

Abstract
Two remarkable features of many siderophores produced by oceanic bacteria are the prevalence of an α-hydroxy-carboxylic acid functionality either in the form of the amino acid β-hydroxy aspartic acid or in the form of citric acid, as well as the predominance of amphiphilic siderophores. This review will provide an overview of the photoreactivity that takes place when siderophores containing β-hydroxy aspartic acid and citric acid are coordinated to iron(III). This photoreactivity raises questions about the role of this photochemistry in microbial iron acquisition as well as upper-ocean iron cycling. The self-assembly of amphiphilic siderophores and the coordination-induced phase-change of the micelle-to-vesicle transformation will also be reviewed. The distinctive photosensitive and self-assembly properties of marine siderophores hint at possibly new microbial iron acquisition mechanisms.
Keywords: Marine siderophores, Photoreactive ferric siderophores, Amphiphilic siderophores
1. Introduction
Iron is a universally required element for bacterial growth, yet iron is generally present in the environment as insoluble iron(III) oxide minerals and thus is not readily available for living organisms to acquire and use. Iron levels are also surprisingly low in surface ocean waters. These low levels of iron limit growth of microorganisms in the open ocean, such as phytoplankton, which are responsible for up to half the world’s fixation of carbon dioxide [1]. To acquire iron, many bacteria growing aerobically, including marine species, produce siderophores, which are low molecular weight compounds produced to facilitate acquisition of iron. Siderophores typically coordinate iron(III) with high affinity and are biosynthesized in response to low iron levels [2,3]. Common functional groups in siderophores that coordinate to Fe(III) include catechols, as in enterobactin, hydroxamic acids, as in the desferrioxamines, and α-hydroxy-carboxylic acids, as in aerobactin (see structures). Siderophores generally form hexadentate, octahedral complexes with iron(III) preferentially, compared to other naturally abundant metal ions such as Zn2+, Cu2+, Ca2+, Mg2+ and Al3+, among others. Siderophore coordination chemistry is governed by the HSAB (hard and soft acids and bases) principle, originally proposed by Pearson [4], that is, with preferential coordination between the hard Fe(III) Lewis acid and the hard anionic oxygen Lewis base donor ligands.
1.1. Tris catecholate siderophores
Enterobactin, bacillibactin and salmochelin are all tris-catechoyl siderophores framed on a cyclic tri-ester scaffold (Fig. 1). Enterobactin, isolated from many different enteric and pathogenic bacteria including Escherichia coli, is a cyclic trimer of 2,3-dihydroxybenzoyl-l-serine. Salmochelin, isolated from Salmonella enterica, and uropathogenic E. coli [5], resembles enterobactin except that up to two of the catechols groups are glucosylated [6]. The glucosylation in salmochelin is proposed to increase bacteria virulence [6]. Bacillibactin, which is isolated from Bacillus subtilis and other Bacilli species, incorporates a scaffold of threonine trilactone and glycine spacers, which elongate the three chelating arms as compared with enterobactin [7].
Fig. 1.

Structures of enterobactin, salmochelin S4 and bacillibactin.
The tris catecholato siderophores complex Fe(III) with remarkably tight affinity. The proton independent stability constant for Fe(enterobactin)3− is 1049 [8], and for Fe(bacillibactin)3− is 1047.6 [7]. In each case, Fe(III) is present in the high-spin electronic configuration. The first (and only to date) X-ray crystal structure of a discrete metal–enterobactin complex is that of bare vanadium(IV)-enterobactin, [V(ent)]2−, which reveals a Δ-configuration at the metal center (Fig. 2) [9,10]. The circular dicroism spectrum of [Fe(III)(ent)]3−, as well as the substitution inert [CrIII (ent)]3− and [RhIII (ent)]3− complexes, are consistent with the right-handed propeller configuration [9,11-13]. While a high resolution X-ray structure of a discrete complex of Fe(ent)3− has not yet been achieved, the X-ray structure of the siderocalin protein with bound Fe(ent)3− has been determined to 2.4 Å resolution [14,15]. Siderocalin is a mammalian protein produced by the immune system and functions to bind Fe(ent)3−, thus limiting its availability for microbial growth in the mammalian host.
Fig. 2.
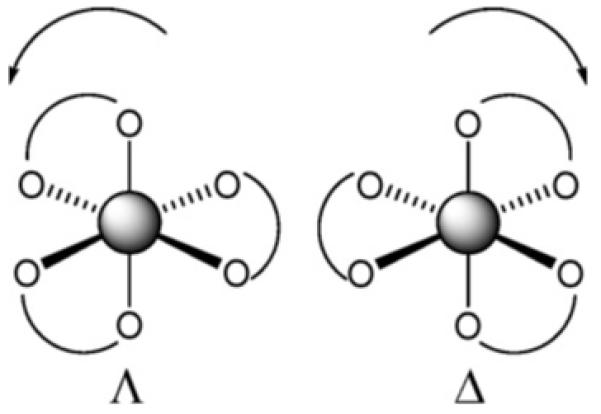
The Δ configuration describes a right-hand propeller formed by three bidentate ligands and the Λ configuration describes a left-hand propeller.
