

Abstract
The conceptual foundation of the genome-wide association study (GWAS) has advanced unchecked since its conception. A revision might seem premature as the potential of GWAS has not been fully realized. Multiple technical and practical limitations need to be overcome before GWAS can be fairly criticized. But with the completion of hundreds of studies and a deeper understanding of the genetic architecture of disease, warnings are being raised. The results compiled to date indicate that risk-associated variants lie predominantly in non-coding regions of the genome. Additionally, alternative methodologies are uncovering large and heterogeneous sets of rare variants underlying disease. The fear is that, even in its fulfilment, the current GWAS paradigm might be incapable of dissecting all kinds of phenotypes. In the following text we review several initiatives that aim to overcome these limitations. The overarching theme of these studies is the inclusion of biological knowledge to both the analysis and interpretation of genotyping data. GWAS is uninformed of biology by design and although there is some virtue in its simplicity it is also its most conspicuous deficiency. We propose a framework in which to integrate these novel approaches, both empirical and theoretical, in the form of a genome-wide regulatory network (GWRN). By processing experimental data into networks, emerging data types based on chromatin-immunoprecipitation are made computationally tractable. This will give GWAS re-analysis efforts the most current and relevant substrates, and root them firmly on our knowledge of human disease.
Genome-wide association studies (GWAS) are the cornerstone of risk-associated variant discovery. To date, thousands of such variants have been associated with a broad variety of phenotypes. Unfortunately, the effects of these variants account for a small proportion of disease incidence and their distribution is at odds with our understanding of disease mechanisms. Technological and methodological advancements in GWAS data collection will help expand the risk-associated variome, that is, the comprehensive collection of risk-associated variants. But it is unclear whether the current paradigm will ever be able to identify all genetic variation underlying human disease. As Clark et al. posit [6], our success with GWAS depends critically on the assumptions we make about the genetic architecture of phenotypes. Half a decade after Clark et al.’s warning we have a better understanding of the genetic architecture of human disease and therefore the ability to shift to a new paradigm. We address two conceptual foundations of GWAS that are suspect and review the novel methods and technologies that have freed themselves of such biases and assumptions. We propose an overarching framework that incorporates these advancements in the form of a genome-wide regulatory network (GWRN). The technology to develop this network with which to enhance the effectiveness of GWAS is now available and affordable. In the following sections we present the reasons why GWAS needs the network, how we can start to build it and how we can use it in the context of association studies.
Why does GWAS need a genome-wide regulatory network?
The importance of considering regulatory elements
Revising the bias toward genes
Johnson and O’Donnell have curated an open access database of genome-wide association results collected from 118 GWAS articles [23]. The database contains approximately sixty thousand significant associations between SNPs and phenotypes. The authors conclude that there is a bias in the distribution of associations toward genes. This is based on the observation that 40% of SNPs lie within the boundaries of a gene and that roughly 70% are within a 60 kb perimeter. This bias is then used to justify associating each variant to a protein-coding gene, which allows for subsequent analyses and interpretations. But the same results can be drastically reinterpreted. Only one percent of the variants lie within the boundaries of exons, making the vast majority non-coding, and more than half lie outside the boundaries of genes. Furthermore, the high percentage of variants falling within a 60 kb perimeter of a gene appears to originate from a bias in the genotyping technology (see figures 3 and 4 in [23]). This interpretation does not support a gene-centric view of risk-associated variation. Variation will ultimately impinge on genes, but the mechanisms of disruption appear to be almost entirely non-coding. Given that exonic variants are not prone to association, what are the next best candidates?
Genetic variation at regulatory elements is associated with disease
The importance of genetic variation at regulatory elements has been shown for colorectal cancer [54, 43, 22, 59] and alpha thalassemia [18]. In the case of alpha thalassemia, De Gobbi et al. identified a gain of function SNP that creates a novel promoter element between the alpha globin genes [18]. Similarly, a SNP associated with colorectal cancer was found to disrupt a regulatory element at the 8q24 cancer-linked locus [54] [43]. The variant allele of the SNP had a significant effect on the expression of the MYC proto-oncogene through the differential recruitment of a transcription factor. These preliminary studies have shown that the disruption of sequence-specific binding by risk-associated variants is relevant to human disease. This interplay between variation, regulation and disease has been reviewed by Lupien and Brown [30]. In an orthogonal approach, Idaghdour et al. present us with the eSNP paradigm that aims to systematically discover variants that affect transcriptional levels of genes [21]. These two approaches have complementary strengths, and it is now feasible to merge them by incorporating regulatory elements to GWAS. With the advent of chromatin immunoprecipitation coupled with deep sequencing (ChIP-Seq), transcription factor binding sites can be determined experimentally at the genome-wide level. Similarly, the chromatin signatures of regulatory elements can also be mapped using variations of the technology. But regardless of the non-coding nature of risk-associated variants and the evidence in favor of prioritizing regulatory variation, the bias toward genes persists. Now that thousands of such elements have been mapped in multiple cell lines, all GWAS strategies should be reformulated to take them into consideration.
