

Abstract
Opioids elicit antinociception in mammals through three distinct types of receptors designated as μ, κ and δ. However, it is not clear what type of opioid receptor mediates antinociception in non-mammalian vertebrates. Radioligand binding techniques were employed to characterize the site(s) of opioid action in the amphibian, Rana pipiens. Naloxone is a general opioid antagonist that has not been characterized in Rana pipiens. Using the non-selective opioid antagonist, [3H]-naloxone, opioid binding sites were characterized in amphibian spinal cord. Competitive binding assays were done using selective opioid agonists and highly-selective opioid antagonists. Naloxone bound to a single-site with an affinity of 11.3 nM and 18.7 nM for kinetic and saturation studies, respectively. A Bmax value of 2725 fmol/mg protein in spinal cord was observed. The competition constants (Ki) of unlabeled μ, κ and δ ranged from 2.58 nM to 84 μM. The highly-selective opioid antagonists yielded similar Ki values ranging from 5.37 to 31.1 nM. These studies are the first to examine opioid binding in amphibian spinal cord. In conjunction with previous behavioral data, these results suggest that non-mammalian vertebrates express a unique opioid receptor which mediates the action of selective μ, κ and δ opioid agonists.
Keywords: Amphibian, [3H]-Naloxone, Antinociception, Opioid, β-FNA, nor-BNI, NTI
1. Introduction
It is known that opioids produce antinociception in mammals and analgesia in humans through the activation of one or more distinct types of opioid receptors. Evidence for the multiplicity of opioid receptors in mammals mediating antinociception originated with behavioral studies [16], was validated by radioligand binding studies [8,14] and was further confirmed with the identification of genes for three distinct types of opioid receptors [26].
Whereas the multiplicity of opioid receptors in mammals is certain, it has not been shown that the opioid actions in non-mammalian vertebrates are mediated by more than one type of opioid receptor. The antinociceptive effects of a number of opioid agonists in amphibians have been well characterized using the acetic acid test [22]. The antinociception produced by opioid agonists in amphibians was shown to be opioid receptor mediated as it was significantly blocked by the general opioid antagonists, naloxone and naltrexone [32,35]. Selective μ, κ, and δ opioid agonists elicit consistent and potent antinociception following systemic or central administration in Rana pipiens [31,32,36]. Interestingly, the relative antinociceptive potency of selective μ, κ and δ opioid ligands in amphibians and rodents is highly correlated in both systemic and intraspinal administration studies [31,32]. Based on these findings, differences in the opioid receptor proteins between mammals and amphibians would not be expected.
Recently, data from behavioral studies in amphibians employing selective μ, κ and δ opioid ligands as well as highly-selective opioid antagonists administered intraspinally produced a surprising finding: highly-selective antagonists for μ, κ, and δ opioid receptors were not selective in amphibians [34]. That is, the μ-selective antagonist, β-funaltrexamine (β-FNA), prevented the antinociceptive effects of μ, κ, and δ opioid agonists with the same unexpected finding observed for the δ-selective antagonist, naltrindole (NTI), and the κ-selective antagonist, nor-binaltorphimine (nor-BNI) [34].
Previous binding studies using amphibian brain tissue have shown predominantly one κ-like opioid binding site with few sites characterized as μ or δ opioid binding sites [1,29]. It has been determined that this opioid binding site in amphibians is so uniquely different from mammalian opioid receptors that some authors call it as a ‘non-μ, non-δ, non-κ’ opioid receptor [17]. No studies thus far have examined a full complement of selective μ, κ, and δ opioid ligands, nor have they used highly selective opioid antagonists in competitive binding assays using an amphibian model.
Recent binding studies in amphibian brain tissue using [3H]-naloxone yielded interesting results with the selective antagonists. All three selective antagonists possessed nearly identical Ki values in their competition for [3H]-naloxone binding [19]. These studies may suggest that either there are three promiscuous receptors that bind several opioid classes or that there is a single binding site mediating antinociception for multiple opioids.
In the present study, a full characterization of [3H]-naloxone binding was performed in amphibian spinal cord tissue using kinetic, saturation and competition analyses to provide a pharmacological correlate to intraspinal behavioral data obtained in Rana pipiens as well as for comparison to [3H]-naloxone binding in brain tissue. A number of μ-, κ- and δ-selective opioid agonists were used to compete with naloxone binding. Finally, the highly-selective μ opioid antagonist, β-FNA [38], the δ-selective antagonist, NTI [25] and the κ-selective antagonist, nor-BNI [39], were assayed against naloxone binding.