Perhaps the most remarkable difference between enterobactin and bacillibactin is the switch in chirality at the metal center when the tris-l-serine ester scaffold of enterobactin and salmochelin is replaced with tris-l-threonine in bacillibactin and a glycine spacer is inserted between threonine and the 2,3-dihydroxybenzoic acid [16].
The tri-serine lactone linkage of enterobactin and salmochelin is quite susceptible to hydrolysis. Initial hydrolysis converts the cyclic lactone scaffold to a linear tri-ester, which is catalyzed in vivo by an esterase in the Fe(III)-release process [17-19], but is also accomplished readily by reduced pH conditions.
1.2. Tris-hydroxamate siderophores
The ferrioxamines are a well-known group of tris-hydroxamate siderophores which are primarily assembled from alternating units of a diamine, generally cadaverine or putrescine and succinic acid, with certain variations (Fig. 3). Desferrioxamines A, B, C, D1, F, and G are linear siderophores comprised of three hydroxamic acid functional groups. Desferrioxamines D2 and E are the cyclic counter parts to the linear desferrioxamines D1 and G. Desferioxamine B (DFOB) is the drug Desferal used to treat iron overload disease. The hydroxamic acid group exists in the E configuration in the apo siderophores, as depicted in Fig. 3, however upon Fe(III) coordination the hydroxamate is switched to the Z configuration for bidentate coordination.
Fig. 3.

Structures of desferrioxamines E, G and B.
To date, the crystal structures of four members of the ferrioxamine family have been determined (i.e., the “ferrioxamine” nomenclature designates Fe(III) coordination): ferrioxamine D1, ferrioxamine E, the retro-isomer of ferrioxamine E [20-22] and more recently Crumbliss and co-workers reported the crystal structure of ferrioxamine B, co-crystallized with ethanolpentaaquomagnesium(II) and perchlorate, of composition Fe(HDFOB)ClO4Mg(H2O)5(EtOH)(ClO4)2 [23]. All ferrioxamines crystallize as a racemic mixture of Λ- and Δ-cis isomers [20-23]. The stability constants reported for DFOB and Fe(III) is 1030.6 [11,24,25].
Desferrichrome is another type of tris-hydroxamate siderophore. It is a cyclic hexapeptide composed of three glycine and three hydroxylated and acetylated ornithine residues [26]. Ferrichrome, one of the first siderophores discovered in nature from the smut fungus, Ustilago sphaerogena, is also produced by fungi of the genera Aspergillus, Ustilago and Penicillium among others [26]. The Fe(III)–desferrichrome complex, more aptly called ferrichrome, assumes the Λ-cis absolute configuration at the Fe(III) center [26], in contrast to the ferrioxamines that exist as a racemic mixture. Variation in the desferrichrome composition comes with substitution of two glycine residues with L-Ser and substitution of three acetic acid residues with β-methyl glutaconic acid as in desferrichrome A (Fig. 4) [27].
Fig. 4.

Structures of selected desferrichrome siderophores.
1.3. α-Hydroxycarboxylate, carboxylate and mixed functional group siderophores
The α-hydroxycarboxylate functional group is another bidentate OO’ donor ligand that is found in many siderophores, but particularly in many marine siderophores (see below). Achromobactin (Fig. 5) is a tris-α-hydroxycarboxylate siderophore whose biosynthesis has recently been reported [28,29]. The citrate backbone of achromobactin contributes one α-hydroxycarboxylate group, and the other two α-hydroxycarboxylates derive from α-ketoglutarate. Bis α-hydroxycarboxylate siderophores, such as vibrioferrin are comprised of one α-hydroxycarboxylate from citrate and one from α-ketoglutarate, and the bis-citrato siderophores staphyloferrin and rhizoferrin are well known, as well as a plethora of siderophores that contain more than one type of functional group moiety, such as aerobactin (Fig. 5). An upper limit on the ferric stability constant of Fe(III)–vibrioferrin is estimated to be poised just below that of Fe(III)–EDTA, ~1025, since EDTA removes all Fe(III) from the Fe(III)–vibrioferrin complex at neutral pH [30]. A remarkable feature of vibrioferrin, discovered during the isolation from of a marine bacterium, is that it binds boron stoichiometrically through the α-hydroxy-carboxylic acid groups, with an appreciable stability constant, 1014.1 [30,31].
Fig. 5.

Structures of selected α-hydroxycarboxylate siderophores.
Even citric acid is believed to be a siderophore [32], functioning in iron uptake as a bis-iron(III), di-citrato complex, which is recognized by a specific citrate outer membrane receptor. The X-ray crystal structure of one example of a di-iron(III), di-citrato complex is shown in Fig. 6 [33], although several other different ferric citrato complexes have been structurally characterized.