The importance of considering regulatory networks
Revising the quest for high-risk variants
Another observation derived from the database of GWAS results is that the individual effects of the discovered variants are low. This phenomenon has been recently reviewed by Ripperger et al [45] This means that each variant is only responsible for a small increment in the risk of developing a disease. The scarcity of high-risk variants could still be due to the technical and practical limitations of GWAS. Of the roughly 24 million SNPs annotated in NCBI dbSNP, at most one million are assayed on any given platform. Furthermore, there is also a severe bias towards particular types of variants, namely SNPs, in the majority of GWAS. To exhaust the GWAS paradigm, the entire spectrum of human variation needs to be considered comprehensively. Ultimately, whole genome and epigenome sequencing in adequately powered studies should allow us to find these much sought-after variants. But even if the GWAS paradigm is realized to its fullest extent, do all diseases have high-risk variants? Is human disease even composed of discrete causal variants or should we start thinking about causal patterns of variation?
Genetic heterogeneity and decentralized patterns of variation in genetic architectures
Genetic heterogeneity poses an ominous threat to the GWAS paradigm (reviewed by McClellan and King [33]) The current assumption that causal variants are common enough within cohorts to be detected individually might not be sound. The genetic architectures of autism and schizophrenia represent two of the most extremely heterogeneous genetics architectures. A substantial portion of autism appears to be caused by rare simple and structural variants [4, 47]. However, these rare mutations disproportionally disrupt genes belonging to networks that are important for brain development [17]. Rare structural mutation also impacts genes in individuals with schizophrenia [57, 33]. In this case, the heterogeneity is so large that in one study each variant disrupted a different set of genes. A genetic architecture in which multiple variants disrupt a single genomic element, such as a gene, is already problematic. But an architecture in which each individual in the cohort has a different set of disrupted genes is simply impervious to GWAS. Autism and schizophrenia might appear to be extreme examples, but how heterogeneous is human disease as a whole? Let us consider cystic fibrosis (CF), a disorder that is primarily attributed to a single gene, CFTR and a single variant ΔF508 [3] In American populations this model accurately reflects reality, but in some populations ΔF508 accounts for as little as 27% of CF incidence. In non-American populations, a disease that is usually considered Mendelian, reveals a shocking amount of heterogeneity: a total of 1600 mutations have been associated with the disease worldwide. It is straightforward to invoke rare variation and mutation to find genetic architectures that are intractable to a conventional GWAS. But can common variation produce similarly insidious architectures?
Non-linear effects and non-contiguous distributions in genetic architectures
Epistasis can produce high-risk combinations of alleles through the non-linear interaction of low-risk variants [36]. Even variants with undetectable effects can yield high-risk combinations. The difficulty in finding such predictors of disease is that variants need to be considered in sets, and to the biologically uninformed GWAS all sets are equally promising, a priory. Unfortunately, the unbiased scan for risk-associated variant sets rapidly becomes computationally intractable. The most naive approach to the search for variant sets is to look at all variants falling within a gene. But, as previously mentioned, even though risk-associated variation is likely to ultimately impinge on genes, the mechanisms by which it does so are probably non-coding. Unfortunately, the regulatory circuitry of the cell is highly distributed and the regulatory elements that affect a gene are unlikely to be contiguous. With the presence of distal enhancers and chromatin loops, physical distance between variants becomes a poor heuristic for interaction. Given the non-coding nature, heterogeneity, non-linear behavior and non-contiguous distribution of the patterns of variation underlying human disease, is it time to update the GWAS paradigm?
Network approaches to GWAS
A robust framework with which to study the genetic architectures of human disease needs to account for (1.) the non-coding classes of genomic elements in addition to genes, (2.) the heterogeneity of variation within cohorts, (3.) the non-linear effects between alleles in determining phenotype and (4.) the non-contiguous distribution of variation across the genome. A variety of methods, referred to here as ’network approaches to GWAS’, are under development to address these issues. These approaches make use of expert knowledge, that is, statistic or experimental data used to prune the daunting search space of variant combinations (reviewed by Moore et al. [35]) Because expert knowledge needs to encode both the elements to be searched and evidence or predictions of their interactions, it is primarily presented in the form of networks. To date, expert knowledge has been applied to GWAS in the form of metabolic pathways [53], immune and inflammatory pathways [13] and protein-protein interaction networks [41, 40, 14]. These methods have produced statistically significant associations between variant sets and phenotypes. These novel approaches are therefore capable, but the networks and annotations that have been used to date have a strong bias towards variants that overlap genes. As we have already mentioned, the wealth of knowledge and data available for regulatory elements and processes is rapidly growing and evolving. Though it has not yet been formalized as an explicit network, we believe it is a propitious time to incorporate the GWRN to the GWAS.
How do we start building the genome-wide regulatory network?
Chromatin immunoprecipitation followed by tiling microarray analysis (ChIP-Chip) or sequencing (ChIP-Seq) has taken the study of transcriptional regulation to the -omics domain. The terms epigenomics and cistromics [31] refer to the study of chromatin modifications and transcription factor binding at the genome-wide scale respectively. These layers of information that sit above the genome are dynamic and context dependent. The interplay between them is intricate and not fully understood. At this time, only a limited number of cell lines have epigenomic and cistromic data that are publicly available. The ENCODE project (ENCyclopedia Of DNA Elements) has spearheaded the data generation process [7]. This data is hosted at the UCSC ENCODE Data Coordination Center (DCC) (http://genome.ucsc.edu/ENCODE/). However, the rate of growth suggests an imminent coverage that will be sufficient for building a comprehensive network for all cell types In this section we will review the projects and techniques that have contributed notably to the study of regulation at the genome-wide level. We will borrow some basic vocabulary from network theory as follows. The term ’node’ will refer to a genomic element (instances of gene, enhancer, promoter…). The term ’edge’ will refer to a relationship between any two genomic elements (instances of A binds to B, A is in linkage disequilibrium with B, A insulates from B…) The term ’state’ will refer to a condition for a node or edge (instances of active, bound, condensed, transcribed…). We would like to emphasize however, that the methods used to define each of these nodes, edges and states are fundamentally empiric. Computational methods are only used for connecting the experimental observations and enhancing the way that the networks are processed and queried.
