

Abstract
Electrophile-mediated disruption of cell signal-ing is involved in the pathogenesis of several diseases including atherosclerosis and cancer. Diffusible and membrane bound lipid electrophiles are known to modify DNA and protein substrates and modulate cellular pathways including ER stress, antioxidant response, DNA damage, heat shock, and apoptosis. Herein we report on a structure−activity relationship for several electrophilic analogues of 4-hydroxynonenal (HNE) and 4-oxononenal (ONE) with regard to toxicity and anti-inflammatory activity. The analogues studied were the oxidation products of HNE and ONE, HNEA/ONEA, the in vivo hydrolysis products of oxidized phosphatidylcholine, COOH-HNE/COOH-ONE, and their methyl esters, COOMe-HNE/ONE. The reactivity of each compound toward N-acetylcysteine was determined and compared to the toxicity toward a human colorectal carcinoma cell line (RKO) and a human monocytic leukemia cell line (THP-1). Further analysis was performed in differentiated THP-1 macrophages to assess changes in macrophage activation and pro-inflammatory signaling in response to each lipid electrophile. HNE/ONE analogues inhibited THP-1 macrophage production of the pro-inflammatory cytokines, IL-6, IL-1β, and TNFα, after lipopolysaccharide (LPS)/IFNγ activation. Inhibition of cytokine production was observed at submicromolar concentrations of several analogues with as little as 30 min of exposure. Phagocytosis of fluorescent beads was also inhibited by lipid electrophile treatment. Lipid electrophiles related to HNE/ONE are both toxic and anti-inflammatory, but the anti-inflammatory effects in human macrophages are observed at nontoxic concentrations. Neither toxicity nor anti-inflammatory activity are strongly correlated to the reactivity of the model nucleophile, N-acetylcysteine.
Introduction
Oxidized phospholipids (oxPL) are generated in vivo as a result of the inflammatory response, mitochondrial respiration, xenobiotic metabolism, and other processes that generate oxidants. The fatty acyl group esterified at the 2 position of glycerophospholipds is mono- or polyunsaturated and is sensitive to oxidation to hydroperoxides and cyclic peroxides. The initial oxidation products decompose to a variety of compounds, some of which are reactive electrophiles.(1) These electrophiles either remain esterified to the phospholipid or can be released from the membrane and react with cellular targets. The most well-studied of these electrophiles is the highly reactive α,β-unsaturated aldehyde, 4-hydroxynonenal (HNE), which can diffuse throughout the cell and modify DNA and protein molecules. Previous work from our laboratory, using mass spectrometry based proteomics and microarray analysis, along with work from other groups, has demonstrated the ability of HNE to modulate cellular pathways including the ER stress response, the antioxidant response, the DNA damage response, the heat shock response, and the induction of apoptosis in human colorectal cancer (RKO) cells.2−10 Additionally, it is known that oxPLs with “HNE-like” carbonyl groups at the sn-2 position can be generated in the plasma membrane and promote macrophage activation and inflammation.(11)
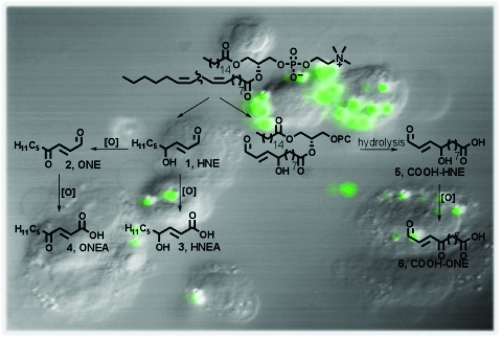
When phospholipids are subjected to oxidative stress, a complex mixture of reactive compounds is formed, and it is often difficult to isolate significant amounts of individual compounds of interest. To evaluate the chemical reactivity and cellular effects of several of these oxidation products, we synthesized a series of electrophiles related to HNE and its oxidation product, ONE (Scheme 1).12−14 Both HNE and ONE can be further oxidized to produce their respective metabolites, HNEA and ONEA, which have also been detected in vivo.15−19 The COOH-HNE and COOH-ONE analogues can be formed in vivo by hydrolysis of the 5-hydroxy-8-oxo-6-octenoic acid ester of 2-lysophophatidylcholine (HOOA-PC) and the 5-keto-8-oxo-6-octenoic acid ester of lysophosphatidylcholine (KOOA-PC), respectively. These forms of oxidized phosphatidylcholine have been identified in oxidized human low-density lipoprotein and are likely hydrolyzed by the enzyme platelet-activating factor-acetylhydrolase (PAF-AH) to produce the carboxylic acid derivative.(20)
Scheme 1. Representative Electrophiles Derived from ω-6 PUFAs.

Each compound was evaluated for electrophilic reactivity toward N-acetylcysteine and toxicity toward the human colon carcinoma cell line, RKO, and the human macrophage cell line, THP-1. HNE and oxPLs are known to inhibit the synthesis of pro-inflammatory cytokines, including IL-6, IL-12, and TNFα, in LPS-activated monocytic cell lines.21−23 Therefore, we performed further analysis in differentiated THP-1 macrophages to assess changes in macrophage activation and pro-inflammatory signaling in response to various lipid electrophiles. Several of the HNE- and ONE-analogues significantly inhibit the THP-1 macrophage production of IL-6, IL-1β, and TNFα after challenge with LPS and interferon gamma (IFNγ). Additionally, it was found that HNE and ONE could inhibit THP-1 macrophage activation as measured by the extent of phagocytosis, under the same conditions as those that inhibit cytokine production.
Experimental Procedures
Synthesis of Lipid Electrophiles
1H and 13C NMR spectra were collected on a Bruker 300 MHz NMR. HRMS experiments were carried out at Notre Dame on a Bruker micrOTOF. All reactions were carried out under an atmosphere of argon. CH2Cl2 was dried using a solvent purification system. Commercial anhydrous Et3N, DMF, and CH3CN were used as received. Purification by column chromatography was carried out on silica gel, and TLC plates were visualized with phosphomolybdic acid. Although HNE is commercially available, the purity was sometimes questionable, and therefore, we chose to synthesize it. The synthesis of HNE (1), its ester precursor 7, and sulfinate 8 has been previously described in the literature.2,24
Synthesis of ONE (2)
Dess-Martin periodinane (1.8 g, 4.3 mmol) was added to a solution of 1 (0.52 g, 3.3 mmol) in CH2Cl2 (16 mL). After 30 min, the reaction mixture was concentrated and purified by column chromatography (10% EtOAc/hexanes). The product (0.37 g) was isolated as a yellow oil in 72% yield. The NMR data was consistent with the literature.(25)1H NMR (CDCl3) δ 9.74 (d, 1H, J = 7.0 Hz), 6.85 (d, 1H, J = 16.2 Hz), 6.73 (dd, 1H, J = 7.0, 16.3 Hz), 2.65 (t, 2H, J = 7.3 Hz), 1.62 (m, 2H), 1.30−1.27 (m, 4H), 0.86 (t, 3H, J = 6.8 Hz); 13C NMR (CDCl3) δ 2 00.1, 193.4, 144.9, 137.2, 41.1, 31.2, 23.3, 22.3, 13.8.
Synthesis of HNEA (3)
Aqueous NaOH (0.25 g, 6.3 mmol, 3 mL) was added to a solution of 7 (0.50 g, 2.5 mmol) in MeOH (12 mL). After stirring overnight, the reaction mixture was acidified with 10% HCl, saturated with NaCl, and extracted with EtOAc. The product (0.25 g, 58%) was isolated as a colorless oil after purification by column chromatography (50% EtOAc/hexanes). The NMR data was consistent with the literature.(26)1H NMR (CDCl3) δ 7.02 (dd, 1H, J = 4.7, 15.6 Hz), 6.36 (br s, 1H), 6.01 (d, 1H, J = 15.6 Hz), 4.31 (dt, 1H, J = 5.1, 5.9 Hz), 1.55 (m, 2H), 1.34−1.22 (m, 6H), 0.86 (t, 3H, J = 5.9 Hz); 13C NMR (CDCl3) δ 171.5, 152.7, 119.3, 71.0, 36.4, 31.6, 24.8, 22.5, 13.9.
Synthesis of ONEA (4)
A solution of CrO3 (1.0 g, 10 mmol) in H2O (9 mL) and H2SO4 (1 mL) was added to a solution of 1 (0.43 g, 2.8 mmol) in acetone (14 mL). After 30 min, the reaction mixture was poured into H2O, saturated with NaCl and extracted with EtOAc. Purification by column chromatography (50% EtOAc/hexanes) yielded 4 (0.19 g, 40%) as a white powder. The NMR data was consistent with the literature.(27)1H NMR (CDCl3) δ 10.3 (br s, 1H), 7.11 (d, 1H, Jn = 15.9 Hz), 6.64 (d, 1H, J = 15.8 Hz), 2.62 (t, 2H, J = 7.3 Hz), 1.62 (m, 2H), 1.30−1.25 (m, 4H), 0.87 (t, 3H, J = 7.3 Hz); 13C NMR (CDCl3) δ 199.8, 170.7, 141.0, 129.7, 41.6, 31.2, 23.3, 22.3, 13.8.
Synthesis of 10-(Tetrahydro-2H-pyran-2-yloxy)decanal
A solution of 3,4-dihydro-2H-pyran (11 mL, 0.12 mol) in CH2Cl2 (100 mL) was added dropwise to a milky solution of 1,10-decanediol (21 g, 0.12 mol) and p-toluenesulfonic acid (4.6 g, 0.20 mol) in CH2Cl2 (300 mL). After 3 h, the reaction mixture was washed with saturated NaHCO3 and dried over MgSO4. Purification by column chromatography (20% EtOAc/hexanes) yielded the product (13 g, 41%) as a colorless liquid. Pyridinium chlorochromate (16 g, 0.074 mol) was added to a solution of 10-(tetrahydro-2H-pyran-2-yloxy)decan-1-ol (13 g, 0.049 mol) in CH2Cl2 (200 mL). After 1 h, the reaction mixture was diluted with ether and filtered through Celite. The product was purified by column chromatography (10% EtOAc/hexanes) and isolated as a colorless liquid (10 g, 81%). The NMR data was consistent with the literature.(28)1H NMR (CDCl3) δ 9.73 (t, 1H, J = 1.9 Hz), 4.54 (t, 1H, J = 2.8 Hz), 3.83 (m, 1H), 3.69 (dt, 1H, J = 6.9, 9.5 Hz), 3.46 (m, 1H), 3.34 (dt, 1H, J = 6.6, 9.5 Hz), 2.38 (dt, 2H, J = 1.9, 7.3 Hz), 1.80−1.68 (m, 4H), 1.56−1.50 (m, 6H), 1.1.35−1.25 (m, 10H); 13C NMR (CDCl3) δ 202.9, 98.8, 67.6, 62.3, 43.8, 30.7, 29.7, 29.32, 29.28, 29.2, 29.1, 26.1, 25.4, 22.0, 19.6.