Fig. 6.

Structure of Fe2(cit)2(H2O)22− adapted from Ref. [33].
2. Marine siderophores
While relatively few siderophore structures from marine bacteria are known compared to many siderophores from terrestrial bacteria that are known, two major structural features characterize the majority of the marine siderophores discovered so far (Figs. 7 and 8). One characteristic is the predominance of siderophores that contain an α-hydroxy-carboxylic acid (e.g., β-hydroxy aspartic acid or citric acid) which are photoreactive when coordinated to Fe(III) [34-38]. The other structural characteristic is the large number of suites of amphiphilic siderophores composed of an iron(III)-binding head-group that is appended by one or two of a series of fatty acids [39-44]. Many of the marine siderophores contain both distinctive structural features, in that they are both produced as suites of amphiphiles and the Fe(III) complexes are photoreactive. Fig. 7 shows the suites of marine amphiphilic siderophores reported so far. Fig. 8 shows the structures of many of the hydrophilic siderophores.
Fig. 7.

Marine amphiphilic siderophores. The α-hydroxy-carboxylic acid is circled in each structure.
Fig. 8.

Hydrophilic marine siderophores. In addition DFOG (Fig. 2), aerobactin, citric acid and vibrioferrin (Fig. 5) have been isolated from marine bacteria. The α-hydroxy-carboxylic acid is circled in each structure.
3. Photoreactivity of Fe(III)–siderophore complexes
3.1. Citrate-containing siderophores
Ferric complexes of α-hydroxy-carboxylic acid siderophores, including aerobactin [34], the ochrobactins [42], the synechobactins [41], and the petrobactins [37,38,40,45] are photoreactive, as is also well known for ferric citrate complexes [46]. Photolysis into the α-hydroxy-acid-to-Fe(III) charge-transfer band, which falls in the near UV wavelength range (Fig. 9) induces ligand oxidation and release of CO2 along with reduction of Fe(III) to Fe(II). Of these siderophores, photolysis of Fe(III)–aerobactin has been studied most extensively.
Fig. 9.

UV–vis absorption spectrum of Fe(III)–aerobactin and after UV photolysis. Reproduced with permission from Ref. [34].
UV photolysis of Fe(III)–aerobactin under aerobic conditions produced a new ligand that also coordinates Fe(III) [34].As Fig. 9 shows, the 300 nm absorption band disappears, which is the α-hydroxy-carboxylic acid-to-Fe(III) charge-transfer band, although the hydroxamate to Fe(III) charge-transfer band at ~440 nm remains after photolysis.
In addition to UV photolysis (450-W mercury arc lamp), natural sunlight and even fluorescent room lights are sufficient to effect the photoreaction. During photolysis, citrate is converted to 3-ketoglutaric acid, as established by 1H and 13C NMR, mass spectral analyses and deuterium exchange in the photoproduct as a result of the keto-enol tautomerization (Fig. 10). The enolate form of the photoproduct prevails in water, which is the also form that coordinates Fe(III). The loss of 46 mass units in the photoproduct relative to aerobactin is accounted for with production of CO2 and release of two protons. The values of the ligand protonation constants of the aerobactin photoproduct are remarkably similar to aerobactin, as are the Fe(III) stability constants of aerobactin, and the photoproduct [34,47].
Fig. 10.

Reaction scheme for the UV photolysis of Fe(III)–aerobactin under aerobic conditions.
UV photolysis of Fe(III)-complexes of the ferric–ochrobactins [39], ferric–synechobactins [41] and ferric–petrobactins [37] produces the same conversion of the citrate backbone to 3-ketoglutarate and coordination of Fe(III) by the enolate form of the photoproduct. Photolysis of Fe(III)–vibrioferrin, however, is different because the two bidentate OO’ donor ligands derive from different kinds of α-hydroxy-carboxylic acids, one from citrate and one from α-ketoglutarate [48]. The photoreaction leads to oxidation of the α-ketoglutarate α-hydroxy-carboxylic acid center and not the citrate α-hydroxy-carboxylic acid (Fig. 11).
Fig. 11.
Photoreaction of Fe(III)–vibrioferrin. Adapted from Ref. [48].
A dimeric ferric citrate complex, Fe(III)2-(cit)22−, which may be the same or similar to the structure shown in Fig. 6 [33], is the form of the ferric siderophore that is thought to bind to the siderophore receptor protein in the bacterium outer membrane. Ferric citrate complexes have been known for almost a century to be photoreactive [49,50]. Abrahamson has shown the photolysis of ferric citrate leads to reduction of two equivalents of Fe(III) per equivalent of citrate decarboxylated, consistent with decarboxylation being a two-electron oxidation reaction (Fig. 12) [46]. The 3-ketoglutarate that is formed initially is not stable in acid and further decomposes, producing acetone. Ferric carboxylate complexes are also quite photoreactive, although with a reactivity somewhat less than that of ferric α-hydroxycarboxylate complexes [46,51].