Nodes
Nodes represent static genomic elements. A gene, for example, is the canonical node. It may be completely inactive in a given cell type, yet we still identify it as being present in the genome. This principle is applied in the same manner to a wide array of heterogeneous genomic elements. For example, we will talk about the set of enhancer nodes as the union of all active enhancers for all cell types. Cell types differ in the subsets of enhancer nodes that are active or inactive. Other classes of nodes in addition to genes and enhancers are silencers, insulators, SNPs, ORFs, ncRNA, miRNA, lincRNA… Many of these node classes do not yet have high-throughput technologies developed for their identification. As these technologies are developed, the network should be updated in order to allow for their incorporation. At this stage however, the foundational node classes of the network are gene, enhancer, promoter, insulator and variant. Comprehensive maps of human genes have long been available. But regulatory elements have been notoriously difficult to identify until recently. Currently we are not only able to locate them, but also to classify them according to a variety of epigenomic signatures. Promoters, the most well known regulatory elements, act on genes in a position and orientation dependent manner and determine the initiation of transcription. Enhancers and silencers are the regulatory elements that act on promotors and modulate their degree of induction or repression in a position and orientation independent manner. Insulators, an additional class, act as barriers between promoters and enhancers [32]. There are several other classes of regulatory elements such as lamina associated domains (LADs), CpG islands… but their interactions have not been characterized to the same extent. A high-resolution map of active promoters was first presented by Kim et al. [25]. An equivalent map of insulator sites [24] followed shortly thereafter and finally a whole genome map of active enhancers [20]. In the later study, the methods previously used to identify each of the three regulatory element classes were used to generate maps in the same group of cell lines, thus allowing the comparison of their distributions. The assessment of the different roles for each of the classes concluded that enhancers have the highest amount of variability between cell-types, whereas promoters and insulators have a narrower variability. Furthermore, the study showed that the cell-type-specific positions of active enhancers correlate spatially with cell-type-specific profiles of gene expression. The validity of the predictions was further tested using reporter assays where active enhancers were shown to drive differential expression in a statistically significant manner. From the number of enhancers identified in the two cell lines assayed they estimated a total number of 10E5 to 10E6 enhancers in the human genome by extrapolating to the roughly 200 cell types of the human body. Given the number of cell types remaining to be assayed and the dwindling cost of sequencing technologies a full map of human regulatory elements should be available in the immediate future.
Edges
Edges represent potential interactions between genomic elements or their expressed products. An interaction between two genes, mediated by their protein products, is the canonical edge. Edges within a genome wide regulatory network are also heterogenous. Determining the edges between nodes requires an array of techniques tailored to each of the many classes of interactions between genomic elements. The following sections present three different classes of edge that are currently available for network approaches.
Edges based on linkage disequilibrium
Linkage disequilibrium (LD) is the non-random association of alleles at two or more genetic loci due to genetic linkage, recombination and mutation rates and several other population genetic factors. LD values for variant pairs are provided by the HapMap Consortium (http://hapmap.ncbi.nlm.nih.gov/), and are already encoded as a collection of explicit edges. In the context of GWAS, LD allows the estimation of allele states at loci that have not been directly assayed. This process is referred to as imputation. The reverse implication of this fact is that causal variants that are in LD with assayed variants can be indirectly detected. Any signal of association in GWAS is therefore not a property of the assayed variant, but of the set of LD variants where it is contained. Due to the low percentage of variants assayed in GWAS, associated variants are more likely to be in LD with causal variants than to be causal themselves. Therefore, the first step in result interpretation or follow up studies to GWAS is to find the set of variants in strong LD with the risk-associated variant. That is, to follow LD edges out of the risk-associated variant (Figure 1A).
Figure 1.

A proposed computational approach for estimating the ramifications of variation. 1A) A variant on chromosome A has been found to associate with TraitX. The association is a property of all variants in strong LD with the initial variant. The first list of candidate variant nodes is obtained by following LD edges. 1B) An LD edge brings us to the E1 enhancer node. Enhancers impinge on the transcription of genes. In order to compile a list of candidate gene nodes, we follow Long-range edges. 1C) A Long-range edge takes us to the P1 promoter node and the Gene1 gene node. This means that empiric evidence exists in favor of a regulatory interaction between E1 and Gene1 via P1. Gene1 is a transcription factor, but is not known to affect TraitX. Stronger evidence might exist and the search continues. In order to find candidate targets of Gene1, cistromic edges are followed. 1D) A cistromic edge reaches the E2 enhancer node on chromosome B. This means that empiric evidence exists supporting binding events of the Gene1 product at E2. Long-range edges are followed in the search for genes. 1E) A long-range edge reveals an interaction between E2 and Gene2 via P2. Gene2 is known to affect TraitX. A circular path closed by a trait node through the the GWRN has now been found. Through this path, each node and edge is supported by some form of empiric evidence. Additionally, the states of all or some of nodes an edges in the path might be validated for the system under study. The variant associated with TraitX, affects the transcriptional output of a gene potentially regulating another gene underlying TraitX. The path is not intended as supporting evidence for the causality of the variant, but directs the investigator to the most likely explanation given available data. The investigator can then choose among the reported paths how to best invest his resources on further experimental validation.