Synthesis of 9
Piperidine (5.0 mL, 0.051 mol) was added to a solution of sulfinate 8 (7.4 g, 0.033 mol) and 10-(tetrahydro-2H-pyran-2-yloxy)decanal (10 g, 0.039 mol) in CH3CN. After stirring overnight, the reaction mixture was diluted with saturated NH4Cl and extracted with EtOAc. The organic layer was washed with brine and dried over MgSO4. The product (8.9 g) was isolated in 79% yield after purification by column chromatography (20 to 30% EtOAc/hexanes). 1H NMR (CDCl3) δ 6.90 (dd, 1H, J = 5.0, 15.7 Hz), 5.98 (dd, 1H, J = 1.6, 15. Hz), 4.53 (t, 1H, J = 2.7 Hz), 4.24 (m, 1H), 4.15 (q, 2H, J = 7.1 Hz), 3.82 (m, 1H), 3.68 (dt, 1H, J = 6.8, 9.5 Hz), 3.45 (m, 1H), 3.33 (dt, 1H, J = 6.6, 9.5 Hz), 2.12 (br d, 1H, J = 4.6 Hz), 1.79−1.70 (m, 2H), 1.54−1.47 (m, 8H), 1.30−1.26 (m, 10H), 1.25 (t, 3H, J = 7.1 Hz); 13C NMR (CDCl3) δ 166.5, 150.3, 120.0, 98.8, 71.0, 67.6, 62.3, 60.4, 36.6, 30.7, 29.6, 29.4, 29.3, 26.1, 25.4, 25.1, 19.6, 14.2, 14.1; HRMS (ESI) calculated 365.2304 (M + Na), observed 365.2285.
Synthesis of 10
TBDMSCl (4.8 g, 0.032 mol) and imidazole (2.7 g, 0.040 mol) were added to a solution of 9 (8.9 g, 0.026 mol) in DMF (65 mL). After stirring overnight, the reaction mixture was diluted with H2O and extracted with EtOAc. The organic layer was washed with brine and dried over MgSO4. The product was purified by column chromatography (10% EtOAc/hexanes) and isolated as a colorless liquid (9.6 g, 81%). 1H NMR (CDCl3) δ 6.88 (dd, 1H, J = 4.7, 15.5 Hz), 5.92 (dd, 1H, J = 1.7, 15.5 Hz), 4.54 (t, 1H, J = 2.7 Hz), 4.25 (m, 1H), 4.16 (dq, 2H, J = 1.7, 7.2 Hz), 3.83 (m, 1H), 3.69 (dt, 1H, J = 6.9, 9.5 Hz), 3.46 (m, 1H), 3.34 (dt, 1H, J = 6.6, 9.6 Hz), 1.73 (m, 2H), 1.56−1.47 (m, 8H), 1.30−1.27 (m, 10H), 1.26 (t, 3H, J = 7.1 Hz), 0.87 (s, 9H), 0.02 (s, 3H), 0.0 (s, 3H); 13C NMR (CDCl3) δ 166.7, 151.1, 119.6, 98.8, 71.5, 67.6, 62.3, 60.2, 37.3, 30.7, 29.7, 29.5, 29.40, 29.35, 26.2, 25.8, 25.6, 25.4, 24.7, 19.6, 18.1, 14.2, −4.7, −5.0; HRMS (ESI) calculated 479.3177 (M + Na), observed 479.3165. To a solution of this product (9.6 g, 0.021 mol) in diethyl ether (100 mL) was added MgBr2-Et2O (17 g, 0.064 mol). After 6 h, the reaction mixture was diluted with H2O and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4 and purified by column chromatography (20 to 30% EtOAc/hexanes). The product (5.2 g) was isolated as a colorless liquid in 66% yield. 1H NMR (CDCl3) δ 6.88 (dd, 1H, J = 4.7, 15.5 Hz), 5.92 (dd, 1H, J = 1.7, 15.5 Hz), 4.25 (m, 1H), 4.15 (dq, 2H, J = 1.6, 7.1 Hz), 3.59 (t, 2H, J = 6.6 Hz), 1.54−1.45 (m, 4H), 1.30−1.26 (m, 10H), 1.25 (t, 3H, J = 7.1 Hz), 0.87 (s, 9H), 0.01 (s, 3H), 0.00 (s, 3H); 13C NMR (CDCl3) δ 166.8, 151.1, 119.6, 71.5, 62.9, 60.3, 37.2, 32.7, 29.44, 29.38, 29.3, 25.8, 25.6, 24.7, 18.1, 14.2, −4.7, −5.0; HRMS (ESI) calculated 373.2769 (M + H), observed 373.2771.
Synthesis of 11
Bleach (30 mL, 0.024 mol) was added to a rapidly stirring solution of 10 (5.2 g, 0.014 mol), Tempo (0.20 g, 0.0013 mol), NaBr (1.1 g, 0.010 mol), Bu4NBr (0.90 g, 0.003 mol), and NaHCO3 (3.0 g, 0.035 mol) in CH2Cl2 (50 mL) and H2O (20 mL). The reaction mixture turned orange as the bleach was added, then the color dissipated. If the reaction was not complete within 30 min, additional bleach was added. The reaction mixture was acidified with 10% HCl, saturated with NaCl, and extracted with EtOAc. The organic layer was dried over MgSO4, filtered, and concentrated. Purification by column chromatography (30% EtOAc/hexanes) yielded 11 (4.1 g) in 75% yield. 1H NMR (CDCl3) δ 6.87 (dd, 1H, J = 4.7, 15.5 Hz), 5.92 (dd, 1H, J =1.7, 15.5 Hz), 4.25 (m, 1H), 4.16 (dq, 2H, J = 1.6, 7.1 Hz), 2.30 (t, 2H, J = 7.4 Hz), 1.57 (m, 2H), 1.46 (m, 2H), 1.30−1.26 (m, 8H), 1.25 (t, 3H, J = 7.1 Hz), 0.86 (s, 9H), 0.01 9s, 3H), −0.01 (s, 3H); 13C NMR (CDCl3) δ 179.6, 166.8, 151.1, 119.6, 71.5, 60.3, 37.2, 33.9, 29.3, 29.0, 28.9, 24.6, 24.5, 18.1, 14.2, −4.7, −5.0; HRMS (ESI) calculated 387.2561 (M + H), observed 387.2551.
Synthesis of 12
DIBAL-H (1 M/toluene, 35 mL, 0.035 mol) was added slowly to a solution of 11 (4.1 g, 0.011 mol) and Et3N (2.3 mL, 0.017 mol) in CH2Cl2 (55 mL) at 0 °C. After 30 min, the reaction mixture was slowly quenched with 10% HCl and saturated with NaCl. The organics were extracted with EtOAc and dried over MgSO4. The product was purified by column chromatography (50% EtOAc/hexanes) and isolated as a pale yellow liquid (1.7 g, 48%). 1H NMR (CDCl3) δ 5.75−5.59 (m, 2H), 4.12−4.05 (m, 3H), 2.30 (t, 2H, J = 7.4 Hz), 1.58 (m, 2H), 1.43 (m, 2H), 1.30−1.25 (m, 8H), 0.85 (s, 9H), 0.01 (s, 3H), −0.01 (s, 3H); 13C NMR (CDCl3) δ 179.1, 134.4, 128.1, 72.7, 63.1, 38.1, 34.0, 29.2, 29.0, 28.7, 25.9, 25.8, 24.8, 24.6, 18.2, 14.2, −4.3, −4.9; HRMS (ESI) calculated 367.2275 (M + Na), observed 367.2253.
Synthesis of 13
Dess-Martin periodinane (1.8 g, 4.2 mmol) was added to a solution of 12 (1.1 g, 3.3 mmol) in CH2Cl2 (16 mL). After 1.5 h, the reaction mixture was diluted with EtOAc and washed with H2O and brine, and dried over MgSO4. Purification by column chromatography (30% EtOAc/hexanes) yielded 13 as a colorless oil (0.64 g, 57%). 1H NMR (CDCl3) δ 9.50 (d, 1H, J = 8.0 Hz), 6.75 (dd, 1H, J = 4.5, 15.5 Hz), 6.20 (ddd, 1H, J = 1.5, 8.0, 15.5 Hz), 4.35 (m, 1H), 2.28 (t, 2H, J = 7.4 Hz), 1.56−1.50 (m, 4H), 1.30−1.20 (m, 8H), 0.85 (s, 9H), 0.00 (s, 3H), −0.03 (s, 3H); 13C NMR (CDCl3) δ 193.8, 179.9, 160.4, 130.5, 71.5, 37.0, 33.9, 29.2, 29.0, 28.8, 25.7, 24.6, 24.5, 18.1, −4.8, −5.0; HRMS (ESI) calculated 343.2299 (M + H), observed 343.2286.
Synthesis of COOH-HNE (5)
Aqueous HF (1 mL, 5% v/v) was added to a solution of 13 (0.64 g, 1.9 mmol) in CH3CN (18 mL). After 1.5 h, the reaction mixture was adjusted to pH 4 with saturated NaHCO3, saturated with NaCl, and extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated. Note: The solution should NOT be concentrated to dryness, or decomposition of the product will result if trace amounts of HF are still present. The product was purified by column chromatography (50% EtOAc/hexanes) and isolated as a white powder (0.17 g, 40%). 1H NMR (CDCl3) δ 9.54 (d, 1H, J = 7.9 Hz), 6.80 (dd, 1H, J = 4.7, 15.7 Hz), 6.27 (ddd, 1H, J = 1.4, 7.9, 15.7 Hz), 4.39 (m, 1H), 2.31 (t, 2H, J = 7.4 Hz), 1.61−1.56 (m, 4H), 1.35−1.25 (m, 8H); 13C NMR (CDCl3) δ 193.8, 179.5, 159.2, 130.6, 71.0, 36.3, 33.9, 29.1, 28.9, 28.8, 25.0, 24.5; HRMS (ESI) calculated 229.1434 (M + H), observed 229.1426.