Fig. 12.

Proposed photoreaction of diferric dicitrate in acid. Derived from Ref. [46].
3.2. β-Hydroxyaspartate-containing siderophores
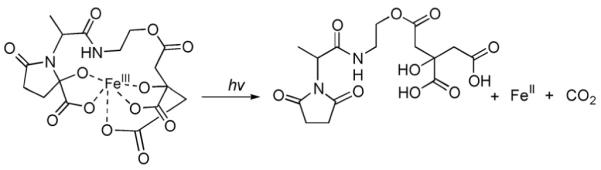
Ferric complexes of β-hydroxyaspartate-containing siderophores, including the aquachelins, loihichelins, marinobactins, and alterobactins are also photoreactive. As shown initially for Fe(III)–aquachelins [35], UV photolysis induces oxidation of the peptide ligand and reduction of Fe(III) (Fig. 13). In the cases of the Fe(III)–siderophore photoreaction, the reaction is likely a radical reaction. The predominant ligand product (m/z 780) is consistent with the loss and oxidation of the β-hydroxyaspartate amino acid and release of the fatty acid. The photoproduct coordinates Fe(III) with the two hydroxamates, however the 5th and 6th ligands that would round out the octahedral coordination have not been determined yet. The conditional stability constants show that the photoproduct has a somewhat lower stability constant for Fe(III), 1011.5 M−1, than Fe(III)–aquachelin C, 1012.2 M−1 [35].
Fig. 13.
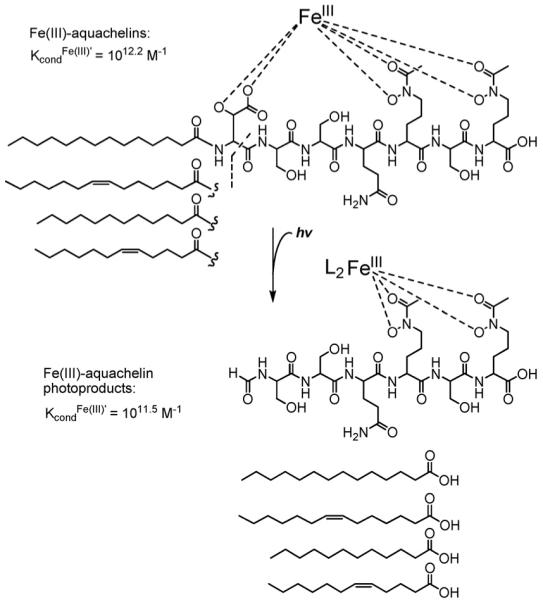
Photoreaction of Fe(III)–aquachelin. Adapted from Ref. [35].
Photolysis of the aquachelins in the presence of substoichiometric or even catalytic amounts of Fe(III) effects complete oxidation of aquachelins. The Fe(II) that is produced is oxidized by O2 in aerobic solution, leading to formation of more Fe(III)–aquachelin that can enter the photochemical cycle again.
The equivalent photoproduct is observed in the photolysis of the Fe(III)–loihichelins [40] as established by mass spectral analysis. UV–vis changes upon photolysis also show the loss of the UV peak corresponding to the β-hydroxyaspartate-to-Fe(III) charge-transfer band in Fe(III)–loihichelin photolysis. Similarly Fe(III)–marinobactins and Fe(III)–alterobactins also display similar UV–vis changes upon photolysis.
4. Coordination-induced self-assembly of amphiphilic siderophores
The suites of amphiphilic siderophores span a fairly broad amphiphilic spectrum, not only within one siderophore family, which arises as a result of fatty acid chain-length variations on a constant head-group, but also across the different families of siderophores as a result of variations in the head-group composition relative to fatty acid chain length. Some of these siderophores are quite hydrophobic, including the amphibactins and ochrobactins that remain cell-associated, whereas others are much more hydrophilic and are isolated from the bacterial culture supernatant. The more hydrophilic amphiphiles, including the loihichelins and aquachelins, have relatively longer peptidic head-groups with seven to eight amino acids, compared to fatty acids of chain length C10–C16. The amphibactins are quite hydrophobic, with only four amino acids in the head-group and fatty acid appendages that are primarily C16 and C18 in chain length. The ochrobactins are also quite hydrophobic with two fatty acids (C8–C10) appending the relatively small head-group comprised of citrate ligated by two lysine residues. The structures of the marinobactins, however, with six amino acids in the head-group ligated predominantly with C16:1 and C16:0 fatty acid (i.e., marinobactins D and E, respectively), place them in the middle of the amphiphilic spectrum of these siderophores. Marinobactins A–E are isolated from the supernatant, however a small amount of marinobactin F with a C18:1 fatty acid tail has been extracted from the bacterial cell mass indicating it is noticeably more hydrophobic [52].
