.0, 113.6, 113.4, 94.9, 78.5, 73.0, 72.2, 70.0, 67.8, 54.4, 52.2, 46.7, 27.7, 25.4, 17.4, −4.4, and −4.9, (peaks corresponding to minor rotamer: δ 134.1, 116.4, 113.5, 95.0, 70.8, −4.5, and −5.1); Anal Calcd for C35H53NO7Si: C, 66.95; H, 8.51; N, 2.23; found: C, 66.91; H, 8.46; N, 2.40.
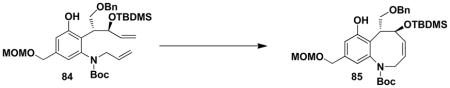
5S,6R-6-benzyloxymethyl-5-(tert-butyldimethysilanyloxy)-7-hydroxy-9- methoxymethoxymethyl-5,6-dihydro-2H-benzo[b]azocine-1-carboxylic acid tert-butyl ester (85)
A 50-mL, recovery flask fitted with a rubber septum and argon inlet needle was charged with a solution of the Grubbs catalyst 60 (0.005 g, 0.00593 mmol, 0.05 equiv) in 9 mL of CH2Cl2. The aniline 84 (0.0745 g, 0.119 mmol, 1.0 equiv) in 3 mL of CH2Cl2 was added to the reaction flask via cannula in one portion and the rubber septum was replaced with a reflux condenser fitted with an argon inlet adapter. The pale pinkcolored reaction mixture was heated at reflux for 5.5 h, allowed to cool to rt, and then concentrated to afford ca. 0.134 g of yellow-brown foam. Column chromatography on 11 g of silica gel (elution with 15% EtOAc-hexanes) yielded 0.057 g (80%) of benzoazocine 85 as a viscous pale yellow oil: [α]21 D − 18.7 (c 0.44, CHCl3); IR (thin film) 3343, 2927, 1694, 1670, 1624, 1573, 1442, 1391, 1258, and 1075 cm−1; 1HNMR (500 MHz, 55 °C, d6-DMSO) δ 9.21 (s, 1H), 7.22–7.28 (m, 5H), 6.77 (s, 1H), 6.49 (s, 1H), 5.38–5.41 (m, 1H), 5.19–5.21 (m, 1H), 5.04 (app t, J = 7.9 Hz, 1H), 4.78 (app d, J = 15.4 Hz, 1H), 4.63 (s, 2H), 4.42 (s, 2H), 4.39–4.46 (m, 2H), 3.76 (br s, 1H), 3.64 (app t, J = 8.4 Hz, 1H), 3.48–3.56 (m, 2H), 3.30 (s, 3H), 1.37 (br s, 9H), 0.87 (s, 9H), 0.04 (s, 3H), and 0.02 (s, 3H); 13C NMR (125 MHz, 55 °C, d6- DMSO) δ 156.0, 139.7, 138.6 137.5, 136.2, 127.8, 127.0, 126.8, 122.7, 118.5, 113.3, 95.1, 79.2, 71.6, 70.4, 70.3, 67.8, 54.6, 48.3, 45.0, 27.9, 25.6, 25.5, 17.6, −4.71, and −5.13; (peaks corresponding to minor rotamer: 156.5, 139.3, 138.4, 130.3, and 72.4). Anal. Calcd for C33H49NO7Si: C, 66.08; H, 8.23; N, 2.34. Found: C, 65.96; H, 8.23; N, 2.42.
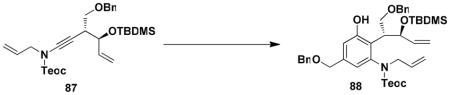
N-Allyl-N-(2-trimethylsilanyloxycarbonyl)-[2-(1R,2S)-{1-(benzyloxy)methyl-2-(tertbutyldimethylsilanyloxy)- but-3-enyl}-3-hydroxy-5-(benzyloxy)methylphenyl]amine (88)
A 25-mL pear flask fitted with a rubber septum and argon inlet needle was charged with a solution of ynamide 87 (0.077 g, 0.145 mmol, 1.0 equiv) and cyclobutenone 80 (0.036 g, 0.189 mmol, 1.3 equiv) in 0.4 mL of toluene. The septum was replaced with a cold finger reflux condenser with an argon inlet and the reaction mixture was heated at 80 °C for 1.5 h and then at reflux for 1.5 h. After cooling to rt, the reaction mixture was concentrated to yield 0.121 g of viscous orange oil. Purification by column chromatography on 10 g of silica gel (gradient elution with 5–10% EtOAc-hexanes) provided 0.086 g (83%) of 88 as a viscous pale yellow oil: [α]21 D −4.05 (c 0.35, CHCl3); IR (thin film) 3272, 2954, 2857, 1703, 1624, 1573, 1454, 1404, 1251, 1075, 924, and 837 cm−1; 1H NMR (400 MHz, 70 °C, d6-DMSO) δ 9.38 (s, 1H), 9.33 (s, minor rotamer), 7.23–7.41 (m, 9H), 7.17–7.19 (m, 1H), 6.81 (s, minor rotamer), 6.79 (s, 1H), 6.58 (s, minor rotamer), 6.51 (s, 1H), 5.98 (dddd, minor rotamer), 5.86 (dddd, J = 5.2, 7.1, 10.2, 17.3 Hz, 1H), 5.68–5.73 (m, minor rotamer), 5.63 (ddd, J = 6.8, 10.3, 17.2 Hz, 1H), 4.77–5.16 (m, 5H), 4.55 (s, minor rotamer), 4.54 (s, 2H), 4.46 (s, minor rotamer), 4.45 (s, 4H), 4.43 (m, 1H), 4.09–4.23 (m, 2H), 3.91–4.05 (m, 2H), 3.86 (dd, J = 7.2, 15.4 Hz, 1H), 3.66 (dd, minor rotamer), 3.11 (dt, J = 4.2, 14.6 Hz, 1H), 0.93 (s, minor rotamer), 0.91 (s, 11H), 0.08 (s, 3H), 0.06 (s, minor rotamer), 0.04 (s, 3H), 0.01 (s, 9H), and −0.07 (s, minor rotamer); 13C NMR (100 MHz, 70 °C, d6-DMSO) δ 156.1, 154.7, 140.1, 138.1, 138.0, 136.9, 133.8, 127.8, 127.7, 127.1, 127.0, 126.9, 126.8, 126.7, 123.6, 119.7, 116.4, 113.7, 113.3, 72.6, 72.2, 71.0, 70.6, 69.8, 62.3, 52.2, 46.8, 25.5, 17.5, 17.0, −1.94, −4.36, and −4.95; peaks corresponding to the minor rotamer: δ 133.9, 116.7, 72.4, 71.8, 71.1, 62.5, 16.9, −2.01, −4.48, and −5.07. Anal. Calcd for C41H59NO6Si2: C, 68.58; H, 8.28; N, 1.95. Found: C, 68.67; H, 8.19; N, 1.94.
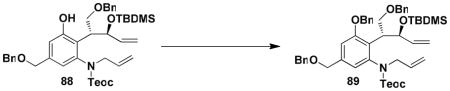
N-Allyl-N-(2-trimethylsilanyloxycarbonyl)-[2-(1R,2S)-{1-(benzyloxy)methyl-2-(tertbutyldimethylsilanyloxy)- but-3-enyl}-3-benzyloxy-5-(benzyloxy)methyl-phenyl]amine (89)
A 25- mL pear flask fitted with a rubber septum and argon inlet needle was charged with a solution of the phenol 88 (0.099 g, 0.138 mmol, 1.0 equiv) in 0.7 mL of DMF. NaH (0.011 g, 0.276 mmol, 2.0 equiv, 60% dispersion in mineral oil) was added to the solution in a single portion. The reaction mixture was stirred at rt for 2–3 min and benzyl bromide (0.025 mL, 0.207 mmol, 1.5 equiv) was added rapidly dropwise via microsyringe. The reaction mixture was stirred at rt for 3 h and then diluted with 10 mL of H2O and 10 mL of Et2O. The aqueous phase was extracted with two 8-mL portions of Et2O and the combined organic phases were washed with 15 mL of brine, dried over MgSO4, filtered, and concentrated to afford 0.152 g of dark yellow oil. Purification by column chromatography on 8 g of silica gel (elution with 10% EtOAc-hexanes) afforded 0.103 g (92%) of 89 as a pale yellow oil: [α]21 D −18.4 (c 0.92, CHCl3); IR (neat) 3032, 2953, 2856, 1700, 1613, 1577, 1454, 1436, 1402, 1361, 1312, 1251, 1094, 920, and 837 cm−1; 1HNMR (500 MHz, 70 °C, d6-DMSO) δ 7.09–7.40 (m, 15H), 7.03 (s, minor rotamer), 7.00 (s, 1H), 6.71 (s, minor rotamer), 6.64 (s, 1H), 5.89–5.98 (m, minor rotamer), 5.84 (ddd, J = 7.0, 12.3, 17.2 Hz, 1H), 5.58 (ddd, minor rotamer), 5.46–5.52 (m, 1H), 4.97–5.11 (m, 4H), 4.85 (dd, J = 10.4, 16.4 Hz, 2H), 4.70 (d, J = 10.4 Hz, 1H), 4.60 (app t, J = 8.4 Hz, 1H), 4.54 (s, minor rotamer), 4.53 (s, 2H), 4.51 (s, minor rotamer), 4.50 (s, 2H), 4.44 (dd, J = 5.0, 15.5 Hz, 1H), 4.36 (AB, J = 12.3 Hz, 1H each), 4.12–4.19 (m, 1H), 4.05–4.11 (m, minor rotamer), 3.97 (app t, J = 9.3 Hz, 1H), 3.86 (dd, J = 7.4, 15.4 Hz, 1H), 3.79–3.83 (m, 1H), 3.58 (dd, minor rotamer), 3.10–3.13 (m, 1H), 0.88 (br s, 2H), 0.80 (s, minor rotamer), 0.78 (s, 9H), −0.03 (s, 9H), −0.12 (s, minor rotamer), −0.18 (s, 3H), −0.22 (s, minor rotamer), and −0.24 (s, 3H); 13C NMR (125 MHz, 55 °C, d6-DMSO) δ 157.3, 154.7, 139.9, 138.1, 137.3, 136.3, 133.7, 128.1, 127.8, 127.74, 127.68, 127.1, 127.03, 126.98, 126.9, 125.4, 121.4, 116.7, 113.7, 110.1, 72.7, 72.1, 71.1, 70.7, 69.8, 62.4, 52.3, 46.9, 25.4, 25.3, 17.3, 17.0, −1.9, −4.6, and −5.3. Anal. calcd for C48H65NO6Si2: C, 71.33; H, 8.11; N, 1.73. Found: C, 71.06; H, 8.19; N, 1.89.