Synthesis of COOH-ONE (6)
Dess-Martin periodinane (0.15 g, 0.35 mmol) was added to a solution of 5 (0.067 g, 0.29 mmol) in CH2Cl2 (2 mL). After 1 h, the reaction mixture was concentrated and purified by column chromatography (50% EtOAc/hexanes). The product was isolated as a white powder (0.052 g, 79%). 1H NMR (CDCl3) δ 9.76 (d, 1H, J = 6.9 Hz), 6.86 (d, 1H, J = 16.2 Hz), 6.75 (dd, 1H, J = 6.6, 16.2 Hz), 2.67 (t, 2H, J = 7.2 Hz), 2.33 (t, 2H, J = 7.4 Hz), 1.66−1.59 (m, 4H), 1.35−1.28 (m, 6H); 13C NMR (CDCl3) δ 200.0, 193.4, 178.7, 144.9, 137.4, 41.1, 33.7, 29.0, 28.83, 28.78, 24.6, 23.5; HRMS (ESI) calculated 249.1097 (M + Na), observed 249.1085.
Synthesis of 14
This procedure was carried out using the mini Diazald diazomethane generator from Aldrich (St. Louis, MO). A solution of Diazald (1.6 g, 7.5 mmol) in ether (35 mL) was added dropwise to a solution of KOH (1.4 g, 25 mmol) in H2O/EtOH (45 mL, 1:1) at 65 °C. The CH2N2 was condensed into a reaction flask containing a solution of 12 (1.6 g, 4.7 mmol) in ether (20 mL) at 0 °C. After the addition was complete, the reaction mixture was stirred for 30 min to dissipate any unreacted CH2N2. The solution was concentrated, and purification by column chromatography (20% EtOAc/hexanes) yielded 14 (1.2 g, 69%). 1H NMR (CDCl3) δ 5.76−5.59 (m, 2H), 4.12−4.05 (m, 3H), 3.63 (s, 3H), 2.27 (t, 2H, J = 7.4 Hz), 1.58 (m, 2H), 1.43 (m, 2H), 1.30−1.20 (m, 8H), 0.86 (s, 9H), 0.01 (s, 3H), −0.01 (s, 3H); 13C NMR (CDCl3) δ 174.3, 135.2, 128.2, 72.6, 63.1, 51.4, 38.1, 34.0, 29.3, 29.1, 29.0, 25.8, 25.0, 24.8, 18.2, −4.3, −4.8; HRMS (ESI) calculated 381.2432 (M + Na), observed 381.2424.
Synthesis of COOMe-HNE (15)
Dess-Martin periodinane (1.4 g, 3.4 mmol) was added to a solution of 14 (1.0 g, 2.8 mmol) in CH2Cl2 (14 mL). After 1 h, the reaction mixture was concentrated and purified by column chromatography (10 to 20% EtOAc/hexanes) yielding the product (0.80 g, 81%) as a colorless liquid. 1H NMR (CDCl3) δ 9.53 (d, 1H, J = 8.0 Hz), 6.76 (dd, 1H, J = 4.5, 15.5 Hz), 6.21 (ddd, 1H, J = 1.5, 8.0, 15.5 Hz), 4.36 (m, 1H), 3.62 (s, 3H), 2.26 (t, 2H, J = 7.4 Hz), 1.60−1.52 (m, 4H), 1.30−1.20 (m, 8H), 0.87 (s, 9H), 0.02 (s, 3H), −0.01 (s, 3H); 13C NMR (CDCl3) δ 193.6, 174.2, 160.2, 130.6, 71.5, 51.4, 37.0, 34.0, 29.3, 29.0, 28.9, 25.7, 24.8, 24.7, 18.1, −4.7, −5.0; HRMS (ESI) calculated 379.2275 (M + Na), observed 379.2287. To a solution of this aldehyde (0.80 g, 2.2 mmol) in CH3CN (14 mL) was added aqueous HF (0.6 mL, 5% v/v). After 1 h, the reaction mixture was neutralized with saturated NaHCO3 and extracted with EtOAc. The organic layer was washed with brine and dried over MgSO4. The product was isolated as a pale yellow liquid (0.41 g, 76%) after purification by column chromatography (30% EtOAc/hexanes). 1H NMR (CDCl3) δ 9.54 (d, 1H, J = 7.9 Hz), 6.79 (dd, 1H, J = 4.7, 15.7 Hz), 6.27 (ddd, 1H, J = 1.5, 7.9, 15.7 Hz), 4.40 (m, 1H), 3.63 (s, 3H), 2.27 (t, 2H, J = 7.4 Hz), 1.62−1.56 (m, 4H), 1.35−1.25 (m, 8H); 13C NMR (CDCl3) δ 193.6, 174.3, 159.1, 130.6, 71.0, 51.5, 36.4, 34.0, 29.2, 29.0, 28.9, 25.1, 24.8; HRMS (ESI) calculated 265.1410 (M + Na), observed 265.1396.
Synthesis of COOMe-ONE (16)
Dess-Martin periodinane (0.43 g, 1.0 mmol) was added to a solution of 15 (0.20 g, 0.84 mmol) in CH2Cl2 (4 mL). After 1 h, the reaction mixture was concentrated and purified by column chromatography (20% EtOAc/hexanes). The product was isolated as a yellow liquid (0.13 g, 64%). 1H NMR (CDCl3) δ 9.73 (d, 1H, J = 7.0 Hz), 6.84 (d, 1H, J = 16.2 Hz), 6.72 (dd, 1H, J = 7.0, 16.2 Hz), 3.61 (s, 3H), 2.64 (t, 2H, J = 7.3 Hz), 2.25 (t, 2H, J = 7.4 Hz), 1.62−1.54 (m, 4H), 1.35−1.25 (m, 6H); 13C NMR (CDCl3) δ 200.0, 193.4, 174.2, 144.8, 137.3, 51.4, 41.1, 33.9, 28.9, 28.8, 28.7, 24.7, 23.4; HRMS (ESI) calculated 263.1254 (M + Na), observed 263.1266.
Cell Culture
Human colorectal carcinoma (RKO) cells were obtained from American Type Culture Collection (ATCC) (Manassas, VA) and maintained in HEPES buffered DMEM + GlutaMax medium from Invitrogen (Carlsbad, CA), supplemented with 10% fetal bovine serum (FBS) from Atlas Biologicals (Fort Collins, CO), 1× MEM vitamins from Mediatech (Herndon, VA), and 1× antibiotic/antimycotic (Invitrogen), and harvested/lysed as described previously.(4)
Human monocytic leukemia (THP-1) cells were obtained from ATCC and maintained in RPMI 1640 medium (Invitrogen) supplemented with 10% FBS (Atlas Biologicals), 1 mM l-glutamine, 1× antibiotic/antimycotic (Invitrogen), and 1× MEM vitamins from (Mediatech). Cells from suspension cultures were plated at approximately 107 cells/plate in 150 mm plates and treated with 100 nM phorbol myristate acetate (PMA) from Sigma-Aldrich (St. Louis, MO) for 72−96 h to induce differentiation. Cells adhered to the plates and were challenged with various concentrations of lipid electrophile or vehicle control. All lipid electrophiles were prepared as a 1000× stock in DMSO and diluted into cell culture medium, which unless otherwise stated, contained FBS for treatment. Thirty minutes postexposure, cells were washed twice with phosphate buffered saline (PBS) and treated with 50 ng/mL of LPS (Sigma) and 10 U/mL IFNγ for various times. Cells were harvested by scraping and were pelleted at 1,000 rpm for 5 min at 4 °C. Cells were washed twice in ice cold PBS and brought up in 0.2 mL/plate of M-PER extraction buffer from Thermo Scientific (Rockford, IL) + 1% protease inhibitor cocktail (Sigma) for mammalian tissue culture. Samples were left on ice for 20 min and then centrifuged at 10,000g for 15 min, whereupon the supernatant was collected, and protein concentration was determined using the BCA assay (Thermo Scientific).
Cell Viability Assay
RKO cells were maintained as described previously and seeded into 96-well plates (7.5 × 103 cells/well). After 24 h, cells were challenged with various concentrations of lipid electrophile (0−500 μM) in the presence of serum. For serum free experiments, RKO cells were seeded normally for 24 h, but electrophile treatment was done with DMEM medium lacking FBS. Following a 24−48 h treatment, the cells were washed with PBS and incubated with 2 μM calcein-AM from Molecular Probes (Carlsbad, CA) for 30 min. In live cells, calcein-AM is cleaved by an endogenous esterase to produce the fluorescent calcein (λabs= 494; λem = 517). Fluorescence was monitored using a Spectramax Multiwell plate reader (Molecular Devices, Silicon Valley, CA), and cell viability was correlated with fluorescent intensity.
THP-1 cells were maintained as described previously, seeded into 96-well plates (3 × 104 cells/well), and treated with 100 nM PMA for 72−96 h to differentiate. Following differentiation, cells were challenged with the lipid electrophile for 24 h before being read using the calcein AM assay (described above).
Reactivity Index
Half-Life Determination
The half-lives for the series of HNE and ONE analogues was determined by the reaction of each compound with N-acetyl cysteine (NAC) at 37 °C. The consumption of the electrophile was monitored by UV (HNE at 225 nm, ONE at 230 nm), which was fitted with a circulating bath to maintain the temperature of the sample holder at 37 °C. Stock solutions of the electrophiles (34 mM) and N-acetyl cysteine (56 mM) were made up in MeOH or sodium phosphate buffer (50 mM, pH 7.4), respectively. NAC (55 μL) was added to phosphate buffer (3 mL) in a quartz cuvette and used as the blank. To this solution, which was pre-equilibrated at 37 °C, was added the electrophile (5 μL). The final concentrations were 0.056 mM electrophile and 1 mM NAC, maintaining pseudofirst-order conditions. UV analysis was started immediately upon the addition of the electrophile. The absorbance of the HNE analogues was measured every 5 min for 2 h, long enough for complete reaction. Measurements of the ONE analogues were taken every 3 s for 5 min, with the exception of ONEA. Since this compound was considerably slower than the other ONE analogues, measurements were taken every 5 min for 2 h. This experiment was carried out in quadruplicate with fresh stock solutions of NAC. The stability of each compound to the reaction conditions was measured in the same manner but in the absence of NAC.
The UV measurement at each time point was corrected for background absorbance based on the absorbance reading upon complete reaction of the electrophile (at least 5 half-lives). A plot of −ln(At/A0) versus time gives a linear correlation for early time points (prior to the consumption of the electrophile). The first order rate constant was determined from the slope, which was then converted to a half-life. These plots are presented in the Supporting Information.