The amphiphilic properties of the marinobactins have been investigated, including, of relevance to this review, the coordination-induced self-assembled structures, both in the presence and absence of Fe(III) and other metal ions. The critical micelle concentration (CMC) of apo-ME (i.e., ME that is not coordinated to Fe(III)) and Fe(III)-ME are relatively low at ~50 and ~75 μM, respectively [44]. At concentrations above the CMC, apo-ME aggregates to form spherical micelles ( ~4.6 nm in diameter) that decrease in size upon coordination of Fe(III) (~3.5 nm in diameter), as analyzed by small-angle neutron scattering (SANS) [53,54]. The decrease in micelle diameter of Fe(III)-ME is attributed to an increase in the head-group area relative to the lipid tail-volume (Fig. 14) [55], which is consistent with molecular modeling results (Fig. 15) [56].
Fig. 14.
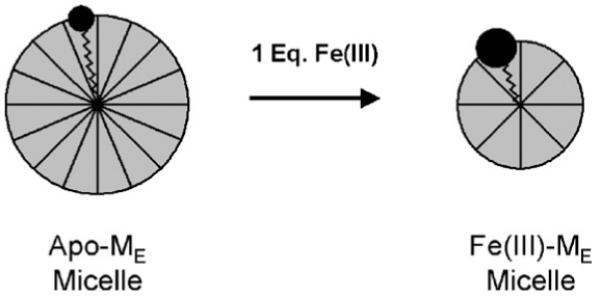
Coordination of Fe(III) could give ME a larger head-group area: tail-volume ratio such that a smaller micelle is formed. Reproduced with permission from Ref. [54].
Fig. 15.
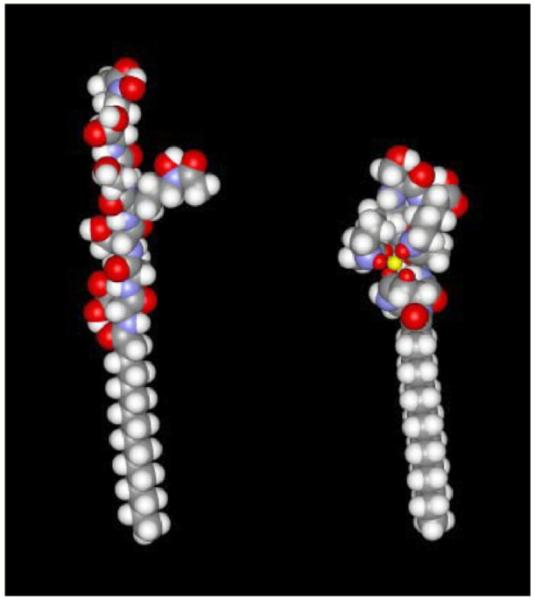
Approximate molecular conformations of apo-ME (left) and Fe-ME (right) derived from MM2 methods using Chem3D and Spartan molecular graphics packages. The color code is standard: C gray, H white, O red, and N blue. The iron coordination core is represented by the yellow octahedral structure (right), in which the small red spheres are the ligating oxygen atoms. Adapted from Ref. [56].
In the presence of excess Fe(III) (such as 0.1–2.0 equivalents of Fe(III) per equivalent of Fe(III)-ME), SANS data is best fit by a decrease in micelle population and a corresponding increase in vesicle population [54]. Dynamic light scattering (DLS) reveals vesicles that are ~190–200 nm in diameter [53,54]. Not only does excess Fe(III) induce the phase-change from micelle-to-vesicle, but so do addition of Zn(II), Cd(II), or La(III) to Fe(III)-ME [53]. However, addition of Zn(II) to apo-ME does not induce the micelle-to-vesicle transition [53]. A Bragg peak develops in the SANS profiles with increasing concentration of Zn(II), Cd(II), La(III) or excess Fe(III), consistent with formation of multilamellar vesicles (Fig. 16).
Fig. 16.
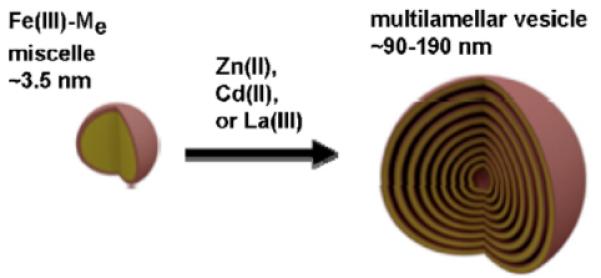
Multilamellar vesicle formation from Fe(III)-ME induced by addition of Zn(II), Cd(II), La(III) or excess Fe(III). Adapted from Ref. [53].
The same micelle-to multilamellar vesicle transition is observed for Zn(II) addition to Fe(III)-MB and Fe(III)-MD [53]. As might be expected, the interlamellar repeat distance is smaller in these vesicles, ~5.0 and ~5.5 nm for Zn(II)-induced Fe(III)-MB vesicles Zn(II)-induced Fe(III)-MD vesicles, respectively, compared to ~6.0 nm for the Zn(II)-induced Fe(III)-ME vesicles [53].