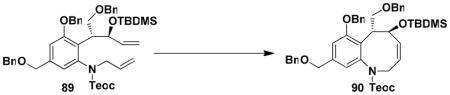
(5S,6R)-7-Benzyloxy-6,9-benzyloxymethyl-5-(tert-butyldimethylsilanyloxy)-5,6-dihydro-2Hbenzo[b]azocine-1-carboxylic acid (2-trimethylsilyl)ethyl ester (90)
A 50-mL, recovery flask fitted with a rubber septum and argon inlet needle was charged with a solution of the Grubbs catalyst 60 (0.005 g, 0.0057 mmol, 0.05 equiv) in 9 mL of CH2Cl2. The aniline 89 (0.092 g, 0.114 mmol, 1.0 equiv) in 2.4 mL of CH2Cl2 was added via cannula in one portion and the septum was replaced with a reflux condenser fitted with an argon inlet adapter. The pale pink-colored reaction mixture was heated at reflux for 4.5 h and then allowed to cool to rt and concentrated to afford ca. 0.110 g of yellow brown oil. Column chromatography on 12 g of silica gel (elution with 10% EtOAc-hexane) yielded 0.082 g (92%) of benzoazocine 90 as a viscous pale yellow oil: [α]22 D −48.6 (c 0.38, CHCl3); IR (thin film) 3031, 2954, 2856, 1699, 1613, 1577, 1453, 1403, 1311, 1250 and 1073 cm−1; 1HNMR (400 MHz, CDCl3) δ 7.32–7.40 (m, 10H), 7.12–7.25 (m, 5H), 6.94 (s, 1H), 6.66 (s, 1H), 5.54 (ddd, J = 3.0, 7.3, 10.5 Hz, 1H), 5.17–5.26 (m, 1H), 5.04 (app AB, J = 11.9 Hz, 3H), 4.57 (s, 2H), 4.52 (s, 2H), 4.39–4.47 (m, 2H), 4.14–4.24 (m, 1H), 3.86–3.96 (m, 1H), 3.83–3.86 (m, 1H), 3.72–3.79 (m, 2H), 3.60 (dd, J = 5.4, 16.6 Hz, 1H), 0.91 (s, 10 H), 0.09 (s, 3H), 0.06 (s, 3H), and −0.08 (s, 9H); 13CNMR (100 MHz, CDCl3) δ 158.1, 138.3, 137.7, 137.3, 128.6, 128.2, 128.0, 127.9, 127.8, 127.5, 127.2, 127.1, 72.3, 72.0, 71.7, 70.9, 70.2, 64.1, 49.1, 45.9, 26.1, 18.4, 18.1, −1.44, −4.26, and −4.73. Anal. Calcd for C46H61NO6Si2: C, 70.82; H, 7.88; N, 1.80. Found: C, 70.71; H, 7.72; N, 1.62.