HPLC Analysis of HNEA Reaction with NAC
The λmax for HNEA shifted from 225 to 210 nm as compared to the parent HNE compound. Since it was possible that NAC or products could absorb in this region, the reaction of HNEA with NAC was also monitored by HPLC. The sample was made up as described above, with the addition of cinnamyl alcohol (0.1 mM) as an internal standard. The reaction was monitored by reverse-phase HPLC (C18 column) using 9:1 MeOH/H2O (0.05% TFA) at a flow rate of 1 mL/min. The amount of HNEA relative to the internal standard was measured at 210 nm every 10 min for 90 min. A plot of the amount of HNEA remaining versus time showed no reaction of the HNEA with NAC, consistent with the UV experiment described above.
Second-Order Rate Constant
The second-order rate constant was determined under the conditions described above except that the reaction was carried out in 100 mM phosphate buffer (pH 7.4). The higher concentration of buffer was necessary to maintain the pH at 7.4 at the higher concentrations of NAC. The consumption of the HNE analogues and ONEA was measured at varying concentrations of NAC ranging from 0.46 to 4.6 mM. Since the ONE analogues react much faster with NAC, these experiments were carried out under more dilute conditions (0.084−0.84 mM NAC) so that the consumption could be more accurately measured. The first order rate constant was determined as described above, then plotted versus the [NAC]. The slope of the latter plot was used to determine the second order rate constant (Figure S3, Supporting Information).
Western Blot
THP-1 cells were grown, differentiated, treated with lipid electrophiles and LPS, and harvested as described above. Samples were quantified by the BCA assay, and 10 μg/well of each sample was run on a 12% (Tris-HCl) SDS gel from Biorad (Hercules, CA). Gels were transferred onto a 0.45 μm nitrocellulose membrane (Biorad), the membrane was blocked for 1 h using 5% nonfat dry milk in 1× TTBS (0.1% Tween 20 + Tris buffered saline), and then reacted with appropriate target antibodies [1:250−1:1000 dilutions in 1× TTBS + 1% nonfat dry milk from Abcam (Cambridge, MA)] overnight at 4 °C. The blot was washed 4 time for 15 min in 1× TTBS, incubated with secondary antibody [1:1,000 dilution from Santa Cruz (Santa Cruz, CA)] for 1 h at room temperature, and washed 3 times for 15 min in 1× TTBS. Blots were visualized using SuperSignal West Pico Chemiluminescent Substrate ECL reagent (Thermo Scientific) and developed on Chemiluminescent-XPosure Clear Blue X-ray Film (Thermo Scientific).
Enyzme-Linked Immunosorbent Assay (ELISA)
All reagents and buffers for the ELISA plate assay were obtained from BD Biosciences (San Jose, CA). For acute exposure experiments, THP-1 cells were treated with DMSO, HNE (0−200 μM), ONE (0−200 μM), HNEA (0−200 μM), or ONEA (0−200 μM) for 30 min, washed twice with PBS, and challenged for various times with LPS (in the absence of electrophiles). For continuous exposure experiments, the experimental design was the same as that with acute exposure; however, lipid electrophiles remained in the media concurrent with LPS activation. One milliliter of conditioned media was removed for sample analysis. All samples were diluted 1:50 or 1:10 in Standard/Sample Diluent Buffer and processed as per BD OptEIA Human IL-6/IL-1β/TNFα ELISA Kit II (#550799, #557966, and #550610) specifications. Samples were quantified using a cytokine specific standard curve (standards provided) that was prepared in parallel. Cytokine levels were determined by measuring absorbance at 450 nm on a Spectramax Multiwell plate reader.
Phagocytosis/Fluorescence Microscopy
THP-1 cells were seeded at 1.5 × 106 cells/well onto glass coverslips (VWR, West Chester, PA) in 6-well tissue culture plates. Cells were differentiated with 100 nM PMA for 72 h, washed, and treated with 10 μM HNE, 0.5 μM ONE, or vehicle for 30 min. Following 30 min of incubation, cells were washed twice with PBS and exposed to opsonized 2.0 μm fluorescent polystyrene latex beads (Fluoresbrite YG Microspheres 2.00 μm, #18338-5; Polysciences Inc., Warrington, PA) in media for 3 or 6 h. One drop (20 μL) of beads was opsonized by incubation with 10% FBS for 60 min at 37 °C. Following exposure to the beads, cells were washed twice with PBS and fixed for 15 min in a 2.5% paraformaldehyde solution. Beads were visualized using a Zeiss Axioplan fluorescence microscope with a green fluorescent protein (GFP) filter.
Results
Synthesis of Lipid Electrophiles
The synthesis of HNE and its ester precursor (7) has been previously described.(2) The more highly oxidized analogues, ONE and ONEA, were synthesized from HNE employing either a Dess-Martin periodinane or Jones oxidation, respectively (Scheme 2).29−32 HNEA was synthesized by simple hydrolysis of ester 7. The carboxy analogues were synthesized utilizing the same strategy as that in HNE synthesis (Scheme 3). Sulfinate 8 was coupled to the appropriate aldehyde to yield α,β-unsaturated ester 9. Protecting group manipulation followed by a Tempo-mediated oxidation yielded the carboxy ester 11. Reduction of the ester with DIBAL-H generated the alcohol 12. This intermediate was used for the synthesis of the carboxy-analogues as well as their corresponding methyl esters. The alcohol was oxidized to the aldehyde using Dess-Martin periodinane. Deprotection with aqueous HF yielded COOH-HNE and further oxidation yielded COOH-ONE. Other common oxidation conditions, such as Swern and PCC, resulted in lower yields of the desired product. In addition, after screening several deprotection conditions, HF deprotection resulted in minimal decomposition of the aldehyde moiety. Intermediate 12 was converted to its corresponding methyl ester using diazomethane. Attempts to esterify using acidic methanol resulted in low yields due to allylic substitution. The same oxidation and deprotection conditions described above were employed to generate the final compounds COOMe-HNE and -ONE.
Scheme 2. Synthesis of HNE, ONE, HNEA, and ONEA.

Reagents: (a) Dess-Martin periodinane, CH2Cl2; (b) NaOH, H2O/MeOH; (c) CrO3, H2O/H2SO4, acetone.
Scheme 3. Synthesis of COOH-HNE, COOH-ONE, and Corresponding Esters.

Reagents: (a) 10-(Tetrahydro-2H-pyran-2-yloxy)decanal, piperidine, CH3CN; (b) TBDMSCl, imidazole, DMF; (c) MgBr2-Et2O, ether; (d) bleach, Tempo, NaBr, Bu4NBr, NaHCO3, CH2Cl2/H2O; (e) DIBAL-H, Et3N, CH2Cl2; (f) Dess-Martin periodinane, CH2Cl2; (g) HFaq, CH3CN; (h) Diazald, KOH, H2O/EtOH, ether.
Lipid Electrophile Toxicity: Cell Viability Assay
The cytotoxicity of each lipid electrophile was determined by performing a cell viability assay and generating a concentration−response curve. Cell viability was measured following a 24−48 h incubation with the compound, using the fluorogenic substrate, calcein-AM. In live cells, calcein-AM is cleaved by an endogenous esterase to produce the fluorescent molecule, calcein. Therefore, cell viability can be directly correlated with fluorescent intensity. IC50 values were calculated from a plot of log of concentration versus the percent of fluorescence as compared to vehicle-treated cells (Figure 1). Each lipid electrophile was assayed for toxicity in two cell types, human colorectal carcinoma, RKO, cells and human acute monocytic leukemia, THP-1, cells. The RKO cells were used in order to compare compound toxicity with previous electrophile studies from our laboratory, and the THP-1 cells were used to focus on the ability of lipid electrophiles to modulate the inflammatory process.
Figure 1.

THP-1 cell viability assay for select lipid electrophiles. THP-1 cells were treated with various concentrations of lipid electrophiles for 24 h, and cell viability was assayed using the fluorescent substrate calcein-AM. IC50 values were determined from a plot of log of electrophile concentration versus percent of live cells as compared to a vehicle (DMSO) control.
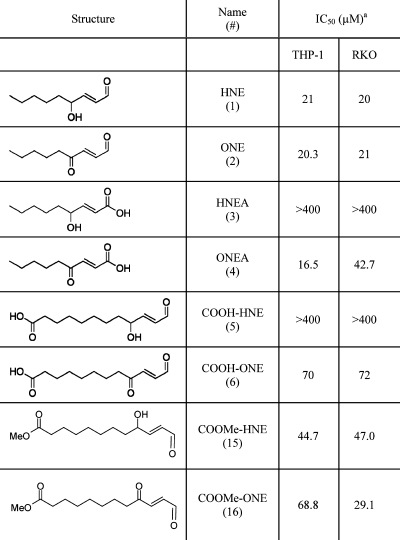
The IC50 values reported in Table 1 indicate that HNE, ONE, ONEA, and COOMe-HNE/ONE all have similar potency with regards to cellular toxicity (IC50 = 20−47 μM) in RKO cells. Previous studies from our laboratory demonstrated the activation of caspase-3 and PARP cleavage, indicative of the induction of apoptosis, at similar concentrations of HNE and ONE.(33) ONEA demonstrates greater than 10-fold higher toxicity than its counterpart HNEA. Additionally, COOH-ONE shows much greater toxicity than its hydroxy HNE analogue COOH-HNE. Conversion of COOH-HNE/ONE to a carboxy-methyl ester led to a decrease in IC50 values for the COOMe derivatives with a more pronounced effect in the HNE derivatives as compared to the ONE derivatives. Increased toxicity of the COOMe derivatives is most likely due to an increased uptake of the neutral molecules, as the charged carboxylic acid moiety can inhibit diffusion across the cell membrane.
Table 1. IC50 Values for Lipid Electrophiles in RKO and THP-1 Cells.
 |
RKO cells were seeded into 96-well plates (7.5 × 103 cells/well). After 24 h, cells were challenged with various concentrations of the lipid electrophile (0−500 μM) in the presence of serum. Following a 24−48 h treatment, the cells were washed with PBS and incubated with 2 μM calcein-AM. Fluorescence was monitored at 517 nm, and cell viability was correlated with fluorescent intensity. THP-1 cells were seeded into 96-well plates (3 × 104 cells/well) and treated with 100 nM PMA for 72−96 h to differentiate. Following differentiation, cells were challenged with the lipid electrophile for 24 h before being read using the calcein AM assay (described above).