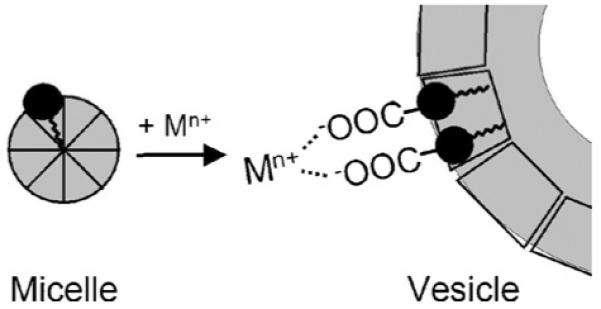
What induces the micelle-to-vesicle phase-change? The terminal carboxylic acid of the marinobactins is not involved in coordination in the monomeric Fe(III)-ME siderophore complex, and thus is available for coordination to the added cations (Fig. 17). EXAFS reveals that the Zn(II), and Cd(II) cations likely crosslink two peptide head-groups [57].
Fig. 17.

Proposed terminal carboxylate crosslinking of marinobactin E by the added cations Zn(II), Cd(II), La(III) or excess Fe(III). The bis-bidentate coordination geometry of the two carboxylates that is depicted here could also be bis-monodentate crosslinking. The identity of ligand, L, has not been established conclusively; EXAFS data is consistent with a light-atom ligand such as H2O or OH−
Relative to a single fatty acid surfactant, a dual fatty acid surfactant would have a smaller head-group area: tail-volume ratio that may favor vesicle formation (Fig. 18) [55].
Fig. 18.
Proposed cation-induced carboxylate-crosslinking of Fe(III)-ME head-groups as a mechanism to promote the micelle-to-vesicle transition. Reproduced with permission from [57]. The resulting “composite surfactant” would have a lower head-group area: tail-volume ratio that may favor vesicle formation [55].
The Zn(II)-induced Fe(III)-ME particles have been shown by dynamic light scattering measurements to break up upon addition of EDTA [53]. EDTA thus competes effectively against the terminal peptide carboxylate for coordination of Zn(II), as one would expect. EDTA does not compete effectively against Fe(III) coordination at near neutral pH, but would only compete at much lower pH conditions. Therefore the dissolution of the vesicles is not a result of removal of Fe(III) from the siderophore, but rather is complexation of the crosslinking Zn(II) cations.
Cations with a propensity to coordinate two or more carboxylate ligands would be expected to induce the multilamelar vesicle formation of the marinobactins, as well as possibly other amphiphilic Fe(III)–siderophore complexes with a terminal carboxylate group. Zn(II), Cd(II), La(III), and Fe(III) do form 2:1 ligand/metal coordination complexes with carboxylic acids such as acetic acid [53,58]. In contrast, metals that do not induce a phase-change, such as Ba(II) and Ca(II), only coordinate a single acetate ligand in aqueous solution [58].
5. Conclusions
Ferric siderophore complexes are classic coordination compounds produced by bacteria to acquire iron. As a result of the bidentate OO’ donor ligands in the vast majority of siderophores, the Fe(III) is present as in a high spin, d5 configuration. Hence, with no ligand field stabilization energy, the ligand substitution rates are relatively fast. The ferric stability constants on the other hand are quite high (1023–1049) accounting for the remarkable selectivity of siderophores for Fe(III) over other cations. However, ferric siderophore complexes are far more reactive than generally reported to date. Given the well recognized photoreactivity of ferric citrate complexes [49] and ferrioxalate, which is an actinometer, the photoreactivity of ferric siderophores that contain an α-hydroxy-carboxylic acid moiety is not unexpected, although it is surprising that the photoreactivity of siderophores was not recognized sooner [34-38]. Ensuing siderophore investigations are likely to reveal, not only the biological significance of suites of amphiphilic siderophores in marine and pathogenic bacteria and the photoreactivity ferric siderophores with α-hydroxy-carboxylic acids, but also the significance of other structural variations in siderophores, such as glycosylation, sulfonation, and amino acid content of a peptide head-group, among other factors. The amphiphilic character of marine siderophores enhances its surface reactivity, a trait that might be useful in particle interaction, such as particles containing iron(III) oxides. The photoreactivity may be useful in reduction of iron(III) oxides as a means to facilitate dissociation of iron out of iron oxide particles, or possibly as to facilitate uptake of ferrous iron directly. Future investigations are directed towards answering the biological significance of structural traits of marine siderophores.
Acknowledgement
AB gratefully acknowledges NIH grant GM38130 for funding.