(5S,6R)-7-Benzyloxy-6,9-benzyloxymethyl-5-hydroxyl-5,6-dihydro-2H-benzo[b]azocine (64)
A 10-mL pear flask fitted with a rubber septum and argon inlet needle was charged with a solution of benzoazocine 90 (0.051 g, 0.0654 mmol, 1.0 equiv) and 0.9 mL of THF. TBAF (0.24 mL, 0.235 mmol, 3.6 equiv) was added rapidly dropwise via syringe and the reaction mixture was sealed with a glass stopper under argon and stirred at rt for 39 h. The resulting mixture was then diluted with 5 mL of Et2O and 5 mL of H2O, and the aqueous layer was extracted with two 5-mL portions of Et2O. The combined organic phases were washed with 10-mL of brine, dried over MgSO4, filtered, and concentrated to afford 0.040 g of orange oil. Purification by column chromatography on 4 g silica gel (elution with 30% EtOAchexanes) provided 0.025 g (74%) of benzazocine 64 as a viscous pale yellow oil: [α]21 D −111.5 (c 0.41, CHCl3); IR (thin film) 3390, 3029, 2860, 1608, 1580, 1496, 1453, 1360, 1311, 1205, 1112, 1074, 1027, 839, 736, and 697 cm−1; 1HNMR (400 MHz, CDCl3) δ 7.25–7.42 (m, 15H), 6.77 (s, 1H), 6.66 (s, 1H), 5.85 (app t, J = 7.4 Hz, 1H), 5.60 (ddd, J = 2.8, 6.7, 9.6 Hz, 1H), 5.24–5.29 (m, 1H), 5.05 (AB, J = 7.3 Hz, 1H each), 4.55 (s, 2H), 4.52 (s, 2H), 4.47 (s, 2H), 3.91–3.94 (m, 1H), 3.85 (dt, J = 3.8, 13.5 Hz, 1H), 3.69–3.76 (m, 2H), and 3.53 (dd, J = 5.6, 16.2 Hz, 1H); 13CNMR (100 MHz, CDCl3) δ 156.8, 148.2, 138.6, 138.3, 138.0, 137.2, 135.2, 128.7, 128.58, 128.57, 127.99, 127.97, 127.9, 127.8, 127.7, 127.3, 126.3, 125.0, 119.2, 107.9, 74.6, 73.5, 73.3, 72.5, 72.0, 70.4, 51.2, and 46.2. Anal. Calcd for C34H35NO4: C, 78.28; H, 6.76; N, 2.69. Found: C, 78.18; H, 6.87; N, 2.77.
Comparison of NMR Data for 64 with Data Reported by Ducray and Ciufolinixxxvi
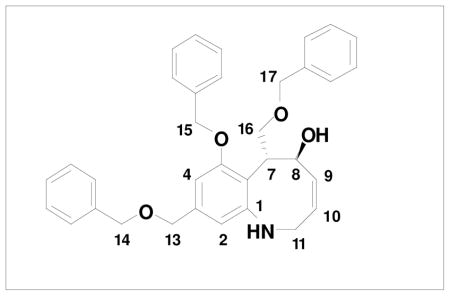
Supplementary Material
Scheme 3.

Allene Byproduct Formation in a Cyclobutenone-Ynamine Benzannulation
Scheme 8.

Conversion of 4-Vinylcyclobutenone 38 to Phenol 36
Scheme 12.

Enantioselective Synthesis of Bromo Alkyne Intermediate 76
TABLE 4.
1H NMR (CDCl3) Spectral Data for Compound 64a
| Ciufolini (300 MHz) | Compound 64 (400 MHz) | |||
|---|---|---|---|---|
| δ | J (Hz) | δ | J (Hz) | |
| H(Ph) | 7.25–7.41 (m, 15H) | - | 7.25–7.42 (m, 15H) | - |
| H(4) | 6.78 (s, 1H) | - | 6.77 (s, 1H) | - |
| H(2) | 6.67 (s, 1H) | - | 6.66 (s, 1H) | - |
| H(10) | 5.83–5.88 (m, 1H) | - | 5.85 (app t, 1H) | 7.4 |
| H(9) | 5.58–5.65 (m, 1H) | - | 5.60 (ddd, 1H) | 2.8, 6.7, 9.6 |
| H(8) | 5.23–5.30 (m, 1H) | - | 5.24–5.29 (m, 1H) | - |
| H(17) | 5.06 (AB, 2H) | - | 5.05 (AB, 2H) | - |
| H(13, 14, 15) | 4.56 (s, 2H) | - | 4.55 (s, 2H) | - |
| 4.53 (s, 2H) | - | 4.52 (s, 2H) | - | |
| 4.48 (s, 2H) | - | 4.47 (s, 2H) | - | |
| H(11,16) | 3.69–3.96 (cplx, 4H) | - | 3.91–3.94 (m, 1H) | - |
| 3.85 (app dt, 1H) | 3.8, 13.5 | |||
| 3.69–3.76 (m, 2H) | - | |||
| H(7) | 3.54 (dd, 1H) | 5.6, 16.2 | 3.53 (dd, 1H) | 5.6, 16.2 |
Chemical shifts are expressed in ppm relative to tetramethylsilane
TABLE 5.
13C NMR (CDCl3) Spectral Data for 64
| Ciufolini (75 MHz) | Compound 64 (100 MHz) |
|---|---|
| δ | δ |
| 156.6 | 156.8 |
| 147.9 | 148.2 |
| 138.4 | 138.6 |
| 138.1 | 138.3 |
| 137.8 | 138.0 |
| 137.0 | 137.2 |
| 135.0 | 135.2 |
| 128.4 | 128.7 |
| 128.3 | 128.58 |
| 127.76 | 128.57 |
| 127.74 | 127.99, 127.97 |
| 127.6 | 127.9 |
| 127.5 | 127.8 |
| 127.4 | 127.7 |
| 127.1 | 127.3 |
| 126.0 | 126.3 |
| 124.8 | 125.0 |
| 118.9 | 119.2 |
| 107.7 | 107.9 |
| 74.2 | 74.5 |
| 73.18 | 73.5 |
| 73.13 | 73.3 |
| 72.2 | 72.5 |
| 71.7 | 72.0 |
| 50.9 | 51.2 |
| 46.0 | 46.2 |
Chemical shifts are expressed in ppm relative to tetramethylsilane
Acknowledgments
We thank the National Institutes of Health (GM 28273), Merck Research Laboratories, and Boehringer Ingelheim Pharmaceuticals for generous financial support.