2. Materials and methods
2.1. Drugs
Drugs used include naltrexone hydrochloride, β-funaltrexamine, morphine and fentanyl which were obtained from the National Institute on Drug Abuse Drug Supply Program (Mr. Robert Walsh of the Research Technology Branch, Rockville, MD). Dermorphin and [d-Pen2, d-Pen5]-enkephalin (DPDPE) were obtained from a commercial source (Bachem Bioscience, Prussia, PA). (5R)-(544α,744α,845β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro [4,5] dec-8yl]-4-benzofuranacetamide monohydrochloride (CI977, Enadoline) was obtained from Ms. Carol Germain of Parke-Davis (Ann Arbor, MI). (±)-6-Ethyl-1, 2, 3, 4, 5, 6-hexahydro-3-[(1-hydroxycyclopropyl)methyl]-11,11-dimethyl-2,6-methano-3-benzazocin-8-ol hydrochloride (bremazocine), 17,17′-bis(Cyclopropylmethyl)-6, 6′, 7, 7′-tetrahydro-4, 5, 4′, 5′-diepoxy-6, 6′-(imino)[7,7′ - bimorphinan]-3,3′,14,14′-tetrol dihydrochloride (nor-binaltorphimine), [D-Ala2]-deltorphin-II, dynorphin A-(1-13) and 17-Cyclopropylmethyl-6,7-dehydro-4,5-epoxy-3,14-dihydroxy-6,7,2′,3′-indolomorphinan hydrochloride (naltrindole) were obtained from Research Biochemicals International (Natick, MA). (+)-4-[(αR)-α-((2S,5R)-4-Allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide (SNC-80) was obtained from Tocris Cookson (Ballwin, MO). [3H]-Naloxone (1.78 TBq/mmol; 48 Ci/mmol) was purchased from Amersham (Arlington Heights, IL). All drugs were mixed with buffer (50 mM Tris HCl with 100 mM NaCl).
2.2. Tissue preparation
Frogs were decapitated and whole spinal cord preparations were obtained by expulsion out the rostral end of the vertebral column using a saline filled syringe inserted into the caudal end. Tissue was stored at −70°C until used in the tissue homogenate binding assay. Spinal cord tissues had a wet weight average of approximately 75 mg. On the day of the experiment, spinal cord tissue was thawed and homogenized in approximately 100 volumes/weight of 50 mM Tris HCl with 1 mM sodium EDTA, pH 7.4. Pellets were obtained by centrifugation of the homogenate at 400 rpm (29 g) at 4°C for 15 min followed by 14,500 rpm (24,000 g) at 4°C for 15 min. The resulting pellet was suspended in 50 mM Tris HCl with 100 mM NaCl, pH 7.4 and rehomogenized for immediate use in the binding assay. This working buffer included 100 mM NaCl for the optimization of [3H]-naloxone binding. Protein analysis was determined according to the Bradford method using bovine serum albumin (BSA) as the standard (BioRad, Richmond, CA).
2.3. Binding assay
Experiments were performed in triplicate and the receptor binding reactions were initiated by adding [3H]-naloxone (50 μl) to 400 μl of tissue homogenate (0.17±0.08 mg of protein) containing either 50 μl of buffer (for total binding) or 50 μl of naltrexone for the determination of nonspecific binding. The components were incubated for 60 min at room temperature in order to equilibrate. Unbound ligand was separated from the receptor–ligand complex and the binding reaction was terminated by rapid filtration using a Brandel 24-cell tissue harvester (Gaithersburg, MD) followed by washing (4×5 ml; 15 s) with cold buffer onto Whatman GF/B glass-fiber filters which were pre-soaked for at least 1 h in 0.3% polyethylenimine (PEI) to decrease nonspecific binding. Radioactivity was counted using a Beckman LS1801 scintillation counter (40–50% efficiency) with Scintiverse scintillation fluid (Fisher, Pittsburgh, PA). Specific binding was defined as the difference between non-specific binding (measured in the presence of excess concentrations (10 μM) of naltrexone to block opioid receptor sites) and total binding.
2.4. Kinetic studies
The association component of kinetic analysis involved the addition of [3H]-naloxone (10 nM) at various time points (10 measurements) where specific binding was measured. Nonspecific binding was defined by a parallel series of tubes containing 10 μM naltrexone. The dissociation component was accomplished by allowing the radioligand and homogenate to bind to equilibrium at which point further binding was blocked by the addition of 10 μM naltrexone at various time points (10 measurements) where specific binding was measured.