References
- [1].Field CB, Behrenfeld MJ, Randerson JT, Falkowski P. Science. 1998;281:237. doi: 10.1126/science.281.5374.237. [DOI] [PubMed] [Google Scholar]
- [2].Albrecht-Gary AM, Crumbliss AL. Metal Ions in Biological Systems. New York; Marcel Dekker: 1998. p. 239. [PubMed] [Google Scholar]
- [3].Boukhalfa H, Crumbliss AL. Biometals. 2002;15:325. doi: 10.1023/a:1020218608266. [DOI] [PubMed] [Google Scholar]
- [4].Pearson RG. J. Am. Chem. Soc. 1963;85:3533. [Google Scholar]
- [5].Reichert J, Sakaitani M, Walsh CT. Protein Sci. 1992;1:549. doi: 10.1002/pro.5560010410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fischbach MA, Lin HN, Liu DR, Walsh CT. Proc. Natl. Acad. Sci. U.S.A. 2005;102:571. doi: 10.1073/pnas.0408463102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dertz EA, Xu JD, Stintzi A, Raymond KN. J. Am. Chem. Soc. 2006;128:22. doi: 10.1021/ja055898c. [DOI] [PubMed] [Google Scholar]
- [8].Loomis LD, Raymond KN. Inorg. Chem. 1991;30:906. [Google Scholar]
- [9].Karpishin TB, Raymond KN. Angew. Chem. Ind. Ed. 1992;31:466. [Google Scholar]
- [10].Karpishin TB, Dewey TM, Raymond KN. J. Am. Chem. Soc. 1993;115:1842. [Google Scholar]
- [11].Raymond KN, Dertz EA. In: Iron Transport in Bacteria. Crosa JH, Mey AR, Payne SM, editors. ASM Press; Washington, DC: 2004. p. 3. [Google Scholar]
- [12].Isied SS, Kuo G, Raymond KN. J. Am. Chem. Soc. 1976;98:1763. doi: 10.1021/ja00423a021. [DOI] [PubMed] [Google Scholar]
- [13].McArdle JV, Sofen SR, Cooper SR, Raymond KN. Inorg. Chem. 1978;17:3075. [Google Scholar]
- [14].Abergel RJ, Clifton MC, Pizarro JC, Warner JA, Shuh DK, Strong RK, Raymond KN. J. Am. Chem. Soc. 2008;130:11524. doi: 10.1021/ja803524w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. Mol. Cell. 2002;10:1033. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- [16].Bluhm ME, Kim SS, Dertz EA, Raymond KN. J. Am. Chem. Soc. 2002;124:2436. doi: 10.1021/ja016651s. [DOI] [PubMed] [Google Scholar]
- [17].Abergel RJ, Zawadzka AM, Hoette TM, Raymond KN. J. Am. Chem. Soc. 2009;131:12682. doi: 10.1021/ja903051q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Brickman TJ, McIntosh MA. J. Biol. Chem. 1992;267:12350. [PubMed] [Google Scholar]
- [19].Lin H, Fiscchbach MA, Liu DR, Walsh CT. J. Am. Chem. Soc. 2005;127:11075. doi: 10.1021/ja0522027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hossain MB, Jalal MAF, van der Helm D, Shimizu K, Akiyama M. J. Chem. Crystallogr. 1998;28:53. [Google Scholar]
- [21].Hossain MB, Jalal MAF, Vanderhelm D. Acta Crystallogr. Sect. C-Cryst. Struct. Commun. 1986;42:1305. [Google Scholar]
- [22].Van Der Helm D, Poling M. J. Am. Chem. Soc. 1976;98:82. doi: 10.1021/ja00417a014. [DOI] [PubMed] [Google Scholar]
- [23].Dhungana S, White PS, Crumbliss AL. J. Biol. Inorg. Chem. 2001;6:810. doi: 10.1007/s007750100259. [DOI] [PubMed] [Google Scholar]
- [24].Schwarzenbach G, Schwarzenbach K. Helv. Chim. Acta. 1963;46:1390. [Google Scholar]
- [25].Crumbliss AL. In: Handbook of Microbial Iron Chelates. Winkelmann GE, editor. CRC Press, Inc.; Boca Raton: 1991. p. 177. [Google Scholar]
- [26].Van Der Helm D, Baker JR, Eng-Wilmot DL, Hossain MB, Loghry RA. J. Am. Chem. Soc. 1980;102:4224. [Google Scholar]
- [27].Zalkin A, Forrester JD, Templeton DH. J. Am. Chem. Soc. 1966;88:1810. doi: 10.1021/ja00960a040. [DOI] [PubMed] [Google Scholar]
- [28].Schmelz S, Kadi N, McMahon SA, Song LJ, Oves-Costales D, Oke M, Liu HT, Johnson KA, Carter LG, Botting CH, White MF, Challis GL, Naismith JH. Nat. Chem. Biol. 2009;5:174. doi: 10.1038/nchembio.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Franza T, Mahe B, Expert D. Mol. Microbiol. 2005;55:261. doi: 10.1111/j.1365-2958.2004.04383.x. [DOI] [PubMed] [Google Scholar]