Footnotes
Supporting Information Available: 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- i.Reviews: Kotha S, Misra S, Halder S. Tetrahedron. 2008;64:10775–10790.Saito S, Yamamoto Y. Chem Rev. 2000;100:2901–2915. doi: 10.1021/cr990281x.
- ii.(a) Danheiser RL, Gee SK. J Org Chem. 1984;49:1672–1674. [Google Scholar]; (b) Danheiser RL, Brisbois RG, Kowalczyk JJ, Miller RF. J Am Chem Soc. 1990;112:3093–3100. [Google Scholar]
- iii.For reviews of the chemistry of vinylketenes, see Danheiser RL, Dudley GB, Austin WF. Alkenylketenes. In: Danheiser RL, editor. Science of Synthesis. Vol. 23. Thieme; Stuttgart: 2006. pp. 493–568.Tidwell TT. Ketenes. 2. John Wiley & Sons; Hoboken, NJ: 2006. pp. 206–214.
- iv.A related, complementary strategy has been reported by Liebeskind and Moore. See ref. 3a and Liebeskind LS, Iyer S, Jewell CF. J Org Chem. 1986;51:3065–3067.Perri ST, Foland LD, Decker OHH, Moore HW. J Org Chem. 1986;51:3067–3068.
- v.Examples: Danheiser RL, Gee SK, Perez JJ. J Am Chem Soc. 1986;108:806–810.Danheiser RL, Cha DD. Tetrahedron Lett. 1990;31:1527–1530.Danheiser RL, Casebier DS, Loebach JL. Tetrahedron Lett. 1992;33:1149–1152.Danheiser RL, Casebier DS, Huboux AH. J Org Chem. 1994;59:4844–4848.Danheiser RL, Helgason AL. J Am Chem Soc. 1994;116:9471–9479.Danheiser RL, Trova MP. Synlett. 1995:573–574.Danheiser RL, Casebier DS, Firooznia F. J Org Chem. 1995;60:8341–8350.Dudley GB, Takaki KS, Cha DD, Danheiser RL. Org Lett. 2000;2:3407–3410. doi: 10.1021/ol006561c.
- vi.For additional examples of the application of this benzannulation strategy in natural product synthesis, see Kowalski CJ, Lal GS. J Am Chem Soc. 1988;110:3693–3695.Smith AB, III, Adams CM, Kozmin SA, Paone DV. J Am Chem Soc. 2001;123:5925–5937. doi: 10.1021/ja0106164.
- vii.For notable recent examples of “aminobenzannulation” approaches to the synthesis of anilines, see Merlic CA, Burns EE. Tetrahedron Lett. 1993;34:5401–5404.Padwa A, Dimitroff M, Waterson AG, Wu T. J Org Chem. 1997;62:4088–4096.Saito S, Uchiyama N, Gevorgyan V, Yamamoto Y. J Org Chem. 2000;65:4338–4341. doi: 10.1021/jo000171m.Barluenga J, López LA, Martínez S, Tomás M. Tetrahedron. 2000;56:4967–4975.Neumann H, von Wangelin AJ, Klaus S, Strübing D, Gördes D, Beller M. Angew Chem, Int Ed. 2003;42:4503–4507. doi: 10.1002/anie.200351484.Tiano M, Belmont P. J Org Chem. 2008;73:4101–4109. doi: 10.1021/jo800249f.Kiren S, Padwa A. J Org Chem. 2009;74:7781–7789. doi: 10.1021/jo9017793.
- viii.Delaunois M, Ghosez L. Angew Chem Int Ed. 1969;8:72–73.See also Ficini J, Pouliquen Tetrahedron Lett. 1972:1135–1138.Himbert G. Liebigs Ann Chem. 1979:829–841.Henn L, Himbert G. Chem Ber. 1981;114:1015–1026.Dötz KH, Mühlemeier J, Trenkle B. J Organomet Chem. 1985;289:257–262.Henn L, Himbert G, Diehl K, Kaftory M. Chem Ber. 1986;119:1953–1963.Barbaro G, Battaglia A, Giorgianni P. J Org Chem. 1987;52:3289–3296.Schulte N, Möller MH, Rodewald U, Würthwein EU. Chem Ber. 1994;127:1287–1293.