2.5. Saturation studies
Saturation analysis was performed by measuring specific binding over increasing concentrations (0.5–70 nM) of [3H]-naloxone to determine receptor density (Bmax) and apparent affinity (KD). Nonspecific binding was defined by 10 μM naltrexone. Binding reactions proceeded as described in the binding assay.
2.6. Competition studies
Competition binding experiments were performed using [3H]-naloxone (10 nM) with increasing concentrations (15) of unlabeled ligand (0.01 nM–100 μM). 10 μM naltrexone was used to define nonspecific binding. Binding reactions proceeded as described in the binding assay.
2.7. Data analysis
Association kinetic analysis involved fitting the data by the one phase exponential association equation or the two phase exponential association equation to determine the best fit. The one phase exponential association equation resulted in the best fit. Dissociation kinetic data were fitted to one and two phase exponential decay to as before determine the best fit for the data. As with the association data, the one phase equation was the best fit. For saturation analysis the data were first fit to the rectangular hyperbolic function followed by linear transformation (Scatchard, bound/free versus bound). Analysis of the rectangular hyperbola was used to obtain apparent affinity (KD) and density (Bmax) data. In competition experiments, the concentrations of unlabeled ligand that bound to half of the binding sites at equilibrium (Ki) were calculated by GraphPad using the correction of Cheng and Prusoff [6] which corrects for the concentration of radioligand as well as the affinity of the radioligand for its binding site. Competition curves were fitted to one- or two-site binding models, to determine to which the data were best fit, using the nonlinear least-squares curve-fitting by GraphPad Prism (version 3.00, San Diego, CA) and are based on the statistical F-test.
3. Results
3.1. Kinetics of [3H]-naloxone binding
Kinetic analysis was performed to determine the time needed to attain the condition of steady-state as well as the rate constants for association and dissociation. Kinetic analyses of [3H]-naloxone (10 nM) binding in Rana pipiens spinal cord homogenates are shown in Fig. 1. Association studies (Fig. 1A) in the spinal cord yielded a kobs (observed association rate) value of 0.3505 min−1 while dissociation (Fig. 1B) results yielded a koff (dissociation rate constant) value of 0.2429 min−1. Nonspecific binding represented 25% of total binding. These rate constants yielded a KD value of 11.29 nM. Statistical analysis of the comparison between one and two site models yielded a best fit for the one site model (see Table 1 for results of F-test and significance).
Fig. 1.

Association kinetics of [3H]-naloxone (10 nM) binding in Rana pipiens spinal cord (A). Dissociation kinetics in Rana pipiens spinal cord (B).
Table 1.
Kinetically and experimentally derived affinity and density parameters for [3H]-naloxone binding in Rana pipiens spinal cord
| Kinetic analysis |
Saturation analysis |
||
|---|---|---|---|
| Parameters | Statistics | Parameters | Statistics |
| kobsa 0.4581±0.1822 |
F value 0.1467, P=0.9523 |
KD 18.75±19.55 nM |
F value 1.591, P=0.2285 |
| koffa 0.2429±0.1607 |
F value 0.06451, P=0.9377 |
Bmaxd 2725±1055 | – |
| konb 0.02152 | – | ||
| KDc 11.29 nM | – | ||
| Bmaxd 1090±145 | – | ||
min−1.
mol−1 min−1.
KD values were calculated from rate constant on/off values where and .
fmol/mg protein.
3.2. Saturation studies
The properties of naloxone binding sites were studied over an extended range of concentrations of [3H]-naloxone (0.5–70 nM) where apparent affinity and density data for [3H]-naloxone were determined. Saturation data for spinal cord tissue is shown in Fig. 2. Scatchard analysis of these data is shown in the inset. The experimentally derived KD and Bmax from saturation analysis were found to be 18.75 nM and 2725 fmol/mg protein, respectively. Kinetic and saturation data for [3H]-naloxone are summarized in Table 1. These data were best fit to a one site binding model as determined by the F-test.
Fig. 2.

Saturation analysis of [3H]-naloxone in spinal cord tissue homogenates. The membrane preparation was incubated with various concentrations of [3H]-naloxone. Measured binding is the difference between total and nonspecific binding. Values represent the mean of three independent determinations, each performed in triplicate. KD and Bmax values were determined by the rectangular hyperbole using GraphPad Prism. Inset shows Scatchard analysis of the saturation data.