- [30].Amin SA, Kupper FC, Green DH, Harris WR, Carrano CJ. J. Am. Chem. Soc. 2007;129:478. doi: 10.1021/ja067369u. [DOI] [PubMed] [Google Scholar]
- [31].Harris WR, Amin SA, Kupper FC, Green DH, Carrano CJ. J. Am. Chem. Soc. 2007;129:12263. doi: 10.1021/ja073788v. [DOI] [PubMed] [Google Scholar]
- [32].Guerinot ML, Meidl EJ, Plessner O. J. Bacteriol. 1990;172:3298. doi: 10.1128/jb.172.6.3298-3303.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shweky I, Bino A, Goldberg DP, Lippard SJ. Inorg. Chem. 1994;33:5161. [Google Scholar]
- [34].Kupper FC, Carrano CJ, Kuhn JU, Butler A. Inorg. Chem. 2006;45:6028. doi: 10.1021/ic0604967. [DOI] [PubMed] [Google Scholar]
- [35].Barbeau K, Rue EL, Bruland KW, Butler A. Nature. 2001;413:409. doi: 10.1038/35096545. [DOI] [PubMed] [Google Scholar]
- [36].Barbeau K, Rue EL, Trick CG, Bruland KT, Butler A. Limnol. Oceanogr. 2003;48:1069. [Google Scholar]
- [37].Barbeau K, Zhang GP, Live DH, Butler A. J. Am. Chem. Soc. 2002;124:378. doi: 10.1021/ja0119088. [DOI] [PubMed] [Google Scholar]
- [38].Hickford SJH, Kupper FC, Zhang GP, Carrano CJ, Blunt JW, Butler A. J. Nat. Prod. 2004;67:1897. doi: 10.1021/np049823i. [DOI] [PubMed] [Google Scholar]
- [39].Butler A, Martin JD. Metal Ions in Biological Systems. Taylor & Francis Ltd.; London: 2005. p. 21. [PubMed] [Google Scholar]
- [40].Homann VV, Sandy M, Tincu JA, Templeton AS, Tebo BM, Butler A. J. Nat. Prod. 2009;72:884. doi: 10.1021/np800640h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ito Y, Butler A. Limnol. Oceanogr. 2005;50:1918. [Google Scholar]
- [42].Martin JD, Ito Y, Homann VV, Haygood MG, Butler A. J. Biol. Inorg. Chem. 2006;11:633. doi: 10.1007/s00775-006-0112-y. [DOI] [PubMed] [Google Scholar]
- [43].Martinez JS, Carter-Franklin JN, Mann EL, Martin JD, Haygood MG, Butler A. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3754. doi: 10.1073/pnas.0637444100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Martinez JS, Zhang GP, Holt PD, Jung HT, Carrano CJ, Haygood MG, Butler A. Science. 2000;287:1245. doi: 10.1126/science.287.5456.1245. [DOI] [PubMed] [Google Scholar]
- [45].Bergeron RJ, Huang GF, Smith RE, Bharti N, McManis JS, Butler A. Tetrahedron. 2003;59:2007. [Google Scholar]
- [46].Abrahamson HB, Rezvani AB, Brushmiller JG. Inorg. Chim. Acta. 1994;226:117. [Google Scholar]
- [47].Harris WR, Carrano CJ, Raymond KN. J. Am. Chem. Soc. 1979;101:2722. [Google Scholar]
- [48].Amin SA, Green DH, Hart MC, Kupper FC, Sunda WG, Carrano CJ. Proc. Natl. Acad. Sci. U.S.A. doi: 10.1073/pnas.0905512106. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fry HS, Gerwe EG. Ind. Eng. Chem. 1928;20:1392. [Google Scholar]
- [50].Krizek DT, Bennett JH, Brown JC, Zaharieva T, Norris KH. J. Plant Nutr. 1982;5:323. [Google Scholar]
- [51].Waite TD, Morel FMM. J. Colloid Interface Sci. 1984;102:121. [Google Scholar]
- [52].Martinez JS, Butler A. J. Inorg. Biochem. 2007;101:1692. doi: 10.1016/j.jinorgbio.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Owen T, Pynn R, Hammouda B, Butler A. Langmuir. 2007;23:9393. doi: 10.1021/la700671p. [DOI] [PubMed] [Google Scholar]
- [54].Owen T, Pynn R, Martinez JS, Butler A. Langmuir. 2005;21:12109. doi: 10.1021/la0519352. [DOI] [PubMed] [Google Scholar]
- [55].Israelachvili JN, Mitchell DJ, Ninham BW. J. Chem. Soc., Faraday Trans. 1976;72:1525. [Google Scholar]
- [56].Xu GF, Martinez JS, Groves JT, Butler A. J. Am. Chem. Soc. 2002;124:13408. doi: 10.1021/ja026768w. [DOI] [PubMed] [Google Scholar]
- [57].Owen T, Webb SM, Butler A. Langmuir. 2008;24:4999. doi: 10.1021/la703833v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Sillen LG, Martell AE. Stability Constants of Metal Ion Complexes. Vol. 1. The Chemical Society; London: 1971. [Google Scholar]