- ix.Koch R, Wentrup C. Org Biomol Chem. 2004;2:195–199. doi: 10.1039/b309549e. [DOI] [PubMed] [Google Scholar]
- x.Ficini observed that the reaction of certain ynamines and cyclobutenones forms strained bicyclo[2.2.0]hexenones that undergo π2s+σ2s+ σ2s rearrangement to generate a vinylcyclobutenone. This vinylcyclobutenone is isomeric with respect to general structure 5, and would yield a phenol of type 16 upon 4-electron electrocyclic opening followed by 6-electron electrocyclic closure. See Ficini J, Falou S, d’Angelo J. Tetrahedron Lett. 1977;18:1931–1934.
- xi.For recent reviews on the chemistry of ynamides, see see DeKorver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP. Chem Rev. 2010;110:5064–5106. doi: 10.1021/cr100003s.Evano G, Coste A, Jouvin K. Angew Chem Int Ed. 2010;49:2840–2859. doi: 10.1002/anie.200905817.
- xii.Kohnen AL, Mak XY, Lam TY, Dunetz JR, Danheiser RL. Tetrahedron. 2006;62:3815–3822. doi: 10.1016/j.tet.2005.11.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xiii.For recent reviews on the application of RCM to the synthesis of heterocyclic compounds, see Phillips AJ, Abell AD. Aldrichimica Acta. 1999;32:75–89.Felpin F-X, Lebreton J Eur J Org Chem. 2003:3693–3712.Deiters A, Martin SF. Chem Rev. 2004;104:2199–2238. doi: 10.1021/cr0200872.Donohoe TJ, Orr AJ, Bingham M. Angew Chem Int Ed. 2006;45:2664–2670. doi: 10.1002/anie.200503512.Compain P. Adv Cat Synth. 2007;349:1829–1846.Villar H, Frings M, Bolm C. Chem Soc Rev. 2007;36:55–66. doi: 10.1039/b508899m.
- xiv.(a) Dunetz JR, Danheiser RL. Org Lett. 2003;5:4011–4014. doi: 10.1021/ol035647d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kohnen AL, Dunetz JR, Danheiser RL. Org Synth. 2007;84:88–101. [PMC free article] [PubMed] [Google Scholar]
- xv.(a) Frederick MO, Mulder JA, Tracey MR, Hsung RP, Huang J, Kurtz KCM, Shen L, Douglas J Am Chem Soc. 2003;125:2368–2369. doi: 10.1021/ja021304j. [DOI] [PubMed] [Google Scholar]; (b) Zhang Y, Hsung RP, Tracey MR, Kurtz KCM, Vera EL. Org Lett. 2004;6:1151–1154. doi: 10.1021/ol049827e. [DOI] [PubMed] [Google Scholar]; (c) Zhang X, Zhang Y, Huang J, Hsung RP, Kurtz KCM, Oppenheimer J, Petersen ME, Sagamanova IK, Shen L, Tracey MR. J Org Chem. 2006;71:4170–4177. doi: 10.1021/jo060230h. [DOI] [PubMed] [Google Scholar]; (d) Sagamanova IK, Kurtz KCM, Hsung RP. Org Synth. 2007;84:359–367. [Google Scholar]
- xvi.For other useful coupling routes to ynamides, see Hamada T, Ye X, Stahl SS. J Am Chem Soc. 2008;130:833–835. doi: 10.1021/ja077406x.Coste A, Karthikeyan G, Couty F, Evano G. Angew Chem Int Ed. 2009;48:4381–4385. doi: 10.1002/anie.200901099.
- xvii.Caruso T, Spinella A. Tetrahedron. 2003;59:7787–7790. [Google Scholar]
- xviii.Danheiser RL, Savariar S, Cha DD. Org Synth. 1990;68:32–40. [Google Scholar]
- xix.(a) Niwayama S, Kallel EA, Sheu C, Houk KN. J Org Chem. 1996;61:2517–2522. doi: 10.1021/jo950884i. [DOI] [PubMed] [Google Scholar]; (b) Niwayama S, Kallel EA, Spellmeyer DC, Sheu C, Houk KN. J Org Chem. 1996;61:2813–2825. doi: 10.1021/jo950884i. [DOI] [PubMed] [Google Scholar]
- xx.Cyclobutenones were prepared via ketene-alkyne cycloadditions. For 35: ref. xviii; 39: via addition of ketene to methoxypropyne (see Supporting Information); 40: ref. va; 41: ref. xviii; 42: ref. xii.
- xxi.For examples of the photocyclization of 2-allylphenols, see Jiménez MC, Márquez F, Miranda MA, Tormos R. J Org Chem. 1994;59:197–202.and references cited therein.
- xxii.(a) Cacchi S, Ciattini PG, Morera E, Ortar G. Tetrahedron Lett. 1986;27:5541–5544. [Google Scholar]; (b) Cabri W, De Bernardinis S, Francalanci F, Penco S, Santi R. J Org Chem. 1990;55:350–353. [Google Scholar]
- xxiii.Hydrogenation of the alkene double bonds was necessary to avoid the formation of byproducts in the triflate reduction step.