Edges based on long-range physical interactions
Linkage disequilibrium is tightly related to genomic sequence proximity. But most regulatory elements and genes interact in a manner that is largely independent of the number of bases between them. The next class of edge attempts to capture the long-range physical interactions between genomic elements. These chromatin structures or loops have been shown to play a role in regulatory interactions between enhancers, silencers and promoters. The family of chromatin conformation capture techniques are designed to identify such edges for a locus of interest, namely 3-C [11], 4-C [61, 49] and 5-C [12]). Hi-C, the latest addition to this family of methods, provides a low resolution map of long-range interactions for the entire genome [29]. Future refinements of this technique will continue to provide valuable insights into the spatial organization of the genome. Long-range edges can be used in addition to LD edges to further expand the repertoire of related variants from a single risk-associated variant. In order to do this or to determine the functional ramifications of a causal variant once it has been identified, long-range edges are followed (Figure 1B).
Edges based on cistromic interactions and insulators
The prototypical regulatory interaction is between a transcription factor and regulatory element affecting gene expression. This class of edge or interaction is independent of genomic or physical distance between genomic elements because it is mediated by diffusing molecules. In order to consider all variation that might impinge on a gene’s expression, or all genes that a transcription factor might impinge upon, cistromic edges are followed (Figure 1C). Cistrome data has advanced the study of genome-wide regulation dramatically by providing comprehensive lists of locations for binding events. But the interplay between bound regulatory elements and genes is still poorly understood. Chan and Song have pioneered the integrative approach to genome-wide regulatory networks by merging a variety of epigenomic and cistromic data sources [5]. In their study, they developed a quantitative model for predicting gene-to-enhancer associations in the context of estrogen induction through its effector transcription factor; estrogen receptor (ER). Their predictive model can account for approximately 70% of estrogen induced gene expression. The initial challenge addressed in the study is that the positions of binding sites alone are not predictive of a genes responsiveness to a transcription factor. What they initially observed was that only a small percentage of genes with ER binding in proximal promoter regions, exons or introns were regulated by ER. This trend contradicted a promoter-centric formulation of genome wide regulation. The first step in their approach was the identification of estrogen-responsive genes in the MCF7 breast cancer cell line. This was achieved through time course expression profiling after estrogen induction. Performing the experiments in the presence of the translation inhibitor cycloheximide allowed for the separation of primary targets from secondary targets, this step proved to be critical in determining the subset of directly responsive genes. Once the up-regulated and down-regulated genes were identified, the genome was partitioned into regulatory units or blocks bounded by insulators. The cistromic signature of insulators is the nuclear protein CTCF, which is ubiquitously expressed across all cell types (reviewed by Phillips and Corces [42]). These insulated blocks are therefore referred to as CTCF blocks. ER binding sites and estrogen-responsive genes were subsequently assigned to CTCF blocks and tested for correlation. In this first assignment they found that only a small percentage (14%) of CTCF blocks containing ER binding sites contained estrogen-responsive genes. However, a large percentage (68%) of up-regulated genes had ER within their CTCF blocks. It is important to note that this remarkable prediction was achieved using CTCF data from a cell line other than MCF7. The predictive accuracy of their model using matched cell line data is yet to be assessed. Additionally, they assessed the degree of insulation achieved by CTCF by testing the correlation between gene expression profiles within and between CTCF blocks. Their findings revealed a significantly higher correlation within blocks as compared to between blocks. In other words, ER activation is mostly confined to blocks with bound ER and does not cross boundaries into adjacent blocks. The study also reported interesting negative results. First, the distance between ER binding events and gene promoters did not correlate with transcriptional induction. Second, the number of ER binding events within a block only slightly correlate with transcriptional induction. Chromatin loops between enhancers and promoters might bypass insulators, but this layer of information was not considered in the study. This approach is an excellent example of how the incorporation of genomic, epigenomic and cistromic layers of information can begin to generate predictive models of transcriptional induction.
States
Having defined nodes and edges as static, states are intended to encode the dynamics of the cell. Genes have activation states that vary between cell types and experimental conditions. Variants have allelic states that vary across individuals. LD edges have strong or weak states that vary across populations. Regulatory elements can be evolutionarily conserved or display acceleration in certain species. And chromatin loops may be present or absent depending on the cell type. The state space of the GWRN is vast and uncharted. But epigenomics, cistromics, personal genomics and spatial biology have already started fathoming its depths.
States of genes based on epigenomics
Post-translational modifications of histones have been shown to correlate with the activation states of genes. The cascade of epigenomic events at transcriptional start sites (TSSs) has been studied extensively (reviewed by Orphanides and Reinberg [39]). Recently, Seila et al. presented a highly detailed picture of a subclass of TSSs through ChIP-seq [48]. Among several interesting insights, they revealed the mechanisms by which cells are able to break the initial symmetry in promoter binding events and initiate transcription in the correct direction. Both promoter-associated RNAPII and trimethylation at histone 3 lysine 4 (H3K4me3) were confirmed as hallmarks of initiation and dimethylation at histone 3 lysine 79 (H3K79me2) for elongation. The asymmetric distribution of these signatures is particularly interesting. The ratio between sense and antisense TSSa-RNA (short, divergent RNA molecules, nascent at TSS), was found to correlate positively with transcriptional induction. And a similarly asymmetric distribution was found for the H3K4me3 signature.