3.3. Competition analysis
In order to clarify drug interaction with particular receptor types, inhibition experiments were performed with selective opioid ligands using [3H]-naloxone as the label. Fig. 3 shows these results with Fig. 3A depicting competition with μ agonists, Fig. 3B showing competition with κ ligands and Fig. 3C, competition with δ receptor agonists. Percent specific binding was measured over a range of concentrations (0.01 nM–100 μM) of cold competitor. For each of these competitors the affinity constant (Ki) was calculated from the complete data set and is shown in Table 2. Fig. 4 shows the correlation between brain and spinal cord Ki values in the amphibian where a correlation value of 0.786 was obtained. Additional competition studies with increasing concentrations (0.01 nM–100 μM) of selective antagonists against [3H]-naloxone (10 nM) were performed. These results are shown in Fig. 5 with a summary of the Ki values shown in Table 3. In the case of all competitive ligands, the data were best fit to a one site model as determined by the F-test.
Fig. 3.

Inhibition of 10 nM [3H]-naloxone binding with various unlabeled opioid receptor ligands in Rana pipiens spinal cord tissue homogenates. (A) depicts competition with μ agonists, (B) shows competition with κ ligands and (C) with δ agonists. Aliquots of tissue homogenates were incubated with radioligand in the presence of various concentrations (0.01 nM–100 μM) of cold competitor. Data was normalized to aid comparisons defining the smallest value in the data set as 0% and the largest value as 100% of specific binding. Ki values for these competitors are shown in Table 2. Data points are the means of one representative experiment in triplicate determinations, which was repeated three times.
Table 2.
Competition of [3H]-naloxone (10 nM) binding by selective opioid receptor ligands in Rana pipiens spinal cord
| Drug | Type | Ki (nM) | 95% CIa | Hillb | 95% CI |
|---|---|---|---|---|---|
| bremazocine | κ | 2.58 | (1.50–4.42) | −0.5461 | (−0.78 to −0.31) |
| naloxone | μ, δ, κ | 15.4 | (7.97–29.95) | −0.2643 | (−0.40 to −0.12) |
| morphine | μ | 728 | (515–1029) | −1.109 | (−1.5 to −0.76) |
| fentanyl | μ | 223 | (137–362) | −0.9327 | (−1.3 to −0.54) |
| dynorphin | κ | 4252 | (2005–9018) | −1.209 | (−2.0 to −0.37) |
| dermorphin | μ | 1870 | (285–12,250) | −0.3797 | (−0.98 to 0.22) |
| CI977 | κ | 7755 | (3207–18,750) | −1.231 | (−2.3 to −0.20) |
| SNC-80 | δ | 25050 | (3064–204,800) | −0.9404 | (−1.4 to −0.66) |
| DPDPE | δ | 12320 | (4673–32,470) | −0.6799 | (−1.2 to −0.13) |
| deltorphin | δ | 84910 | (32,000–225,300) | −0.3432 | (−0.90 to 0.21) |
95% confidence interval.
Hill slope.
Fig. 4.
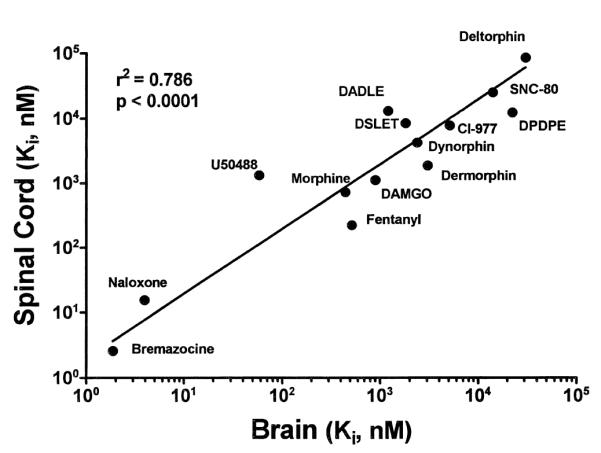
Correlation plot of Rana pipiens spinal cord Ki values versus brain Ki values against [3H]-naloxone. Numerical results of regression analysis are shown in the figure.
Fig. 5.

Competition of 10 nM [3H]-naloxone binding with increasing concentrations (0.01 nM–10 μM) of selective antagonists in Rana pipiens spinal cord. β-funaltrexamine is a μ-selective antagonist, naltrindole is δ-selective and nor-binaltorphimine is a κ-selective antagonist. Data was normalized to aid comparisons defining the smallest value in the data set as 0% and the largest value as 100% of specific binding. Data points are the means of one representative experiment in triplicate determinations, which was repeated three times.
Table 3.