- xxiv.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- xxv.For a discussion of alkene isomerization in RCM reactions, see Schmidt B. Eur J Org Chem. 2004:1865–1880.
- xxvi.Reviews: Mori M. Adv Synth Catal. 2007;349:121–135.Diver ST, Giessert AJ. Chem Rev. 2004;104:1317–1382. doi: 10.1021/cr020009e.
- xxvii.Derivatives examined include the corresponding N-tosyl sulfonamide corresponding to 49 and the (desilylated) terminal alkyne, as well as its TBDMS phenol ether derivative.
- xxviii.(a) Uchida I, Takase S, Kayakiri H, Kiyoto S, Hashimoto M, Tada T, Koda S, Morimoto Y. J Am Chem Soc. 1987;109:4108–4109. [Google Scholar]; (b) Terano H, Takase S, Hosoda J, Kohsaka M. J Antibiot. 1989;42:145–148. doi: 10.7164/antibiotics.42.145. [DOI] [PubMed] [Google Scholar]
- xxix.Rajski WR, Williams RM. Chem Rev. 1998;98:2723–2796. doi: 10.1021/cr9800199. [DOI] [PubMed] [Google Scholar]
- xxx.(a) Fukuyama T, Xu L, Goto S. J Am Chem Soc. 1992;114:383–385. [Google Scholar]; (b) (a) Suzuki M, Kambe M, Tokuyama H, Fukuyama T. Angew Chem Int Ed. 2002;41:4686–4688. doi: 10.1002/anie.200290016. [DOI] [PubMed] [Google Scholar]; (b) Suzuki M, Kambe M, Tokuyama H, Fukuyama T. J Org Chem. 2004;69:2831–2843. doi: 10.1021/jo049862z. [DOI] [PubMed] [Google Scholar]
- xxxi.Schkeryantz JM, Danishefsky SJ. J Am Chem Soc. 1995;117:4722–4723. [Google Scholar]
- xxxii.(a) Katoh T, Itoh E, Yoshino T, Terashima S. Tetrahedron. 1997;53:10229–10238. [Google Scholar]; (b) Yoshino T, Nagata Y, Itoh E, Hashimoto M, Katoh T, Terashima S. Tetrahedron. 1997;53:10239–10252. [Google Scholar]; (c) Katoh T, Nagata Y, Yoshino T, Nakatani S, Terashima S. Tetrahedron. 1997;53:10253–10270. [Google Scholar]
- xxxiii.(a) Judd TC, Williams RM. Angew Chem Int Ed. 2002;41:4683–4685. doi: 10.1002/anie.200290015. [DOI] [PubMed] [Google Scholar]; (b) Judd TC, Williams RM. J Org Chem. 2004;69:2825–2830. doi: 10.1021/jo035828t. [DOI] [PubMed] [Google Scholar]; (c) Ducept P, Gubler DA, Williams RM. Heterocycles. 2006;67:597–619. [Google Scholar]
- xxxiv.For a recent very efficient route to 7-epi (+)-FR900482, see Trost BM, O’Boyle BM. Org Lett. 2008;10:1369–1372. doi: 10.1021/ol800127a.
- xxxv.Fellows IM, Kaelin DE, Martin SF. J Am Chem Soc. 2000;122:10781–10787. [Google Scholar]
- xxxvi.Ducray R, Ciufolini MA. Angew Chem Int Ed. 2002;41:4688–4691. doi: 10.1002/anie.200290017. [DOI] [PubMed] [Google Scholar]
- xxxvii.Paleo MR, Aurrecoechea N, Jung KY, Rapoport H. J Org Chem. 2003;68:130–138. doi: 10.1021/jo0206521. [DOI] [PubMed] [Google Scholar]
- xxxviii.Iwasawa N, Huang H, Mukaiyama T. Chem Lett. 1985:1045–1048. [Google Scholar]
- xxxix.(a) Nagao Y, Hagiwara Y, Kumagai T, Ochiai M, Inoue T, Hashimoto K, Fujita E. J Org Chem. 1986;51:2391–2393. [Google Scholar]; (b) Nagao Y, Nagase Y, Kumagai T, Matsunaga H, Abe T, Shimada O, Hayashi T, Inoue Y. J Org Chem. 1992;57:4243–4249. [Google Scholar]
- xl.For a review on the use of oxazolidinonethione and thiazolidinethione chiral auxiliaries see: Velázquez F, Olivo HF. Curr Org Chem. 2002;6:303–340.
- xli.Delaunay D, Toupet L, Le Corre M. J Org Chem. 1995;60:6604–6607. [Google Scholar]
- xlii.Davis FA, Kasu PVN, Sundarababu G, Qi H. J Org Chem. 1997;62:7546–7547. [Google Scholar]
- xliii.For details on the assignment of the relative and absolute stereochemistry of 73, see the Supporting Information.