The overall trend for the viability assays is that the ONE analogues are more toxic than the HNE analogues. These data corroborate previous literature reports, which argue that ONE is more reactive than HNE. However, in our hands the parent compounds themselves, ONE versus HNE, show little difference in toxicity. Prior reports indicate that HNE and ONE are equitoxic in RKO cells or that ONE is 5-fold more toxic than HNE in human neuroblastoma cells.33,34 To address this issue, we repeated the viability assay for HNE and ONE in the absence of serum to determine if ONE’s apparent toxicity was lowered because of its higher reactivity with serum proteins, as compared to HNE. In RKO cells treated in the absence of serum, HNE and ONE both exhibited greater toxicity with IC50 values of 5 and 9 μM, respectively (data not shown). However, HNE and ONE exhibit similar toxicity in the absence of serum suggesting that serum reactivity does not explain why two electrophiles with very divergent chemical reactivity have nearly identical toxicities.
The IC50 values in THP-1 cells show remarkable concordance with those values obtained for the RKO cells. The toxicity data in THP-1 recapitulates the overall trend observed in the RKO cells with the ONE analogues being more toxic than their respective HNE analogues.
Relative Reactivity of Electrophiles
In order to determine the relative reactivity of the lipid electrophiles, the reaction of each compound with N-acetyl cysteine (NAC) was investigated. Since the reactive moiety of most of the compounds was essentially unchanged from the parent HNE and ONE, we anticipated that these compounds would also undergo a Michael addition with NAC. Upon reaction, the characteristic chromophore due to the α,β-unsaturated carbonyl would be lost, and the consumption could therefore be followed by UV.
The reaction of each electrophile with NAC was carried out in phosphate buffer (pH 7.4) at 37 °C under pseudofirst-order conditions. In addition, the stability of each electrophile to the reaction conditions was investigated in the absence of NAC. The reaction of the electrophile was monitored by UV at 225 or 230 nm for HNE or ONE, respectively. Upon reaction with NAC, the absorbance at these wavelengths decreased until the electrophile had been completely consumed. The reaction was continued until at least 5 half-lives had been reached (see Supporting Information). In most cases, some background absorbance remained; therefore, the data were corrected for this residual absorbance.
The UV absorbance for most of the analogues was similar to those of their parent compound. However, the λmax for HNEA was shifted from 225 to 210 nm. Since it is possible that the NAC or Michael adducts could also absorb in that region, the reaction of HNEA with NAC was also analyzed by HPLC. The sample was made up in phosphate buffer as described above, with the addition of an internal standard. The amount of HNEA relative to the standard was measured over time.
The stability and reactivity of each electrophile is presented in Figure 2. All of the compounds were stable to the reaction conditions, represented by the open symbols in the figure. Upon addition of NAC, all of the electrophiles except HNEA reacted, resulting in a decrease in the UV absorbance (closed symbols). COOMe-HNE and COOMe-ONE (squares) exhibited reactivities similar to those of their parent compounds (triangles), while COOH-HNE and COOH-ONE (circles) were slightly less reactive. Substitution of the aldehyde moiety with a carboxylic acid had a significant impact on the reactivity of the HNEA and ONEA with NAC. HNEA, as analyzed by UV and HPLC, showed no reaction. Although the ONEA reacted with NAC, its reactivity was much slower than that of ONE and was comparable to that of the HNE analogues.
Figure 2.

Stability (open symbols) and reactivity (closed symbols) of HNE and ONE analogues. (A) HNE with (filled red triangle) and without (open red triangle) NAC, COOH-HNE with (filled blue circle) and without (open blue circle) NAC, COOMe-HNE with (filled black square) and without (open black square) NAC; (B) ONE with (filled red triangle) and without (open red triangle) NAC, COOH-ONE with (filled blue circle) and without (open blue circle) NAC, COOMe-ONE with (filled black square) and without (open black square) NAC; (C) HNEA with (filled blue circle) and without (open red triangle) NAC; (D) ONEA with (filled blue circle) and without (open red triangle) NAC. Reactions were carried out with 0.056 mM electrophile and 1 mM NAC in phosphate buffer (50 mM, pH 7.4) at 37 °C. The consumption of the electrophile was monitored by UV absorbance at 225 and 230 nm for HNE and ONE, respectively.
First-order plots (see Supporting Information) of the data in Figure 2 were used to calculate the half-lives for the series of HNE and ONE analogues at 1 mM NAC. The second-order rate constant for each electrophile was determined from plots of the k1 at varying concentrations of NAC (data presented in Supporting Information). The half-lives and second-order rate constants for each electrophile are presented in Table 2.
Table 2. Half-Lives and Second Order Rate Constants for the Reaction of HNE, ONE, and their Analogues with N-Acetyl Cysteine.
| electrophile | half-life (s)a | k (M−1 s−1)b |
|---|---|---|
| HNE | 732 (±44) | 1.22 (±0.08) |
| HNEA | NR | NR |
| COOH-HNE | 1213 (±116) | 0.79 (±0.11) |
| COOMe-HNE | 794 (±48) | 1.18 (±0.08) |
| ONE | 7.1 (±0.6) | 238 (±13) |
| ONEA | 580 (±40) | 1.72 (±0.28) |
| COOH-ONE | 13.9 (±0.7) | 169 (±7) |
| COOMe-ONE | 7.1 (±0.5) | 301 (±9) |
Half-life determined in the presence of 1 mM NAC in phosphate buffer (50 mM, pH 7.4) at 37 °C.
Rate constant determined in the presence of varying concentrations of NAC in phosphate buffer (100 mM, pH 7.4) at 37 °C.
Effect of Continuous HNE Exposure on Cytokine Expression
Previous literature reports have shown that low concentrations of HNE (1−10 μM) can inhibit the production of various pro-inflammatory cytokines, IL-6, TNFα, and IL-1β, in several macrophage cell lines.22,35 To recapitulate these experiments in our model system, THP-1 macrophages were pretreated with various concentrations of HNE for 30 min before the addition of LPS/IFNγ and 6 h of activation. Medium was collected from the treated cells and assayed by ELISA for the expression of the pro-inflammatory cytokines IL-1β and IL-6. Under continuous HNE exposure, IL-1β exhibited a biphasic decline of expression whereby at low concentrations of HNE (0.01−0.5 μM), there was a gradual loss of cytokine expression to about 80% of DMSO control. At higher concentrations, there was a dramatic loss in IL-1β expression with complete inhibition at concentrations greater than 25 μM (Figure 3A). Measurement of IL-6 expression showed similar results with minimal loss of IL-6 expression at low concentrations of HNE (0.1−1 μM) followed by a sharp decline in cytokine expression above 5 μM HNE (Figure 3B). The sharp decline in cytokine expression at doses exceeding 5 μM HNE cannot be explained by a loss in cell viability. Previous studies from our laboratory evaluating HNE induction of apoptosis as measured by PARP cleavage and caspase-3 activation showed no evidence of apoptosis up to 8 h after treatment with 45 μM HNE.(33)
Figure 3.
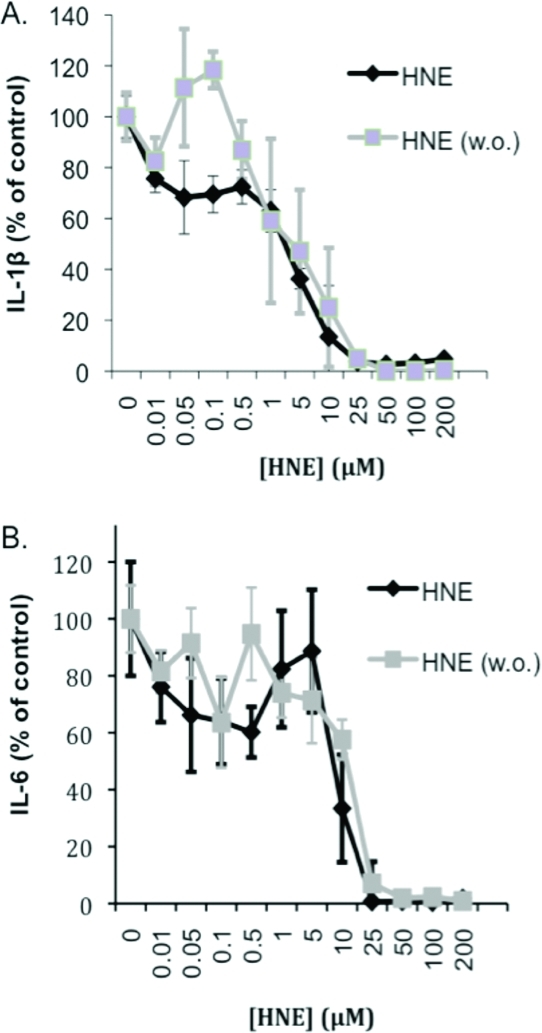
Effect of continuous vs transient HNE treatment on cytokine expression during LPS/IFNγ challenge. THP-1 cells were pretreated with various concentrations of HNE for 30 min, followed by either leaving in or washing out (w.o.) the electrophile, and 6 h of LPS challenge. HNE was assayed for its ability to inhibit IL-1β (A) and IL-6 (B) expression.
Inhibition of Pro-Inflammatory Cytokines after Acute HNE or ONE Exposure
Although no evidence of cell death was found which could account for the loss of cytokine expression during continuous HNE exposure, we chose to modify our protocol to eliminate any potential for toxicity during the course of the experiment. To this end, all cytokine experiments were conducted by treating THP-1 cells for short periods of time with various concentrations of lipid electrophile, washing with PBS to remove any electrophile, and then challenging the cells with LPS/IFNγ. We monitored the cytokines IL-6, IL-1β, and TNFα because of their key role in the progression/activation of the macrophage inflammatory response and because of previous literature reports which indicate a role for HNE and oxPLs in their inhibition.21−23 ELISAs specific for each of the pro-inflammatory cytokines were used to analyze the conditioned medium from THP-1 macrophages treated with various concentrations of HNE, ONE, and vehicle control. Briefly, the assay was a solid phase sandwich ELISA in which a monoclonal antibody, specific to the cytokine of interest, was coated on a 96-well plate. Any target protein in the conditioned medium was immobilized by binding to the antibody on the plate. The plate was then washed and developed by adding a second biotinylated antibody and a streptavidin−horseradish peroxidase conjugate. The ELISA is sensitive for an individual cytokine and utilizes a standard curve for quantification of the specific cytokine. Preliminary studies using both ELISAs and Western blotting were used to assay THP-1 macrophages exposed to subcytotoxic doses of HNE and ONE for varying periods of time (5 min−1 h) before washout and LPS/IFNγ challenge. From this work, it was determined that subcytotoxic doses of electrophiles could efficiently inhibit cytokine production in as little as 30 min of pretreatment (data not shown). Therefore, 30 min of pretreatment with electrophile was used for our cytokine inhibition experiments.
