

Abstract
The highly efficient synthesis of a novel heat shock protein 90 (Hsp90)-based anticancer agent, triazole-cycloproparadicicol (5), is described. The key step involves a fragment coupling using “click chemistry.” The preliminary biological evaluation of triazole-cycloproparadicicol is also reported.
Keywords: heat shock protein 90, triazole-cycloproparadicicol, total synthesis, “click chemistry”, antitumor agent
Heat shock protein 90 (Hsp90), a molecular chaperone that mediates the folding of a number of oncogenic proteins (cf. Raf-1 and Her-2),[i] is considered to be an attractive target for inhibition by anticancer agents.[ii] Recent investigations have argued that, compared to normal cell Hsp90s, the tumor cell variations exist in activated, high-affinity, multi-chaperone form, with particularly heightened affinities for their inhibitors. These increased inhibitor affinities – up to one hundred fold higher than those of normal cell Hsp90s – provide a basis for the selective targeting of tumor cells.[iii] Importantly, it has been suggested that drug-resistant proteins are particularly dependent on association with Hsp90 in order to properly fold and function. Thus, the selective inhibition of tumor Hsp90 could ultimately provide a means by which to counter the growing problem of drug resistance in cancer treatment.[iv]
The interest in the development of selective Hsp90 inhibitors is evidenced by a grouping menu of candidates which have been targeted for development directed to this proposition. In particular, the natural product Hsp90 inhibitors, radicicol (1)[v] and geldanamycin (2)[vi] have attracted a great deal of attention (Figure 1).[vii] Currently, the geldanamycin derivative, 17-AAG (3), is in Phase II clinical trials for breast cancer.[vii]
Figure 1.

Hsp90 inhibitors.
In 1999, our laboratory initiated a program directed toward the diverted total synthesis-based discovery of novel Hsp90-based anticancer agents. We were particularly attracted to radicicol (1) as an initial target. The absence of a quinine moiety in 1 seems to be advantageous relative to compounds in the geldanamycin series, such as 17-AAG (3). Radicicol (1), a metabolite of Monocillium nordinii,[v] exhibits potent in vitro inhibitory acitivity against Hsp90 (IC50 = 20 nM)[viii] and has been shown to be significantly less hepatotoxic than 17-AAG (3).[ix] Even more interestingly, radicicol (1) and its analogs demonstrate potent cytotoxicity against Rb (retinoblastoma)-negative cells which are resistant to 17-AAG.[x]
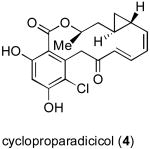
Upon completion of the first total synthesis of radicicol, we were able to confirm its reported in vitro activity, but we found the compound to be inactive in in vivo settings. We postulated that the disappointing lack of in vivo activity may be attributable to the presence of the chemically vulnerable epoxide functionality of radicicol.[xi] Through application of a diverted total synthesis (DTS)[xii] approach, we successfully prepared the congener, cycloproparadicicol (4, Figure 2), in which the epoxide was replaced with a more robust cyclopropyl functionality.[xiii]
Figure 2.

Diverted total synthesis of cycloproparadicicol.
Preliminary investigations revealed that the epoxide functionality of radicicol was not crucial for inhibitory activity, as cycloproparadicicol (4) inhibited Hsp90 at levels as low as 160 nM. Furthermore, cycloproparadicicol exhibited significant inhibitory activity against MCF-7 breast cancer cells (IC50 = 49 nM).[xiii] In a preliminary in vivo study against mice implanted with human colon carcinoma (HCT-116), cycloproparadicicol (75 mg/kg, QDx7) achieved 68% tumor cell growth suppression. Its stereoisomer SAR profile tracked very closely with that of the corresponding epoxide, suggesting a common recognition pattern with Hsp90.