States of regulatory elements based on epigenomics
Epigenomic signatures of function have also been found for enhancers and promoters. Lupien et al. revealed how the pioneering factor FoxA1 acts in combination with mono and dimethylation at histone 3 lysines 4 (H3K4me1 and H3K4me2) to remodel chromatin [31]. This remodeling subsequently allows for the binding of sequence-specific transcription factors. In the model presented, the chromatin modification precedes the recruitment of the pioneering factor, thus revealing further insight into the temporal succession of genomic events in the regulatory cascade. These results were confirmed and extended to the genome-wide level by the work of Heintzman et al. [20]. Several other epigenomic marks have also been found to play important roles in regulation. Histone 3 acetylation (AcH3) is typically associated with chromatin accessibility and transcriptional activity.
Trimethylation at histone 3 lysines 27 and 36 (H3K27me3 & H3K36me3) have been associated with transcriptional silencing and transcriptional elongation respectively [58]. The complete cascade of events that leads to the activation or repression of a genomic element is still unclear. But as these layers of genomic annotation are understood we will increasingly be able to predict and estimate the states of the GWRN as a function of cell type and stimuli.
How can we use the genome-wide regulatory network to enhance GWAS?
Promoting GWAS results: from association to putative-causality
Assigning causality to a variant requires extensive experimental validation in the target cohort. A step prior to assigning causality to a variant is to show that it is capable of disrupting the normal function of the cell. This is usually achieved by finding a correlation between genomic variation and a significant change in gene expression. This degree of functional disruption is also referred to a putative-causality. In this section we claim that the promotion of risk-associated variants to putative-causal variants is a process that can be automated through the use of the GWRN.
A systematic experimental approach for a risk-associated variant
The elucidation of the biological mechanisms underlying the non-protein-coding risk variant rs6983267 at 8q24 is one of the first instances of this process. Homozygosity for the G allele has been found to increase colorectal cancer (CRC) risk by 1.5 fold [54] [43]. In the case of rs6983267, the variant was finally attributed to differential binding at a region harboring a transcriptional enhancer. Despite its specificity, the methodology they developed can be generalized to any risk-associated variant produced in a GWAS. The starting point was a strong association between the variant and CRC [52] [60] [19]. The region containing the candidate variant was then re-sequenced and linkage disequilibrium was determined between the neighboring SNPs in order to identify linked variants. Further experimental data was generated in order to determine the function of the variants within the block of LD. Due to the variants location in a gene desert approximately 335 kb from the MYC proto-oncogene and the lack of any kind of transcriptional activity (mRNA, miRNA, ncRNA…), the most likely function was that of a transcriptional enhancer. Epigenomic and cistromic ChIP data were generated for enhancer signatures (H3K4me1 and H3K4me3, and p300 respectively) in the CRC cell line Colo205 in order to confirm the functional assignment and select the most promising variant. Indeed, the region overlapping rs6983267 displayed all the characteristics of an enhancer. In this instance, the risk-associated SNP was chosen as the most likely variant to be causal. However, this will not necessarily be the case for all follow up studies, as discussed previously. Knowing that the region has enhancer activity, they then looked for sequence motifs that the polymorphisms alleles could be disrupting. The motif found corresponded to the TCF7L2 sequence-specific DNA-binding protein. Conveniently, Colo205 is heterozygous for rs6983267, allowing them to rapidly probe for differential binding affinity between major and minor alleles. Binding was tested both in vivo and in vitro, revealing significant differences in transcriptional induction. Having shown differential binding and enhancer activity, the next step was to find the downstream targets of this enhancer. MYC was the closest gene to the enhancer and a known mediator of Wnt signaling. Given that TCF7L2 is the main transcriptional effector of Wnt signaling, they set out to test the interaction between the enhancer region and the MYC gene. Through the use of chromosome conformation capture they provided evidence of a long-range physical interaction between rs6983267 and MYC regions. The small scale interactions revealed in this study were further bolstered by the fact that Wnt and MYC play important roles in CRC pathogenesis [38] [26]. The effect of rs6983267 on MYC induction was confirmed by Wright et al. [59]. Additionally they showed that the formation of the physical interaction did not vary between the SNPs alleles