After 30 min of pretreatment with 5 or 10 μM HNE or ONE followed by washout, there was a 20−60% decrease in the levels of IL-6 in the medium following 6 h of LPS challenge (Figure 4A). It is interesting to note that ONE afforded less cytokine inhibition than HNE at both the 5 and 10 μM concentrations, even though both compounds exhibit similar toxicity profiles. Under similar conditions, HNE inhibited LPS-induced TNFα by 30% at 5 μM and 40% at 10 μM (Figure 4C). HNE was also assayed for its ability to inhibit IL-1β over a 24 h time course. HNE repressed LPS induction of IL-1β expression by up to 80% 24 h after HNE pretreatment and washing (Figure 4B). These data suggest that HNE works rapidly to modify a putative target(s), which mediates the inhibition of IL-1β expression. This inhibitory effect persists for 24 h after HNE is removed from the cells. These data indicate that HNE has the strongest inhibitory effect on IL-1β production, followed by IL-6, and to a lesser extent TNFα.
Figure 4.

Inhibition of cytokine expression by HNE and ONE. IL-6 ELISA of conditioned media from THP-1 macrophages pretreated with HNE or ONE (30 min), washed, and challenged with LPS/IFNγ for 6 h (A). IL-1β (B) or TNFα (C) ELISA of media from THP-1 cells pretreated with HNE for 30 min, washed, and challenged with LPS for the times indicated (6 h LPS challenge for TNFα ELISA).
A dose−response curve was generated for IL-1β and IL-6 expression using the 30 min electrophile treatment protocol to compare with the dose−response curve resulting from continuous HNE exposure (Figure 3A and B). Unlike continuous HNE exposure, the IL-1β levels in the acute HNE treatment condition showed no decrease at low concentrations (0.01−0.5 μM) of HNE. However, at higher concentrations of HNE there was comparable loss of cytokine expression between the continuous and transient treatment conditions. Transient HNE exposure produced a similar curve for the inhibition of IL-6 synthesis as compared to continuous exposure whereby lower concentrations of HNE (0.01−5 μM) afforded minimal inhibition of expression, and higher concentrations (10 μM or higher) produced a steep decline in levels of IL-6.
Viability assays were performed using comparable conditions to 30 min of electrophile treatment to determine if cell death caused the cytokine inhibition. THP-1 macrophages were treated for 30 min with HNE, washed, and their viability assayed after a 24 h incubation. No toxicity was observed at 5−10 μM HNE and less than 25% cell death was observed at concentrations of HNE >100 μM. Therefore, cell death during the 6 h of LPS activation cannot account for the dramatic decrease in cytokine expression that is observed with higher concentrations of HNE (data not shown).
Inhibition of IL-6 Expression by HNE
ELISA assays measure only secreted proteins; therefore, it was necessary to recapitulate these results by using Western blot to assay cytokines from cell lysates. THP-1 cells were treated with HNE at concentrations of 0−10 μM for 5−30 min time periods before being removed by washing twice with PBS. Cells were then challenged with LPS/IFNγ for 6 h to induce an inflammatory response. Western blots were used to probe cell lysates for lipid electrophile-mediated changes in the pro-inflammatory cytokine, IL-6. Partial inhibition of IL-6 induction by LPS was observed at the 5 μM dose of HNE, and complete inhibition was observed at the 10 μM dose for 30 min (Figure 5). These results confirm that the changes in IL-6 cytokine levels, seen in the ELISA assays, are due to the inhibition of cytokine expression rather than the electrophile-mediated inhibition of cytokine secretion.
Figure 5.

IL-6 Western blot of cell lysate from THP-1 macrophages pretreated with HNE, washed, and challenged with LPS for 6 h. The lysate was probed with either anti-IL-6 or antiactin antibodies.
HNE Inhibition of IL-6 Is Dependent on the Duration of Incubation Between Electrophile Washout and LPS challenge
To examine whether the effect of HNE on cytokine inhibition was transient, THP-1 macrophages were treated with 10 μM HNE or vehicle control for 30 min, and then, the electrophile was washed out. The cells were either immediately challenged with LPS/IFNγ or incubated for 30 min or 3 h before challenge. After 6 h of LPS treatment, the cell lysate was examined by Western blot for levels of IL-6. When cells were pretreated with HNE, washed, and immediately challenged with LPS for 6 h, there was a complete repression of IL-6 expression. However, if LPS challenge was delayed for 30 min there was a 50% reduction in IL-6 repression, and after delaying LPS challenge for 3 h, there was no observable repression of IL-6 synthesis (Figure 6).
Figure 6.

Recovery of IL-6 synthesis by delaying the time between LPS challenge and HNE treatment. Western blot for IL-6 from THP-1 macrophage cell lysate. Cells were pretreated with 10 μM HNE or the vehicle control for 30 min followed by washing with PBS. Following washout of the electrophile, cells were challenged with LPS/IFN-γ after an incubation time of 0, 0.5, or 3 h. The cell lysate was probed with either anti-IL-6 or anti-actin antibodies.
Dependence of Cytokine Inhibition on Lipid Electrophile Reactivity
To test whether there was a correlation between electrophile reactivity and cytokine inhibition, several pairs of HNE/ONE analogues were assayed for their ability to inhibit cytokine synthesis over a broad concentration range. The most divergent pair of analogues in terms of reactivity, HNEA and ONEA, were assayed for their ability to inhibit IL-1β, IL-6, and TNFα. THP-1 macrophages were treated with HNEA/ONEA for 30 min, the compounds were removed by washing, and then the cells were challenged for 6 h with LPS/IFNγ. At low concentrations, 0−1 μM HNEA or ONEA, there was a decrease in IL-1β levels to approximately 40% of the vehicle control (Figure 7 A). However, at concentrations of 5 μM and above the compounds diverge in terms of their ability to inhibit IL-1β production. Increasing concentrations of ONEA led to a complete loss of IL-1β expression at a concentration of 50 μM, whereas increasing concentrations of HNEA showed no effect as IL-1β returned to control levels. Levels of IL-6 remained relatively unaffected by both HNEA and ONEA at concentrations of 0−1 μM (Figure 7B). At concentrations of ONEA greater than 5 μM, there was a decline in IL-6 expression with a complete loss of expression at 200 μM. No change in IL-6 expression was apparent with concentrations of HNEA up to 200 μM. At low concentrations of HNEA (0−1 μM), TNFα expression was inhibited in a manner similar to that in IL-1β, with loss of expression to approximately 50% of the control (Figure 7C). However, at higher concentrations of HNEA (>5 μM) there was no apparent inhibition of TNFα expression. ONEA afforded no inhibition of TNFα at concentrations up to 50 μM but produced a steep decline in expression at concentrations above 50 μM. For these two compounds, there seems to be a correlation between electrophile reactivity and cytokine inhibition. At concentrations of 5 μM and greater, the reactive ONEA had a greater inhibitory effect on pro-inflammatory cytokines as compared to HNEA, which exhibited little cytokine inhibition and no reactivity.
Figure 7.

Inhibition of cytokine expression following acute exposure to HNEA and ONEA. IL-1β ELISA (A), IL-6 ELISA (B), and TNFα ELISA (C) of conditioned media from THP-1 macrophages pretreated with HNEA or ONEA (30 min), washed, and challenged with LPS/IFNγ for 6 h.
COOMe-ONE is approximately 100-fold more reactive than its analogue COOMe-HNE. However, both compounds display reactivity and toxicity nearly identical to those of their respective parent compounds. Therefore, these compounds were assayed for their ability to inhibit cytokine synthesis to determine if their inhibitory properties mimicked their respective parent compounds and to determine if there was a similar correlation between reactivity and cytokine inhibition as seen with the HNEA and ONEA compounds. With the exception of TNFα, the COOMe-HNE and COOMe-ONE compounds gave similar cytokine inhibition profiles. There was minimal loss of cytokine expression (less than 20%) at concentrations of 0−10 μM, and at higher concentrations, there was a steep decline in cytokine expression whereby cytokine expression was nearly zero at concentrations of electrophile greater than 25 μM (Figure 8A and B). In the case of TNFα, COOMe-HNE was more active than COOMe-ONE and afforded greater than 40% inhibition at concentrations of 0.5 μM and higher, while COOMe-ONE required 10-fold higher concentrations to produce a similar effect (Figure 8C). Strictly on the basis of reactivity profiles, there does not appear to be any correlation between reactivity and cytokine inhibition as the COOMe-ONE is 100-fold more reactive than COOMe-HNE, but both compounds exhibit similar cytokine inhibitory properties. Additionally, both the COOMe derivatives are less effective at inhibiting cytokines as compared to their respective parent compounds under comparable conditions. At 10 μM HNE, cytokine inhibition for all three cytokines is 40−60%, whereas at 10 μM COOMe-HNE, there is only 10−20% cytokine inhibition. Taken as a whole, these data suggest that there is no simple correlation between electrophile reactivity, toxicity, or anti-inflammatory activity.
Figure 8.

Inhibition of cytokine expression following treatment with COOMe-HNE and COOMe-ONE. IL-1β ELISA (A), IL-6 ELISA (B), and TNFα ELISA (C) of conditioned media from THP-1 macrophages pretreated with COOMe-HNE or COOMe-ONE (30 min), washed, and challenged with LPS/IFNγ for 6 h.
Inhibition of THP-1 Macrophage Phagocytosis by HNE and ONE
Cytokine expression is only one method of assessing macrophage activation and the inflammatory response; therefore, we also analyzed changes in the ability of THP-1 macrophages to phagocytose fluorescent beads following pretreatment with HNE or ONE. Phagocytosis is critical to the activation of the immune response because it leads to antigen presentation on the cell surface of the ingesting macrophage and subsequent activation of other macrophages and immune cells. Macrophage activation was assayed by using fluorescence microscopy to visualize the phagocytosis of fluorescent beads by differentiated THP-1 macrophages following 30 min of lipid electrophile treatment and removal (Figure 9 A). HNE and ONE inhibited phagocytosis by 20−40% after 3 h; however by 6 h, inhibition of phagocytosis was observed only in those cells pretreated with HNE (Figure 9 B). Importantly, the conditions of electrophile dosage and treatment duration used in these experiments are identical to those conditions found to inhibit cytokine production.
Figure 9.

Inhibition of phagocytosis by THP-1 macrophages following pretreatment with lipid electrophiles. Fluorescent microscopy images (GFP Filter) of THP-1 macrophage phagocytosis following 30 min of pretreatment with the DMSO vehicle, 10 μM HNE, or 0.5 μM ONE and washout with PBS. Phagocytosis of fluorescent polystyrene latex beads after 3 h (A). Quantification of fluorescent bead uptake by THP-1 macrophages following pretreatment with electrophiles, washout, and exposure to fluorescent beads for 3 or 6 h (B). * p-value <0.05; **p-value <0.01 as compared to the 3 h and 6 h vehicle (DMSO) control, respectively.