Our synthesis of cycloproparadicicol suffered from several low-yielding and difficultly scalable steps that significantly limit comfortable access for further preclinical studies.[xiii] Furthermore, we suspected that the dienone moiety of cycloproparadicicol may be responsible for toxicity and loss of in vivo activity.[xiv] We thus sought to design novel analogs based on the cycloproparadicicol framework, which might exhibit a more exploitable therapeutic index than the parent compound and which might be accessible through an efficient and scalable synthetic route.
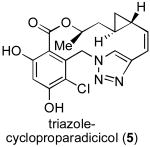
Given these considerations, we chose as our target compound a novel triazole-containing analog of cycloproparadicicol (5). As outlined in Figure 3, we hoped to execute a highly convergent and scalable route to 5, which would rely on a Cu(I)-mediated triazole formation – “click chemistry”[xv] – to achieve the rapid buildup of the backbone (cf. 7 + 8 → 6). We also anticipated that replacement of the original dienone moiety with a triazole functionality might offer a means by which to overcome toxicity and erosion of in vivo activity.[xvi] The 14-membered macrolactone would be constructed through intramolecular transesterification of alcohol 6. We proposed accessing Fragment A (7) from the commercially available orcinol 9. Fragment B (8) would be prepared from the commercially available “chiral non-racemic” β-hydroxylester 10. As a consequence of its convergence and scalability, we anticipated that this proposed route would be highly amenable to the preparation of analogs through diverted total synthesis.
Figure 3.

Proposed route to triazole-cycloproparadicicol (5).
The synthesis of fragment A (7) began with commercially available orcinol 9. (Scheme 1). Phenol 11 was efficiently prepared in accordance with literature reported methods[xvii] in 70% overall yield. TBS-protection of 11 afforded compound 12. Selective chlorination of the aromatic ring, followed by selective bromination of the methyl group under radical conditions smoothly generated benzylbromide 13 in one pot (72% yield). Treatment of the latter with sodium azide at room temperature, followed by phenol protection with MOMCl afforded desired fragment A (7) in 90% yield for 2 steps.
Scheme 1.

Synthesis of Fragment A (7)
Our synthesis of fragment B (8) commenced with the readily available alcohol 14 which was itself prepared from the commercially available, chiral non-racemic compound 10 in 90% overall yield (Scheme 2).[xiii] Oxidation of 14 cleanly afforded aldehyde 15.[xviii] Next, sulfone 16 was provided from 17 according to the literature reported method.[xix] Julia olefination of aldehyde 15 with sulfone 16, using KHMDS as base, smoothly afforded the desired enyne compound 18 in 60% yield with excellent Z-selectivity (>20:1).[xix] Global desilylation of 18 cleanly provided the desired fragment B (8) in 94% yield.
Scheme 2.

Synthesis of Fragment B (8).
With fragments A (7) and B (8) in hand, we were able to assemble the triazole 6 through application of the highly efficient Cu(I)-mediated “click chemistry” protocol[xx] (Scheme 3). As hoped, the intramolecular macrolactonization of 6 proceeded readily under basic conditions to provide the macrolactone 19 in 70% yield. Finally, HCl-mediated MOM deprotection served to complete the total synthesis of triazole-cycloproparadicicol 5. The structure of 5 was unambiguously confirmed by X-ray crystal structure analysis.[xxi]
Scheme 3.
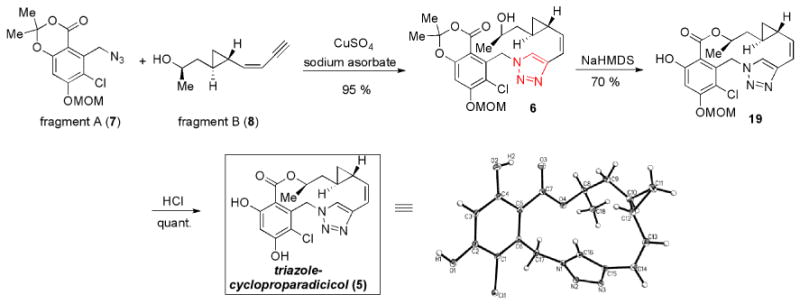
Synthesis of triazole-cycloproparadicicol (5).