Competition of [3H]-naloxone (10 nM) binding by selective antagonists in Rana pipiens spinal cord
| Drug | Type | Ki (nM)a | 95% CIb | Hillc | 95% CI |
|---|---|---|---|---|---|
| β-FNA | μ | 5.37 | (3.88–7.08) | −0.7632 | (−0.96 to −0.56) |
| nor-BNI | κ | 9.97 | (5.31–20.48) | −0.4951 | (−0.83 to −0.15) |
| NTI | δ | 31.1 | (10.15–42.90) | −0.3780 | (−0.64 to −0.11) |
Apparent Ki value.
95% confidence interval.
Hill slope.
4. Discussion
4.1. Opioid action in amphibians
The study of opioid receptor expression in phylogenetically different species has played a significant role in the understanding of opioid receptor pharmacology [5,20]. It is widely recognized that three distinct receptors mediate the effects of opioids in mammals. However, previous behavioral and binding studies in Rana pipiens suggest the possibility of a single opioid receptor which may mediate the actions of μ, κ and δ opioids [19,34]. The present results are the first to document the binding characteristics of [3H]-naloxone in Rana pipiens spinal cord homogenates. Furthermore, the present data are the first to use highly-selective opioid antagonists in a competitive binding assay using central nervous system tissue from a non-mammalian vertebrate species.
4.2. Naloxone binding affinity and density
Numerous mammalian binding studies have shown that opioid agonists elicit antinociception through μ, κ and δ opioid receptors in both brain and spinal cord tissue homogenates [3,9,11,15,21,24,33,41]. Kinetic analysis of [3H]-naloxone in Rana pipiens spinal cord tissue resulted in a KD value of 11.29 nM. [3H]-Naloxone binding was saturable in amphibian spinal cord tissue, yielding a KD value of 18.75 nM. The kinetic and saturation KD values were not statistically different. This similarity in KD values, together with the linear transformation of the binding data, is suggestive of binding to a single, noninteractive site but does not rule out binding to several different sites with a similar affinity. However, analysis of the data show a best fit to a single site as indicated by the F-test. Comparable high affinity for [3H]-diprenorphine, a general opioid antagonist, binding to a single site was also seen in Rana pipiens brain tissue homogenates [18]. The density of opioid binding sites was 2725 fmol/mg protein in the amphibian spinal cord. This density value in Rana pipiens brain tissue studies as well as values in other amphibian species using brain tissue are higher than those in the mammal where Bmax values range from 13 to 177 fmol/mg protein [11,28,37].
4.3. Competitive binding with selective μ, κ and δ opioid ligands
In assays of expressed mammalian opioid receptors, naloxone preferentially interacts with μ binding sites (KD = 3.9 nM) but also has significant affinity for κ-opioid receptors (KD = 16 nM) and a lesser affinity for δ-opioid receptors (KD = 95 nM) [27]. [3H]-Naloxone, through competition analysis, has been useful in the determination of receptor affinities of type-selective opioids in mammals [23,28,37]. The Ki values for [3H]-naloxone binding in frog spinal cord ranged from 2.58 nM for bremazocine to 84 μM for deltorphin. As is shown in Table 2, δ receptor ligands and most κ receptor agents were weak competitors of [3H]-naloxone binding. The strong competition of [3H]-naloxone binding by bremazocine is interesting as it is classified as a κ-selective agonist in mammalian studies [10], but has been considered a non-selective antagonist in previous binding studies [4,40]. Additionally, in behavioral studies, bremazocine has been shown to attenuate morphine analgesia in frog spinal cord without demonstrating agonist activity [2]. This potent competition of [3H]-naloxone binding by bremazocine was also seen in other amphibian binding studies using [3H]-naloxone [7,19,30] as well as in this lab with Chinese hamster ovary (CHO) membrane preparations expressing the human μ-opioid receptor (unpublished data). Affinity values for the competitors in spinal cord tissue were similar to those observed in brain where the corresponding Ki values were found to be highly correlated (see Fig. 4). Additionally, in behavioral studies in this lab, systematically administered bremazocine showed partial agonist/antagonist properties as it significantly blocked the antinociception produced by fentanyl following systemic administration (data not shown). In examining average Ki values, the overall trend of binding in Rana pipiens shows an affinity series of μ>κ>δ. This affinity profile is consistent with the relative affinity of naloxone for μ, κ and δ receptors [27]. The agonist Ki values showed a high degree of correlation between brain and spinal cord as is shown in Fig. 4. This has consistency in Ki values between brain and spinal cord also been observed in the rat [15].