Discussion
Lipid electrophiles, produced enzymatically or under conditions of oxidative stress, are significant mediators of intracellular signaling. The best characterized of these electrophile mediators is HNE, which activates and inhibits complex networks of cell signaling and gene expression, induces the intrinsic and extrinsic pathways of apoptosis, and modulates the inflammatory response at the cellular level. HNE has been shown to inhibit production of the pro-inflammatory cytokines, TNFα, IL-1β, and IL-6 in several monocytic/macrophage cell lines.21−23 Here, we defined the toxicity profile, the relative reactivity, and the anti-inflammatory properties of a number of previously uncharacterized HNE analogues resulting from phospholipid oxidation. Additionally, we characterized the anti-inflammatory properties of another reactive electrophile, ONE, and its analogues.
Viability data for HNE, ONE, and their analogues indicate that with the exception of the parent compounds, the ONE series of derivatives are more toxic than their respective HNE analogues. HNEA exhibited no toxicity under any conditions, which is consistent with previous studies in neuroblastoma cells where the compound did not produce cell death when used in concentrations exceeding 300 μM.(18) In contrast, ONEA had toxicity comparable to that of the parent compound ONE. This loss of toxicity results from the replacement of the aldehyde with the more electron-rich carboxylate anion, which deactivates the C2=C3 double bond of HNEA toward Michael additions with thiol or amino groups.(36) ONEA is less affected by this substitution because it retains an α,β-unsaturated ketone which serves as a Michael acceptor.(19) COOH-HNE and COOH-ONE exhibit a similar divergence in toxicity as compared to the HNEA/ONEA analogues. Both compounds exhibit lower toxicity overall relative to the parent compounds, primarily due to the charge on the carboxylic acid moiety, which inhibits entry of the compounds into the cell. While it is unclear whether charge state completely explains the divergence in toxicity or if the difference in toxicity is due to COOH-ONE mediating its toxicity via a unique mechanism at the cell surface, there is strong evidence that charge state plays a critical role in compound toxicity as the methyl ester derivatives of ONE and HNE have toxicity similar to that of each other and the parent compounds.
The reactivity profiles for each electrophile with N-acetyl cysteine followed trends similar to those of the toxicity data and are consistent with those values reported in the literature.37,38 In general, the ONE compounds were both more reactive and toxic than their HNE analogues. Additionally, the COOMe-HNE/ONE derivatives demonstrated comparable reactivity to those of the parent compounds, which is in agreement with the similarity in toxicity data between these analogues. HNEA exhibited no measurable activity with NAC, which is also consistent with its lack of toxicity and previous data showing that HNEA does not form protein adducts.(18) The one main inconsistency between the toxicity data and the relative reactivity data is for the parent compounds, HNE and ONE. ONE is greater than 100-fold more reactive than HNE, and yet both compounds are identical in terms of IC50 values. Previous work in neuroblastoma cells has shown ONE to be as much as 31 times more reactive with proteins than HNE and has also demonstrated 4−5 times greater toxicity with ONE as compared to HNE.(34) The higher reactivity but similar toxicity of ONE to HNE in our system could be caused by a number of factors. It has been suggested that ONE is highly reactive such that its effective concentration could be reduced (i.e., by reacting with FBS in the media) before ever entering the cell. Viability assays conducted in our laboratory using RKO cells treated with HNE and ONE in the presence or absence of serum demonstrate that this is not the case, as both HNE and ONE treated cells exhibit a similar 5-fold decrease in their IC 50 values in the absence of serum, suggesting that serum reactivity does not account for differences in reactivity but similarities of toxicity. Other possible explanations for these results are (1) ONE is removed more effectively by detoxifying mechanisms within the cell, such as spontaneous or catalyzed reactions with glutathione or via electrophile metabolizing enzymes. There is precedent in the literature for detoxifying enzymes such as aldehyde dehydrogenase that demonstrate selectivity in their ability to neutralize reactive electrophiles.(39) (2) In any lipid electrophile-mediated signaling event, the biological activity represents the rate-limiting step. For many signaling events, only a subpopulation of proteins must be modified in order to mediate downstream effects. The reaction of HNE and ONE with a given protein, while a 100-fold different in rate, is likely to occur more rapidly than the expression of transcription factors and changes in downstream gene expression. Thus, the rate-limiting step in the cellular response is likely to be biological rather than chemical. (3) ONE and HNE have greater than 100-fold difference in reactivity toward cysteine residues. However, this difference in the ratio of reactivity is less dramatic when considering reactivity with histidine and lysine residues. Additionally, there is no correlation between rapid electrophile−protein residue reactivity and toxicity.(40) The toxicity is dependent more on what role the modified residue has in the proper folding and functioning of the protein.(40)
All of the electrophiles assayed (with the exception of HNEA) were able to inhibit the production of the pro-inflammatory cytokines IL-1β, IL-6, and TNFα to varying degrees. These data are consistent with literature reports that describe inhibition of the production of the same cytokines in LPS-activated monocytes treated with HNE or oxPLs. There was also a general trend toward greater cytokine inhibition in cells treated with the more toxic/reactive electrophiles (i.e., HNE, ONEA, and COOMe-ONE). With regards to individual cytokines, IL-1β expression was most affected by treatment with lipid electrophiles, whereas TNFα expression was least affected by electrophile treatment. Inhibition of IL-1β expression could be observed with lower concentrations of the electrophile, as in the cases of ONEA, HNE, and COOMe-ONE where 20−60% inhibition was observed following treatment with 1 μM or less of the compound. Taken as a whole, these data that characterize diffusible lipid electrophiles provide an interesting contrast to membrane bound oxidized phospholipids, which can promote pro-inflammatory signaling. Oxidized PLs with HNE/ONE-like carbonyl groups at the sn-2 position can be generated in the plasma membrane and promote macrophage activation and inflammation. These oxPLs undergo a conformational change that shifts hydrophobic portions of their fatty acyl chains from the interior lipid bilayer to the external aqueous environment. This phenomenon is referred to as “growing (lipid) whiskers”.20,41,42 OxPL whiskers may permit an interaction with macrophage CD36 scavenger receptors leading to their activation and a pro-inflammatory response. At this time, it is unclear whether the pro- vs anti-inflammatory behavior of lipid peroxidation products is related to their location in the membrane or ability to diffuse throughout the cell.
Dose−response curves were generated to assay cytokine expression in response to acute or chronic HNE exposure. Consistent with our other findings, IL-1β expression shows the most sensitivity to electrophile treatment conditions (acute vs chronic). Under conditions of continuous treatment with the electrophile, there appear to be two phases in the concentration dependence for inhibition of cytokine production, one at lower concentrations (0.01−1 μM) and another at higher concentrations (greater than 5 μM) that is not seen with transient treatment conditions or with IL-6 under transient or continuous conditions. These data suggest the possibility of HNE reversibly binding to a high-affinity target associated with IL-1β inhibition. Such binding/inhibition could be readily reversible and thereby be lost upon removal of electrophile during washing. At higher concentrations of HNE, a second lower affinity and more slowly reversible target may be accessed which could play a greater role in inhibiting IL-1β expression. The fact that HNEA inhibits cytokine expression at low concentrations, especially for TNFα expression, suggests that covalent modification of a protein target may not be essential to inhibit expression.
For transient HNE exposure, IL-6 cytokine expression is impacted by the duration of incubation between electrophile washout and LPS challenge. Loss of IL-6 repression by delaying LPS challenge suggests that the HNE-protein adduct(s) responsible for the inhibition of IL-6 have a short half-life. Whether this short half-life is due to active removal/degradation of the HNE-protein adduct by cellular machinery or rapid reversibility of the HNE-protein adduct is unclear.
There are several unanswered questions that need to be explored relating to the mechanism(s) by which lipid electrophiles mediate the inhibition of pro-inflammatory cytokines and whether individual electrophiles mediate their effects by activating the same cellular signaling pathways. To address these issues, we and our collaborators are using a click chemistry based approach whereby THP-1 cells are treated with alkynylated electrophiles and reacted with an azido compound containing an affinity tag to enrich for protein targets of individual electrophiles. This method will allow us to generate an inventory of proteins that are modified by individual electrophiles. Inventories from electrophiles with different biological properties may provide insights into the molecular targets responsible for their biological activities.
Acknowledgments
We thank Dr. W. Gray Jerome for assistance with the fluorescent microscopy studies.
Glossary
Abbreviations:
- OxPL
oxidized phospholipids
- SAR
structure−activity relationship
- HNE
4-hydroxynonenal
- ONE
4-oxononenal
- NAC
N-acetyl cysteine
- COOH-HNE/ONE
carboxy-HNE/ONE
- COOMe-HNE/ONE
carboxy methyl-HNE/ONE
- HNEA/ONEA
HNE/ONE acid
- LPS
lipopolysaccharide
- IFNγ
interferon gamma
- IL-6/1β
interleukin-6/1β
- TNFα
tumor necrosis factor alpha
- PC
phosphatidylcholine
- PUFA
polyunsaturated fatty acid.
This work was supported, in whole or in part, by National Institutes of Health Grant P01ES013125 from NIEHS (Program Project Grant) and by the National Institutes of Health Training Grant in Environmental Toxicology T32-ES007028.