A proposed computational approach for estimating the ramifications of variation
The systematic experimental approach just described can be reformulated in a rudimentary, network theory vocabulary. Once abstracted, the computational approach that results has a high potential for being scaled and automated. From the starting point of the trait node for CRC, an association edge was followed to a SNP node (rs6983267). From there, spatial edges were followed in order to locate adjacent nodes. In this instance, other SNP nodes were found following LD edges and an enhancer node was found via and overlap edge. Because the nodes are static as we have defined them, the enhancer state had to be validated as being active in the Colo205 cell line. As no other functional nodes of any type could be found in the region, the most likely mechanism of action of the SNP was that of a distal regulatory element or enhancer. Once this was established, they tried to elucidate the mechanistic ramifications of the different SNP states (alleles). To this end, the functional edges and spatial edges were followed out of the enhancer node to look for target promoter nodes within the regulatory block defined by insulator nodes. By doing this they found the gene node for MYC as the solitary gene to be regulated within the block. The combination of both functional edges (distance to TSS and absence of insulator) and a spatial edge (long-range physical interaction) between rs6983267 and MYC provided strong evidence for the putative-causality of the variant. In order to gain further understanding of the mechanism underlying the associated risk, trans-acting edges (signaling) were followed downstream of MYC and cistromic edges were followed upstream of the enhancer node. Both paths ultimately re-encountered the trait node for CRC, and the process terminated successfully. The next step would have been to test for correlations between the allelic states of the SNP node and the states of all nodes in the sub-network or pathway identified. A simplified and further abstracted version of this process is presented in Figure 1. The objective of this study was to systematically evaluate the possible roles of regions within the genomic risk interval. This was achieved not only by following edges within the GWRN, but by traversing the tree of all known biological functions and successively pruning unlikely possibilities as the evidence was collected. The first branching point was whether the causal variants were protein-coding, followed by whether there were any transcripts associated with the region, and so on and so forth. At the time the study was conducted the researchers had to generate the majority of the data at each step in order to traverse the tree of possible functions. But as the volume of publicly available data continues to grow it will become possible to replicate this process entirely by querying the appropriate databases. With nothing but the location of a risk-associated variant it will become possible to trace forward and backward and generate the sub-network of nodes that are affected. To illustrate the complexity and scale of publicly available data, Figure 2 shows the cistromes of three transcription factors available through the ENCODE project. The question then is: assuming that we are able to formalize and consolidate the growing body of regulatory data, what computational strategies can we adapt or develop to re-analyze GWAS results?
Figure 2.
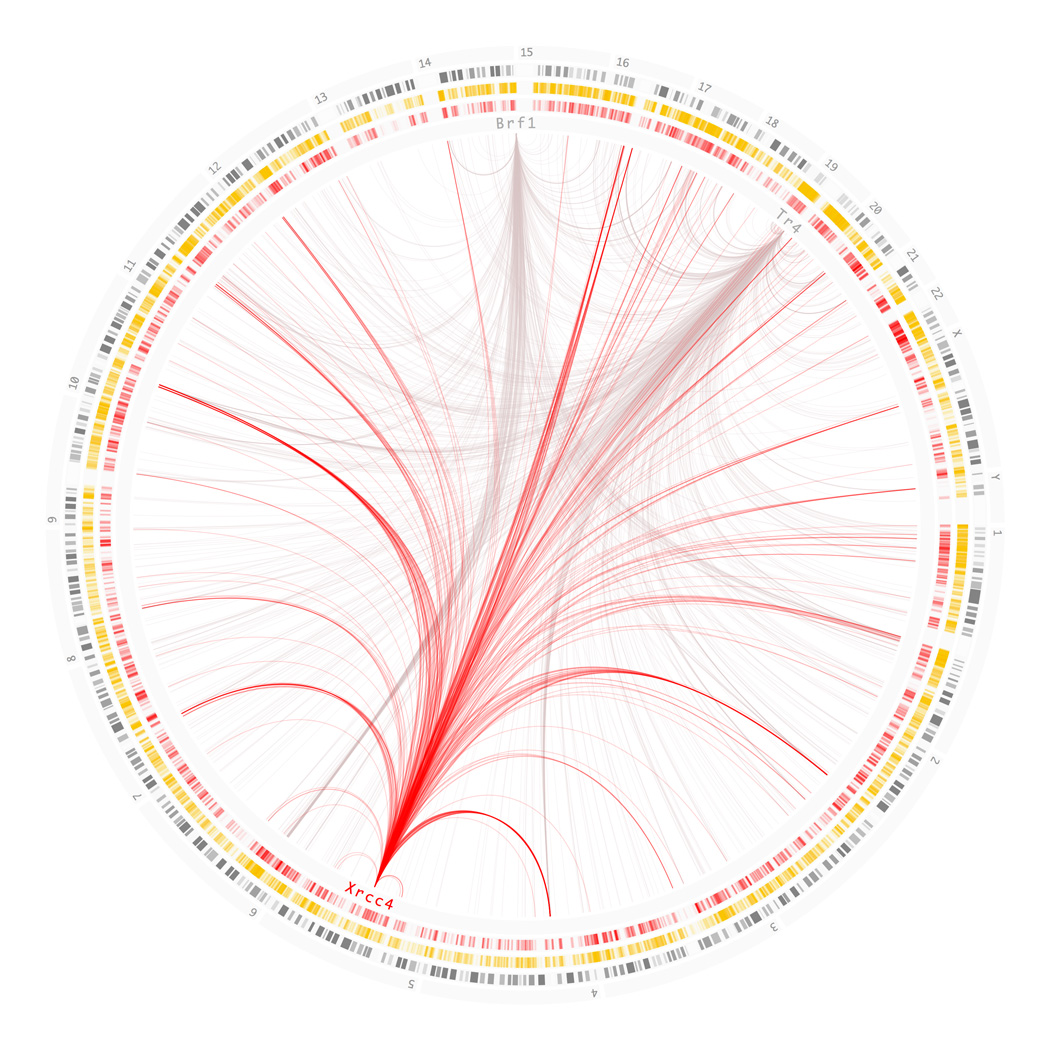
A meagre sliver of the GWRN for the K562 cell line. The two outermost circles show chromosome numbers and ideograms. The following two circles show all genes in orange followed by all active enhancers in red. Edges within the circle show the cistromes for three arbitrary transcription factors. The genomic locations of the genes are marked with text in the innermost circle. These locations act as tethers from which cistromic edges extend towards all positions in the genome where the gene product is bound. Xrcc4, highlighted in red, illustrates the one to many relationship that cistromic edges encode. The figure is intended to glean some insight into the scale and complexity of the GWRN, but also into its feasibility. The technology is currently available, but many cistromes and cell types remain to be assayed. The relation between what remains to be done and what has already been achieved does not exceed a couple of orders of magnitude. Larger technological hurdles have been overcome within the last ten years. All data shown was produced using the K562 cell line. The binding sites were determined through ChIP-seq and were obtained from the ENCODE DCC at UCSC. Data for active regulatory elements was obtained from the supplementary data in Heintzman et al. 2009 [20]. Chromosome ideograms and gene annotations were obtained from the UCSC Genome Browser. The graphic was generated using an in-house Processing program adhering to the Circos information aesthetic [28].