4.4. Competition binding with highly-selective opioid antagonists
The finding that naloxone bound to a single high-affinity site in amphibian spinal cord and that μ, κ and δ opioids could complete with naloxone may be suggestive of a single-type of opioid receptor binding site. To further test this hypothesis, the selective opioid antagonists were employed. In mammals these highly selective μ, κ and δ antagonists affect the binding of opioid agonists only at their respective receptors [38,39]. As mentioned above, behavioral studies revealed a lack of selectivity of these antagonists in Rana pipiens and binding studies with [3H]-naloxone in brain yielded nearly identical Ki values for the selective antagonists [19,34]. Interestingly, the three selective antagonists also yielded similar Ki values in Rana pipiens spinal cord tissue (Fig. 5). Ki values for the selective antagonists in mammals have been determined using selective ligands for the μ, κ and δ cloned opioid receptors. In cell lines expressing the μ-opioid receptor, selective antagonists had Ki values of 0.33 (β-FNA), 2.2 (nor-BNI) and 64 nM (NTI) against [3H]-DAMGO. At the κ receptor, 2.8 (β-FNA), 0.027 (nor-BNI) and 66 nM (NTI) against [3H]-U65953 and at δ receptors, Ki values were 48 (β-FNA), 65 (nor-BNI) and 0.02 nM (NTI) [3H]-naltrindole. Thus each selective antagonist against possessed high affinity binding to its respective receptor and a much lesser affinity for the other two opioid receptors [26]. The similar affinities of the selective antagonists in Rana pipiens in their competition with [3H]-naloxone would suggest that β-FNA, NTI and nor-BNI may not bind to separate sites.
The present results, together with the previous behavioral data (see above) suggest that opioids may act on a single receptor binding site in amphibians, which has been termed the unireceptor [34]. Overall, there is little doubt that the opioid receptors of the amphibian differ from those of the mammal. However, alternative explanations may include a unique physical arrangement of multiple receptors as opioid receptor heterodimers have been reported [12]. Additionally, in another amphibian (Rana catesbiana), partial μ-, κ- and δ-like opioid receptor sequences were cloned [13].
In conclusion, Rana pipiens represents a unique nonmammalian model for which there is a well-established behavioral assay for testing antinociception elicited by opioid ligands. Further studies employing radiolabeled selective agonists are needed to fully characterize the sites of opioid binding in the amphibian and are near completion. Finally, the ultimate determination of the number and type of distinct opioid receptors in amphibians will come from receptor cloning studies that are currently in progress in our lab and elsewhere.
Acknowledgements
Support for this research was provided by the National Institutes of Health-National Institute of Drug Abuse grant (DA 12448). Portions of these studies were previously reported at the International Narcotic Research Conference, July, 1999.
References
- [1].Benyhe S, Varga E, Hepp J, Magyar A, Borsodi A, Wollemann M. Characterization of kappa1 and kappa2 opioid binding sites in frog (Rana esculenta) brain membrane. Neurochem. Res. 1990;15:899–904. doi: 10.1007/BF00965909. [DOI] [PubMed] [Google Scholar]
- [2].Benyhe S, Wollemann M. Ethylketocyclazocine and N-cyclopropylmethyl-norazidomorphine are antagonists of morphine-induced analgesia in frog spinal cord. Biochem. Pharmacol. 1988;37:555–556. doi: 10.1016/0006-2952(88)90229-8. [DOI] [PubMed] [Google Scholar]
- [3].Blurton PA, Broadhurst AM, Wood MD, Wyllie MG. Is there a common, high-affinity opioid binding site in rat brain? J. Recept. Res. 1986;6:85–93. doi: 10.3109/10799898609073926. [DOI] [PubMed] [Google Scholar]
- [4].Broadbear JH, Negus SS, Butelman ER, De Costa BR, Woods JH. Differential effects of systemically administered nor-binaltorphimine (nor-BNI) on kappa-opioid agonists in the mouse writhing assay. Psychopharmacology. 1994;115:311–319. doi: 10.1007/BF02245071. [DOI] [PubMed] [Google Scholar]