Supporting Information Available
Reaction of HNE, ONE, and their analogues with NAC; first order plots for HNE, ONE, and their analogues at 1 mM NAC (n = 4); second order plots for HNE, ONE, and their analogues (n = 3). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Both authors contributed equally to this work.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Niki E. (2009) Lipid peroxidation: Physiological levels and dual biological effects. Free Radical Biol. Med. 47, 469–484. [DOI] [PubMed] [Google Scholar]
- Vila A.; Tallman K. A.; Jacobs A. T.; Liebler D. C.; Porter N. A.; Marnett L. J. (2008) Identification of protein targets of 4-hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem. Res. Toxicol. 21, 432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A. T.; Marnett L. J. (2007) Heat shock factor 1 attenuates 4-hydroxynonenal-mediated apoptosis: Critical role for heat shock protein 70 induction and stabilization of Bcl-XL. J. Biol. Chem. 282, 33412–33420. [DOI] [PubMed] [Google Scholar]
- Jacobs A. T.; Marnett L. J. (2009) HSF1-mediated BAG3 expression attenuates apoptosis in 4-hydroxynonenal-treated colon cancer cells via stabilization of anti-apoptotic Bcl-2 proteins. J. Biol. Chem. 284, 9176–9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J. D.; Marnett L. J. (2005) Alterations in gene expression induced by the lipid peroxidation product, 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 18, 1642–1653. [DOI] [PubMed] [Google Scholar]
- Uchida K.; Shiraishi M.; Naito Y.; Torli Y.; Nakamura Y.; Osawa T. (1999) Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J. Biol. Chem. 274, 2234–2242. [DOI] [PubMed] [Google Scholar]
- Nakashima I.; Liu W.; Akhand A.; Takeda A.; Kawamoto Y.; Kato M.; Suzuki H. (2003) 4-Hydroxynonenal triggers multistep signal transduction cascades for suppression of cellular functions. Mol. Aspects Med. 24, 231–238. [DOI] [PubMed] [Google Scholar]
- Awasthi Y. C.; Sharma R.; Sharma A.; Yadav S.; Singhal S. S.; Chaudhary P.; Awasthi S. (2008) Self-regulatory role of 4-hydroxynonenal in signaling for stress-induced programmed cell death. Free Radical Biol. Med. 45, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone D. L.; Doorn J. A.; Kiebler Z.; Ickes B. R.; Petersen D. R. (2005) Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J. Pharmacol. Exp. Ther. 315, 8–15. [DOI] [PubMed] [Google Scholar]
- Carbone D. L.; Doorn J. A.; Kiebler Z.; Sampey B. P.; Petersen D. R. (2004) Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 17, 1459–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S.; Zhang R.; Greenberg M. E.; Sun M.; Chen X.; Levison B. S.; Salomon R. G.; Hazen S. L. (2006) Phospholipid hydroxyalkenals, a subset of recently discovered endogenous CD36 ligands, spontaneously generate novel furan-containing phospholipids lacking CD36 binding activity in vivo. J. Biol. Chem. 281, 21298–31308. [DOI] [PubMed] [Google Scholar]
- Schaur R. J. (2003) Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol. Aspects Med. 4−5, 149–159. [DOI] [PubMed] [Google Scholar]
- Esterbauer H.; Schaur R. J.; Zollner H. (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biol. Med. 11, 81–128. [DOI] [PubMed] [Google Scholar]
- Lee S. H.; Blair I. A. (2000) Characterization of 4-oxo-2-nonenal as a novel product of lipid peroxidation. Chem. Res. Toxicol. 13, 698–702. [DOI] [PubMed] [Google Scholar]
- Siems W. G.; Pimenov A. M.; Esterbauer H.; Grune T. (1998) Metabolism of 4-hydroxynonenal, a cytotoxic lipid peroxidation product, in thymocytes as an effective secondary antioxidative defense mechanism. J. Biochem. 123, 534–539. [DOI] [PubMed] [Google Scholar]
- Ullrich O.; Grune T.; Henke W.; Esterbauer H.; Siems W. G. (1994) Identification of metabolic pathways of the lipid peroxidation product 4-hydroxynonenal isolated from rat kidney cortex. FEBS Lett. 352, 84–86. [DOI] [PubMed] [Google Scholar]
- Murphy T. C.; Poppe C.; Porter J. E.; Montine T. J.; Picklo M. J. (2004) 4-Hydroxy-trans-2-nonenoic acid is a γ-hydroxybutyrate receptor ligand in the cerebral cortex and hippocampus. J. Neurochem. 89, 1462–1470. [DOI] [PubMed] [Google Scholar]
- Murphy T. C.; Amarnath V.; Picklo M. J. (2003) Mitochondrial oxidation of 4-hydroxy-2-nonenal in rat cerebral cortex. J. Neurochem. 84, 1313–1321. [DOI] [PubMed] [Google Scholar]
- Doorn J. A.; Hurley T. D.; Petersen D. R. (2006) Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon-2-enal and 4-oxonon-2-enal. Chem. Res. Toxicol. 19, 102–110. [DOI] [PubMed] [Google Scholar]
- Podrez E. A.; Poliakov E.; Shen Z.; Zhang R.; Deng Y.; Sun M.; Finton P. J.; Shan L.; Febbraio M.; Hajjar D. P.; Silverstein R. L.; Hoff H. F.; Salomon R. G.; Hazen S. L. (2002) A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J. Biol. Chem. 277, 38517–38523. [DOI] [PubMed] [Google Scholar]
- Page S.; Fischer C.; Baumgartner B.; Haas M.; Kreusel U.; Loid B.; Hayn M.; Ziegler-Heitbrock H. W. L.; Neumeier D.; Brand K. (1999) 4-Hydroxynonenal prevents NF-kappaB activation and tumor necrosis factor expression by inhibiting IkappaB phosphorylation and subsequent proteolysis. J. Biol. Chem. 274, 11611–11618. [DOI] [PubMed] [Google Scholar]
- Marantos C.; Mukaro V.; Ferrante J.; Hii C.; Ferrante A. (2008) Inhibition of the lipopolysaccharide-induced stimulation of the members of the MAPK family in human monocytes/macrophages by 4-hydroxynonenal, a product of oxidized omega-6 fatty acids. Am. J. Pathol. 173, 1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C.; Kennedy S.; Spickett C. M.; Webb D. J. (2008) Oxidized phospholipid inhibition of toll-like receptor (TLR) signaling is restricted to TLR2 and TLR4. J. Biol. Chem. 283, 24748–24759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero M. R.; Carretero J. C. (2003) Intramolecular Pauson-Khand reactions of α,β-unsaturated esters and related electron-deficient olefins. J. Org. Chem. 68, 2975–2978. [DOI] [PubMed] [Google Scholar]
- Kurangi R. F.; Tilve S. G.; Blair I. A. (2006) Convenient and efficient syntheses of 4-hydroxy-2(E)-nonenal and 4-oxo-2(E)-nonenal. Lipids 41, 877–880. [DOI] [PubMed] [Google Scholar]
- Soulère L.; Queneau Y.; Doutheau A. (2007) An expeditious synthesis of 4-hydroxy-2(E)-nonenal (4-HNE), its dimethyl acetal and of related compounds. Chem. Phys. Lipids 150, 239–243. [DOI] [PubMed] [Google Scholar]
- Ballini R.; Bosica G. (1998) Synthesis of (E)-4-oxonon-2-enoic acid, a natural antibiotic produced by streptomyces olivaceus. J. Nat. Prod. 61, 673–674. [DOI] [PubMed] [Google Scholar]
- Cryle M. J.; Ortiz de Montellano P. R.; De Voss J. J. (2005) Cyclopropyl containing fatty acids as mechanistic probes for cytochromes P450. J. Org. Chem. 70, 2455–2469. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Salomon R. G. (1998) Total synthesis of γ-hydroxy-α,β-unsaturated aldehydic esters of cholesterol and 2-lysophosphatidylcholine. J. Org. Chem. 63. [Google Scholar]
- Deng Y.; Salomon R. G. (2000) Total synthesis of oxidized phospholipids.3. The (11E)-9-hydroxy-13-oxotridec-11-enoate ester of 2-lysophosphatidylcholine. J. Org. Chem. 65, 6660–6665. [DOI] [PubMed] [Google Scholar]
- Sun M.; Deng Y.; Batyreva E.; Sha W.; Salomon R. G. (2002) Novel bioactive phospholipids: practical total syntheses of products from the oxidation of arachidonic and linoleic esters of 2-lysophosphatidylcholine. J. Org. Chem. 67. [DOI] [PubMed] [Google Scholar]
- Gu X.; Gugiu M.; Hazen S. L.; Crabb J. W.; Salomon R. G. (2003) Oxidatively truncated docosahexaenoate phospholipids: total synthesis, generation, and peptide adduction chemistry. J. Org. Chem. 68. [DOI] [PubMed] [Google Scholar]
- West J. D.; Ji C.; Duncan S. T.; Amarnath V.; Schneider C.; Rizzon C. J.; Brash A. R.; Marnett L. J. (2004) Induction of apoptosis in colorectal carcinoma cells treated with 4-hydroxy-2-nonenal and structurally related aldehyde products of lipid peroxidation. Chem. Res. Toxicol. 17, 453–462. [DOI] [PubMed] [Google Scholar]
- Lin D.; Lee H. G.; Liu Q.; Perry G.; Smith M. A.; Sayre L. M. (2005) 4-Oxo-2-nonenal is both more neurotoxic and more protein reactive than 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 18, 1219–1231. [DOI] [PubMed] [Google Scholar]
- Luckey S. W.; Taylor M.; Sampey B. P.; Scheinman R. I.; Petersen D. R. (2002) 4-Hydroxynonenal decreases interleukin-6 expression and protein production in primary rat kupffer cells by inhibiting nuclear factor-kappaB activation. J. Pharmacol. Exp. Ther. 302, 296–303. [DOI] [PubMed] [Google Scholar]
- Haynes R. L.; Szweda L.; Pickin K.; Welker M. E.; Townsend A. J. (2000) Structure-activity relationships for growth inhibition and induction of apoptosis by 4-hydroxy-2-nonenal in Raw 264.7 cells. Mol. Pharmacol. 58, 788–794. [DOI] [PubMed] [Google Scholar]
- Aldini G.; Vistoli G.; Regazzoni L.; Gamberoni L.; Facino R. M.; Yamaguchi S.; Uchida K.; Carini M. (2008) Albumin is themain nucleophilic target of human plasma: a protective role against pro-atherogenic electrophilic reactive carbonyl species?. Chem. Res. Toxicol. 21. [DOI] [PubMed] [Google Scholar]
- Doorn J. A.; Petersen D. R. (2002) Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 15, 1445–1450. [DOI] [PubMed] [Google Scholar]
- Townsend A. J.; Leone-Kabler S.; Haynes R. L.; Wu Y.; Szweda L.; Bunting K. D. (2001) Selective protection by stably transfected human ALD3A1 (but not human ALDH1A1) against toxicity of aliphatic aldehydes in V79 cells. Chem.-Biol. Interact 130−132, 261–273. [DOI] [PubMed] [Google Scholar]
- LoPachin R. M.; Gavin T.; Petersen D. R.; Barber D. S. (2009) Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chem. Res. Toxicol. 22, 1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg M. E.; Li X.; Gugiu B. G.; Gu X.; Qin J.; Salomon R. G.; Hazen S. L. (2008) The lipid whisker model of the structure of oxidized cell membranes. J. Biol. Chem. 283, 2385–2396. [DOI] [PubMed] [Google Scholar]
- Hazen S. L. (2008) Oxidized phospholipids as endogenous pattern recognition ligands in innate immunity. J. Biol. Chem. 283, 15527–15531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.