Preliminary biological evaluations reveal triazole-cycloproparadicicol 5 to be a reasonably potent Hsp90 inhibitor, with IC50 = 400 nM. (Table 1). Triazole-cycloproparadicicol 5 also displayed significant in vitro inhibitory activity, comparable to 17-AAG, against the leukemia cell line Kasumi-1 (IC50 = 650 nM).[xxii] These preliminary results show that triazole-cycloproparadicicol is indeed a promising candidate worthy of further in vitro and in vivo preclinical studies.
Table 1.
In vitro activity of triazole-cycloproparadicicol. (Preliminary biological evaluations were performed by Dr. Gabriela Chiosis and coworkers at MSKCC.)
 |
 |
 |
|---|---|---|
| compound | Hsp90 inhibition (IC50) | Inhibition of Kasumi-1 leukemia cell line (IC50) |
| cycloproparadicicol (4) | 160 nM | -- |
| triazole-cycloproparadicicol (5) | 400 nM | 650 nM |
| 17-AAG (3) | 600 nM | 620 nM |
In conclusion, we have developed a highly convergent and robust synthetic route (only 11 steps in total based on the longest linear sequence, with 20 % overall yield) for the facile synthesis of a novel analog of cycloproparadicicol. The key step involved an efficient cross coupling reaction using a “click” type construction. The current synthesis should produce substantial amounts of triazole-cycloproparadicicol for further in vivo studies and could also provide an excellent platform for further analog synthesis and evaluation.[xxiii]
Experimental Section
General Methods
All non-aqueous reactions were carried out in oven-dried glassware under a slight positive pressure of argon unless otherwise noted. All reagents were commercially available and used without further purification from Sigma-Aldrich and TCI America, unless indicated otherwise. Solvents were reagent grade and purified by standard techniques: THF was distilled from Na-benzophenone or filtered through a dry-solvent system; CH2Cl2 was distilled from CaH2 or filtered through a dry-solvent system; all other solvents were Aldrich “anhydrous” grade solvents, unless indicated otherwise. Reactions were magnetically stirred and monitored by thin layer chromatography on Merck silica gel 60-F254 coated 0.25 mm plates. Preparative thin layer chromatography was performed with Merck silica gel 60-F254 coated 0.50 mm plates. Flash chromatography was performed with Sorbent Technology silica gel 60 (particle size 32-63 μm), unless indicated otherwise. Yields reported are for isolated, spectroscopically pure compounds. Melting points are uncorrected. NMR spectra were recorded on Bruker DRX 300 or 400 MHz and DMX 500MHz spectrometers. Proton and carbon chemical shifts were referenced to residual solvent peaks. Abbreviations for 1H NMR: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, or br = broad. High resolution mass spectra were acquired in the Columbia University Mass Spectral Core facility on a JEOL HX110 spectrometer. Optical rotations were measured on a JASCO DIP-1000 spectrometer.