- [5].Buatti MC, Pasternak GW. Multiple opiate receptors: phylogenetic differences. Brain Res. 1981;218:400–405. doi: 10.1016/0006-8993(81)91319-6. [DOI] [PubMed] [Google Scholar]
- [6].Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- [7].Deviche P, Murray TF, Moore FL. Effects of sodium and temperature on naloxone binding in brain tissues of a urodele amphibian. Comp. Biochem. Physiol. 1990;96C:393–398. doi: 10.1016/0742-8413(90)90028-8. [DOI] [PubMed] [Google Scholar]
- [8].Gillan MGC, Kosterlitz HW, Paterson SJ. Comparison of the binding characteristics of tritiated opiates and opioid peptides. Br. J. Pharmacol. 1980;70:481–490. doi: 10.1111/j.1476-5381.1980.tb08727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gouarderes C, Cros J. Opioid binding sites in different levels of rat spinal cord. Neuropeptides. 1984;5:113–116. doi: 10.1016/0143-4179(84)90040-4. [DOI] [PubMed] [Google Scholar]
- [10].Horan PJ, De Costa BR, Rice K, Haaseth RC, Hruby VJ, Porreca F. Differential antagonism of bremazocine and U69,593-induced antinociception by quadazocine: further functional evidence of opioid kappa receptor multiplicity in the mouse. J. Pharmacol. Exp. Ther. 1993;266:926–933. [PubMed] [Google Scholar]
- [11].Jacobson W, Wilkinson M. Opiate ([3H]-naloxone) binding to hypothalamic and cerebral cortical slices of mouse brain. Brain Res. Bull. 1984;13:481–485. doi: 10.1016/0361-9230(84)90028-5. [DOI] [PubMed] [Google Scholar]
- [12].Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li X, Keith DE, Jr., Evans CJ. Multiple opioid receptor-like genes are identified in diverse vertebrate phyla. FEBS Lett. 1996;397:25–29. doi: 10.1016/s0014-5793(96)01126-x. [DOI] [PubMed] [Google Scholar]
- [14].Lord JAH, Waterfield AA, Hughes J, Kosterlitz HW. Endogenous opioid peptides: multiple agonists and receptors. Nature. 1977;267:495–499. doi: 10.1038/267495a0. [DOI] [PubMed] [Google Scholar]
- [15].Mack KJ, Killian A, Weyhenmeyer JA. Comparison of mu, delta, and kappa opiate binding sites in rat brain and spinal cord. Life Sci. 1983;34:281–285. doi: 10.1016/0024-3205(84)90600-3. [DOI] [PubMed] [Google Scholar]
- [16].Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine-and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976;197:517–532. [PubMed] [Google Scholar]
- [17].Mollereau C, Pascaud A, Baillat G, Mazarguil H, Puget A, Meunier JC. Evidence for a new type of opioid binding site in the brain of the frog Rana ridibunda. Eur. J. Pharmacol. 1988;150:75–84. doi: 10.1016/0014-2999(88)90752-2. [DOI] [PubMed] [Google Scholar]
- [18].Newman LC, Wallace DR, Stevens CW. Characterization of 3[H]-diprenorphine binding in Rana pipiens: Observations of filter binding enhanced by naltrexone. J. Pharmacol. Toxicol. Meth. 1999;41:43–48. doi: 10.1016/s1056-8719(99)00020-9. [DOI] [PubMed] [Google Scholar]
- [19].Newman LC, Wallace DR, Stevens CW. Selective opioid agonist and antagonist displacement of [3H]-naloxone binding in amphibian brain. Eur. J. Pharmacol. 2000;397:255–262. doi: 10.1016/s0014-2999(00)00265-x. [DOI] [PubMed] [Google Scholar]
- [20].Pert CB, Aposhian D, Snyder SH. Phylogenetic distribution of opiate binding. Brain Res. 1974;75:356–361. doi: 10.1016/0006-8993(74)90761-6. [DOI] [PubMed] [Google Scholar]
- [21].Pert CB, Snyder SH. Properties of opiate-receptor binding in rat brain. Proc. Natl. Acad. Sci. USA. 1973;70:2243–2247. doi: 10.1073/pnas.70.8.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pezalla PD. Morphine-induced analgesia and explosive motor behavior in an amphibian. Brain Res. 1983;273:297–305. doi: 10.1016/0006-8993(83)90854-5. [DOI] [PubMed] [Google Scholar]
- [23].Pfeiffer A, Herz A. Discrimination of three opiate receptor binding sites with the use of a computerized curve-fitting technique. Mol. Pharmacol. 1982;21:266–271. [PubMed] [Google Scholar]