- xliv.(a) Crimmins MT, King BW, Tabet EA. J Am Chem Soc. 1997;119:7883–7884. [Google Scholar]; (b) Crimmins T, Chaudhary K. Org Lett. 2000;2:775–777. doi: 10.1021/ol9913901. [DOI] [PubMed] [Google Scholar]; (c) Crimmins MT, King BW, Tabet EA, Chaudhary K. J Org Chem. 2001;66:894–902. doi: 10.1021/jo001387r. [DOI] [PubMed] [Google Scholar]; (d) Crimmins MT, She J. Synlett. 2004:1371–1374. [Google Scholar]
- xlv.Michel P, Gennet D, Rassat A. Tetrahedron Lett. 1999;40:8575–8578. [Google Scholar]
- xlvi.Ylide 75 was generated in situ by treatment of dibromomethyl(triphenyl)phosphonium bromide with KOt-Bu (rt, 5 min). For best results it is crucial to employ solvent-free phosphonium salt prepared by the method of Lerm M, Gais HJ, Cheng K, Vermeeren C. J Am Chem Soc. 2003;125:9654–9667. doi: 10.1021/ja030200l.
- xlvii.Prepared by addition of ketene to ethoxyacetylene as described by Wasserman HH, Piper JU, Dehmlow EV. J Org Chem. 1973;38:1451–1455.
- xlviii.Synthesis of 77: Kaufman TS. Synlett. 1997:1377–1378.Synthesis of 78: Danheiser RL, Romines KR, Koyama H, Gee SK, Johnson CR, Medich JR. Org Synth. 1993;71:133–139.
- xlix.Liebeskind LS, Wirtz KR. J Org Chem. 1990;55:5350–5358. [Google Scholar]
- l.(a) Watson SC, Eastham JF. J Organomet Chem. 1967;9:165–168. [Google Scholar]; (b) Ellison RA, Griffin R, Kotsonis FN. J Organomet Chem. 1972;36:209–213. [Google Scholar]
- li.Prepared from the reaction of allylamine and methyl chloroformate according to the procedure of: Kozmin SA, Iwama T, Huang Y, Rawal VH. J Am Chem Soc. 2002;124:4628–4641. doi: 10.1021/ja017863s.
- lii.Carbamate 19 was prepared following the general method of Capson TL, Poulter DC. Tetrahedron Lett. 1984;25:3515–3518.
- liii.Williams JW, Hurd CD. J Org Chem. 1940;5:122.See also Hanford WE, Sauer JC. Preparation of Ketenes and Ketene Dimers. Org React. 1958;3:108–140.
- liv.Methoxypropyne was prepared according to: Hine J, Mahone LG, Liotta CL. J Org Chem. 1967;32:2600–2609.
- lv.Davis FA, Kasu PVN, Sundarababu G, Qi H. J Org Chem. 1997;62:7546–7547. [Google Scholar]
- lvi.Prepared from (S)-valinol as reported in Delaunay D, Toupet L, Le Corre M. J Org Chem. 1995;60:6604–6607.
- lvii.Tin (II) trifluoromethanesulfonate was prepared according to the procedure reported in Evans DA, Weber AE. J Am Chem Soc. 1986;108:6757.What was estimated to be the desired amount of Sn(OTf)2 was transferred to the tared 200-mL recovery flask as a suspension in 50 mL of anhydrous Et2O via cannula. Et2O was removed from the flask via cannula by applying a positive pressure of argon, and the Sn(OTf)2 was washed with three 40-mL portions of Et2O while under argon. Following the addition of each portion of Et2O, the resulting suspension was stirred vigorously for 1–2 min and then allowed to stand for 1–2 min before removing the Et2O via cannula. After the final wash, the Sn(OTf)2 was dried overnight at 0.3–0.5 mmHg at 100 °C for ca. 16 h.
- lviii.Breit B, Zahn SK. J Org Chem. 2001;66:4870–4877. doi: 10.1021/jo015634i. [DOI] [PubMed] [Google Scholar]
- lix.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- lx.The phosphonium bromide 75 was prepared according to the procedure reported in ref. 46.
- lxi.Prepared via the reaction of allylamine with di-tert-butyl dicarbonate as reported in: Kirkland TA, Lynn DM, Grubbs RH. J Org Chem. 1998;63:9904–9909.
- lxii.Shu L, Schäfer A, Schlüter AD. Macromolecules. 2000;33:4321–4328.(b) Carbamate 86 used in this reaction was prepared in near quantitative yield from the reaction of 2- trimethylsilylethanol with 1.2 equiv allyl isocyanate in the presence of 2.0 equiv of i-Pr2NEt in toluene at 65 °C for 20 h.
- lxiii.Further elution with 10% EtOAc-hexanes resulted in the recovery of 2.1 equiv of unreacted carbamate 86.
- lxiv.Still CW. J Am Chem Soc. 1978;100:1481.(b) Tributyl(benzyloxymethyl)stannane was prepared according to ref. 48a.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.