Procedure for the Synthesis of Representative Compounds
Compound 13
To a solution of compound 12 (2.0 g, 6.2 mmol, 1.0 eq.) in anhydrous chlorobenzene (10.0 mL) was added N-chlorosuccinimide (915 mg, 6.8 mmol, 1.1 eq.). The reaction mixture was stirred at 100 °C for 12 h. After the reaction mixture was cooled to rt, freshly purified N-bromosuccinimide (1.65g, 9.3 mmol, 1.5 eq.) and benzoyl peroxide (750 mg, 3.1 mmol, 0.5 eq.) were added. The reaction mixture was stirred at 100 °C for 16 h. Then the reaction mixture was cooled to rt, and quenched with sat. NaHSO3 (10 mL), extracted with ether (3 × 20 mL). The combined organic phase was washed with brine, dried over MgSO4, concentrated and purified by flash chromatography (Hexane/EtOAc: 10/1) to afford product 13 (1.94 g, 4.4 mmol, 72%) as a white solid. m.p. 102-103 °C
Compound 18
To a solution of aldehyde 15 (2.3 g, 6.3 mmol, 1.0 eq) and sulfone 16 (2.5 g, 8.2 mmol, 1.3 eq.) in anhydrous THF (30 mL) was slowly added KHMDS (0.5 M in toluene, 16.4 mL, 8.2 mmol, 1.3 eq.) at −78 °C under argon during 1 hour. The reaction mixture was stirred at −50 °C for 6 hours, before it was quenched by sat. NH4Cl. The mixture was diluted with Et2O, washed with water and brine, dried over MgSO4, concentrated and purified via flash chromatography (Hexane/EtOAc: 20/1) to afford enyne 18 (1.7 g, 3.8 mmol, 60%) as a yellow oil.
Compound 6
To a solution of benzylazide 7 (730 mg, 2.2 mmol, 1.0 eq.) and alkyne 8 (370 mg, 2.4 mmol, 1.1 eq.) in t-BuOH (2.0 mL) and H2O (2.0 mL) was added CuSO4 (8.0 mg, 0.05 mmol, 0.02 eq.) followed by sodium asorbate (90 mg, 0.5 mmol, 0.5 eq.) at rt. The reaction mixture was vigorously stirred at rt for 12 h. The resulting mixture was diluted with EtOAc (50 mL), washed with water and brine, dried over MgSO4, concentrated and purified via flash chromatography (Hexane/EtOAc: 1/1) to afford triazole 6 (1.01 g, 2.1 mmol, 95 %) as a colorless oil.
Compound 5
To the solution of phenol 19 (460 mg, 1.1 mmol) in MeOH (3 mL) was added 2M HCl (1 mL) at room temperature. The reaction mixture was stirred at 50 °C for 8 hours. The solution was concentrated in vacuo and the residue was dissolved with EtOAc (50 mL). The solution was washed with water and brine, dried over anhydrous MgSO4, and filtrated. After evaporation of the solvent, the product was collected and further recrystalized by CHCl3/EtOH (10:1) to afford the pure triazole-cycloproparadicicol 5 (410 mg, 1.1 mmol, quant.) as a white solid. m.p. 240-242 °C
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health (CA28824). We thank Professor Gabriela Chiosis and coworkers (MSKCC) for the preliminary biological evaluation of compound 5, and Ms. Daniela Buccella (Columbia University) for X-ray crystal structure analysis.
Footnotes
Dedication: This manuscript is dedicated in honor of the 60th birthday of Professor Chi-Huey Wong for his historic contributions at the intersection of chemistry and biology.
Supporting Information. Additional experimental procedures and spectra data for new compounds and intermediates are available as Supporting Information free of charge via the Internet.