- [24].Pollack AE, Wooten GF. Effects of sodium on cell surface and intracellular [3H]-naloxone binding sites. Life Sci. 1987;41:385–390. doi: 10.1016/0024-3205(87)90212-8. [DOI] [PubMed] [Google Scholar]
- [25].Portoghese PS, Sultana M, Takemori AE. Naltrindole, a highly selective and potent non-peptide delta opioid receptor antagonist. Eur. J. Pharmacol. 1988;146:185–186. doi: 10.1016/0014-2999(88)90502-x. [DOI] [PubMed] [Google Scholar]
- [26].Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. Pharmacological characterization of the cloned κ-, δ-, and μ-opioid receptors. Mol. Pharmacol. 1994;45:330–334. [PubMed] [Google Scholar]
- [27].Satoh M, Minami M. Molecular pharmacology of the opioid receptors. Pharmacol. Ther. 1995;68:343–364. doi: 10.1016/0163-7258(95)02011-x. [DOI] [PubMed] [Google Scholar]
- [28].Schnittler M, Liebmann C, Schrader U, Schulze HP, Neubert K, Repke H. [3H]-Naloxone as an opioid receptor label: Analysis of binding site heterogeneity and use for determination of opioid affinities of casomorphin analogues. Biomed. Biochim. Acta. 1990;49:209–218. [PubMed] [Google Scholar]
- [29].Simon EJ, Hiller JM, Groth J, Itzhak Y, Holland MJ, Beck SG. The nature of opiate receptors in toad brain. Life Sci. 1982;31:1367–1370. doi: 10.1016/0024-3205(82)90383-6. [DOI] [PubMed] [Google Scholar]
- [30].Simon J, Szucs M, Benyhe S, Borsodi A, Zemlan FP, Wollemann M. Solubilization and characterization of opioid binding sites from frog (Rana esculenta) brain. J. Neurochem. 1984;43:957–963. doi: 10.1111/j.1471-4159.1984.tb12830.x. [DOI] [PubMed] [Google Scholar]
- [31].Stevens CW. Relative analgesic potency of mu, delta and kappa opioids after spinal administration in amphibians. J. Pharmacol. Exp. Ther. 1996;276:440–448. [PubMed] [Google Scholar]
- [32].Stevens CW, Klopp AJ, Facello JA. Analgesic potency of mu and kappa opioids after systemic administration in amphibians. J. Pharmacol. Exp. Ther. 1994;269:1086–1093. [PubMed] [Google Scholar]
- [33].Stevens CW, Lacey CB, Miller KE, Elde RP, Seybold VS. Biochemical characterization and regional quantification of mu, delta, and kappa opioid binding sites in rat spinal cord. Brain Res. 1991;550:77–85. doi: 10.1016/0006-8993(91)90407-m. [DOI] [PubMed] [Google Scholar]
- [34].Stevens CW, Newman LC. Spinal administration of selective opioid antagonists in amphibians: evidence for an opioid unireceptor. Life Sci. 1999;64:PL125–PL130. doi: 10.1016/s0024-3205(99)00013-2. [DOI] [PubMed] [Google Scholar]
- [35].Stevens CW, Pezalla PD. Naloxone blocks the analgesic action of levorphanol but not of dextrorphan in the leopard frog. Brain Res. 1984;301:171–174. doi: 10.1016/0006-8993(84)90418-9. [DOI] [PubMed] [Google Scholar]
- [36].Stevens CW, Rothe-Skinner K. Supraspinal administration of opioids with selectivity for μ-, δ-, and κ-opioid receptors produces analgesia in amphibians. Eur. J. Pharmacol. 1997;331:15–21. doi: 10.1016/s0014-2999(97)01026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Szucs M, Borsodi A, Bogdany A, Gaal J, Batke J, Toth G. Detailed analysis of heterogeneity of [3H]-naloxone binding sites in rat brain synaptosomes. Neurochem. Res. 1987;12:581–587. doi: 10.1007/BF00971005. [DOI] [PubMed] [Google Scholar]
- [38].Takemori AE. Affinity labels for opioid receptors. Ann. Rev. Pharmacol. Toxicol. 1985;25:193–223. doi: 10.1146/annurev.pa.25.040185.001205. [DOI] [PubMed] [Google Scholar]
- [39].Takemori AE, Portoghese PS. Selective naltrexone-derived opioid receptor antagonists. Ann. Rev. Pharmacol. Toxicol. 1992;32:239–269. doi: 10.1146/annurev.pa.32.040192.001323. [DOI] [PubMed] [Google Scholar]
- [40].Wood MS, Traynor JR. [3H]-diprenorphine binding to kappa sites in guinea-pig and rat brain: evidence for apparent heterogeneity. J. Neurochem. 1989;53:173–178. doi: 10.1111/j.1471-4159.1989.tb07310.x. [DOI] [PubMed] [Google Scholar]
- [41].Zarr GD, Werling LL, Brown SR, Cox BM. Opioid ligand binding sites in the spinal cord of the guinea-pig. Neuropeptides. 1986;25:471–480. doi: 10.1016/0028-3908(86)90170-x. [DOI] [PubMed] [Google Scholar]