Aggregating GWAS results: discovering sets of risk-associated variants
Approaches that evaluate additive sets of variants as risk factors
[Analytical strategies that consider sets, pathways or networks of variants as opposed to individual variants have already been applied to GWASs. Gene set enrichment analysis [37] [51] (GSEA) is a method that tests for differential expression of sets of protein-coding genes. The approach is completely independent of how these sets are constructed and can therefore support a wide variety of clustering and grouping criteria. Exploratory Visual Analysis [44] (EVA) performs similar tests of significance but is not restricted to genes and can accommodate any type of variant. This approach uses a permutation testing strategy to assess the significance of the statistical results within a set of variants. Askland et al. have applied EVA to a GWAS of bipolar disorder [2]. In their study, they find only a modest overlap in the particular genes driving the significance of gene sets. But their observations strongly suggest that variation in ion channel genes contributes to the susceptibility of the disease. Furthermore, the lack of overlap may indicate that the genetic architecture of bipolar disorder is highly heterogeneous. Torkamani et al. present one of the most thorough pathway analysis to date [53]. Their study attempts to characterize the polygenic basis of seven common diseases using the Wellcome Trust Case Control Consortium (WTCCC) GWAS results [8]. The WTCCC data sets consist of genotype data for seven common diseases. The collection includes bipolar disorder (BD), coronary artery disease (CAD), Crohns disease (CD), hypertension (HT), rheumatoid arthritis (RA), type 1 diabetes (T1D) and type 2 diabetes(T2D). The approach builds from the strategy defined by Kotti et al. for identifying the additive effects of low-penetrance variants [27]. This is achieved by determining if the variants that are associated with a disease have a tendency to cluster in biological pathways. Namely, the pathway analysis tools provided in the MetaCore software suite developed by GeneGo (http://www.genego.com/). A hypergeometric model was then applied to determine the significance of enrichment for each pathway. Statistically significant pathogenic pathways were discovered for each of the seven diseases, all of which were considered biologically plausible. Interestingly, the pathways for each of the diseases had a significant amount of overlap. Eleftherohorinou et al. also re-analyzed the WTCCC data through a pathway-based approach [13]. By summing the Armitage trend test statistic over all variants in a pathway, they derived a cumulative trend test statistic. Associations were obtained after estimating the null distribution of the statistic through the random permutation of case/control labels. SNPs within associated pathways were subsequently used to build predictive models of disease risk by fitting a logistic regression model with variable selection. This study provides two important insights into the genetic architectures of WTCCC diseases in the context of inflammatory pathways. First, an estimate of the number of variants and genes within each of the architectures. They report and average of 205 SNPs and 149 genes in T1D, 350 SNPs and 189 genes in RA and 493 SNPs and 277 genes in CD. Second, a comparison of the predictive power of variants within risk-associated pathways that are not risk-associated individually and those that are. The models in which significant associations are excluded have a comparable predictive power to those in which they are included. In addition to novel associations with pathways and variants, this study highlights the limitations of single variant approaches to GWAS. The same general approaches used by Askland et al., Torkamani et al. and Eleftherohorinou el al. are applicable to any type of network structure. An important limitation of these approaches is that they have all made use of gene networks and have ignored regulatory elements.