References
- [i].Banerji U, Judson I, Workman P. Curr Cancer Drug Targets. 2003;3:385–390. doi: 10.2174/1568009033481813. [DOI] [PubMed] [Google Scholar]
- [ii].(a) Xiao L, Lu X, Ruden DM. Mini-Reviews in Medicinal Chemistry. 2006;6:1137–1143. doi: 10.2174/138955706778560166. [DOI] [PubMed] [Google Scholar]; (b) Dymock BW, Drysdale MJ, McDonald E, Workman P. Expert Opinion on Therapeutic Patents. 2004;14:837–847. [Google Scholar]; (c) Workman P. Curr Cancer Drug Targets. 2003;3:297–300. doi: 10.2174/1568009033481868. [DOI] [PubMed] [Google Scholar]
- [iii].Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- [iv].Jarvis LM. Chem & Eng News. 2007;9:15–23. [Google Scholar]
- [v].Delmotte P, Delmotte-Plaquee J. Nature. 1953;171:344. doi: 10.1038/171344a0. [DOI] [PubMed] [Google Scholar]
- [vi].(a) Rinehart KL, Jr, Sasaki K, Slomp G, Grostic MF, Olson EC. J Am Chem Soc. 1970;92:7591–7593. doi: 10.1021/ja00729a018. [DOI] [PubMed] [Google Scholar]; (b) Uehara Y, Hori M, Takeuchi T, Umezawa H. Japan J Can Res. 1985;76:672–675. [PubMed] [Google Scholar]
- [vii].Newman DJ, Cragg GM, Holbeck S, Sausville EA. Curr Cancer Drug Target. 2002;2:279–308. doi: 10.2174/1568009023333791. [DOI] [PubMed] [Google Scholar]
- [viii].Sharma SV, Agatsuma T, Nakano H. Oncogene. 1998;16:2639–2645. doi: 10.1038/sj.onc.1201790. [DOI] [PubMed] [Google Scholar]
- [ix].Chiosis G, Lucas B, Huezo H, Solit D, Basso A, Rosen N. Curr Cancer Drug Targets. 2003;3:371–376. doi: 10.2174/1568009033481778. [DOI] [PubMed] [Google Scholar]
- [x].Yamamoto K, Garbaccio RM, Stachel SJ, Solit DB, Chiosis G, Rosen N, Danishefsky SJ. Angew Chem, Int Ed. 2003;42:1280–1284. doi: 10.1002/anie.200390329. [DOI] [PubMed] [Google Scholar]
- [xi].Greene JF, Newman JW, Williamson KC, Hammock BD. Chem Res Toxicol. 2000;13:217–226. doi: 10.1021/tx990162c. [DOI] [PubMed] [Google Scholar]
- [xii].Wilson RM, Danishefsky SJ. J Org Chem. 2006;71:8329–8351. doi: 10.1021/jo0610053. [DOI] [PubMed] [Google Scholar]
- [xiii].(a) Yamamoto K, Garbaccio RM, Stachel SJ, Solit DB, Chiosis G, Rosen N, Danishefsky SJ. Angew Chem, Int Ed. 2003;42:1280–1284. doi: 10.1002/anie.200390329. [DOI] [PubMed] [Google Scholar]; (b) Yang ZQ, Geng X, Solit D, Pratilas CA, Rosen N, Danishefsky SJ. J Am Chem Soc. 2004;126:7881–7889. doi: 10.1021/ja0484348. [DOI] [PubMed] [Google Scholar]
- [xiv].Turbyville TJ, Wijeratne EMK, Liu MX, Burns AM, Seliga CJ, Luevano LA, David CL, Faeth SH, Whitesell L, Gunatilaka AAL. J Nat Prod. 2006;69:178–184. doi: 10.1021/np058095b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [xv].For a review, see: Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5.
- [xvi].For a review, see: Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7.
- [xvii].Islam MS, Kitagawa M, Imoto M, Kitahara T, Watanabe H. Bioscience, Biotechnology, and Biochemistry. 2006;70:2523–2528. doi: 10.1271/bbb.60287. [DOI] [PubMed] [Google Scholar]
- [xviii].Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis. 1994:639–666. [Google Scholar]
- [xix].Bonini C, Chiummiento L, Videtta V. Synlett. 2006:2079–2082. [Google Scholar]
- [xx].Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem, Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- [xxi].CCDC 682086 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033
- [xxii].Yu W, Rao Q, Wang M, Tian Z, Lin D, Liu X, Wang J. Leukemia Res. 2006;30:575–582. doi: 10.1016/j.leukres.2005.08.028. [DOI] [PubMed] [Google Scholar]
- [xxiii].Limited in vivo studies have been performed with triazole-cycloproparadicicol. As yet, it has not demonstrated appreciable in vivo activity in mouse xenograft systems, presumably due to solubility issues. We are currently evaluating related analogs in an effort to improve the pharmacokinetic properties of the constructs.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.