Approaches that evaluate interacting sets of variants as risk factors
The methods described thus far make the strong assumption that the effects of the variants are additive (or linear) and that the interaction-based (or epistatic) effects between them are negligible. To be more specific, statistical epistasis was defined by Fisher as a deviation from additivity in a linear model [15]. The likelihood of epistatic behavior in the context of regulatory networks has been evaluated by Gertz and colleagues [16]. In their study they claim that the majority of genotype to phenotype mappings are expected to behave in a non-linear or epistatic fashion. The pervasiveness of interactions in biological systems has been reviewed in Tyler et al. [55]. And the modeling and analysis of interactions has been reviewed in Cordell et al. [10]. Epistatic analysis makes fewer assumptions about the underlying genetic architecture of traits and diseases. However, approaches that consider interactions suffer greatly from what is known as the curse of dimensionality. First, the combinatorial explosion of variant sets makes exhaustively testing all possible epistatic models with more than three variants computationally intractable. Second, the explosion of combinations entails the dilution of data points as the same number of samples need to be divided among increasing numbers of the combinations. Third, multiple testing correction is based on the effective number of variant pairs, further reducing the threshold for significance. Multifactor Dimensionality Reduction (MDR) overcomes part of this burden through the use of dimensionality reduction [46]. In this method, combinations are merged into low and high risk classes to prevent sample dilution. Additionally, a streamlined design of the algorithm allows for the fast evaluation of millions of combinations. This process of defining new attributes from sets of attributes is referred to as constructive induction or attribute construction [34]. The MDR approach has been applied to a wide range genetic association studies. For example, Andrew et al. used MDR on variants within DNA repair genes to predict susceptibility to bladder cancer [1]. They found a highly significant non-additive interaction in the xeroderma pigmentosum group D gene (XPD). The epistatic model composed of two SNPs was a better predictor of bladder cancer than smoking. Importantly, these results have been independently replicated [50]. But despite the success of MDR, the unbiased exploration of epistatic models with four or more variants is still computationally intractable. Exploring the space of epistatic models must be therefore performed in a directed manner. Prior knowledge can be used to prioritize sets of variants under a variety of criteria (reviewed by Moore et al. [35]). Emily et al. present one of the first genome-wide scans for epistasis based on biological networks [14]. The study’s network substrate is the STRING database of protein-protein interactions [56]. And the analytical methodology is based on that proposed by Pattin et al. [41] [40]. In their approach, only high-confidence STRING source scores were used to scan the WTCCC data for two-locus epistatic models. SNPs were associated to interacting proteins by sequence proximity, and the window size was intended to include the variants in LD with the gene and in nearby regulatory elements. Interactions between pairs of SNPs were tested by using a likelihood ratio test proposed by Cordell [9]. The difference between logistic regression models, built with and without interaction coefficients, is taken as a measure of interaction. Significant results were reported for gene pairs in four out of the seven diseases. Most of these were found to be one or two interactions away from genes involved in the etiologies of the corresponding diseases. The final tally of results did not surpass what would be expected for a conventional GWAS.
Conclusion
The methods described thus far represent the infancy of network approaches to GWAS. Even though we have focused on regulatory networks; metabolic and signaling pathways also need to be included. Furthermore, transcription factors and DNA binding events only represent a fraction of cellular regulation. Genome-wide technologies for microRNAs (miRNA), large intergenic non-coding RNAs (lincRNA), nuclear export and other mechanisms are under development. As they become available, they will hopefully be incorporated into a generalized network approach to GWAS. These networks represent a bridge between cutting-edge high-throughput genomic technology and state-of-the-art bioinformatic algorithms and programs. But most importantly they represent a common ground between biomedical researchers and computer scientists. In order to elucidate the complex mechanisms of common disease the expertise of both fields is indispensable.
As these methods develop, they will be able to address most of the issues raised in the introduction of this review. Non-coding variants will be incorporated, non-linear effects will be considered and the non-contiguous relationships between elements will be encoded as networks. However, the genetic heterogeneity of human disease will remain a challenge. Network approaches to GWAS will be less susceptible than conventional GWAS. In this new paradigm, low-risk variants will be able to additively make up a high-risk model. Even variants that are only significant in particular cohorts and do not replicate will be able to cluster in the same nodes or sub-networks. But one critical limitation remains: the substrate for all these re-analyses is the collection of GWAS p-values calculated on individual variants. The aggregate statistical significance for sets of variants is only computed after the fact and only for significant variants. In order to overcome this last hurdle a new statistical foundation must be built. A method for the ab initio calculation of significance for large, heterogeneous patterns of variation needs to be conceived. Until this open question is solved, genetic association studies will continue to be undermined by genetic heterogeneity.
Acknowledgements
This study was funded by NIH grants LM010098, LM009012, AI59694 and ES007373. We would like to thank the anonymous reviewers for their very helpful comments.
Contributor Information
Richard Cowper-Sal·lari, Department of Genetics, Dartmouth Medical School, Lebanon, NH 03756.
Michael D. Cole, Departments of Pharmacology and Toxicology and Genetics, Dartmouth Medical School, Lebanon, NH 03756, USA
Margaret R. Karagas, Section of Biostatistics and Epidemiology, Department of Community and Family Medicine, Dartmouth Medical School, Lebanon, NH, USA
Mathieu Lupien, Norris Cotton Cancer Center, Department of Genetics, Dartmouth Medical School, Lebanon, New Hampshire 03756, USA.
Jason H. Moore, Email: Jason.H.Moore@Dartmouth.EDU, Institute for Quantitative Biomedical Sciences, Norris Cotton Cancer Center, Departments of Genetics and Community and Family Medicine, Dartmouth Medical School, Lebanon, NH 03756.
References
- 1.Andrew AS, Nelson HH, Kelsey KT, Moore JH, Meng AC, Casella DP, Tosteson TD, Schned AR, Karagas MR. Concordance of multiple analytical approaches demonstrates a complex relationship between dna repair gene snps, smoking and bladder cancer susceptibility. Carcinogenesis. 2006;27(5):1030–1037. doi: 10.1093/carcin/bgi284. [DOI] [PubMed] [Google Scholar]
- 2.Askland K, Read C, Moore J. Pathways-based analyses of whole-genome association study data in bipolar disorder reveal genes mediating ion channel activity and synaptic neurotransmission. Hum Genet. 2009;125(1):63–79. doi: 10.1007/s00439-008-0600-y. [DOI] [PubMed] [Google Scholar]
- 3.Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of cftr mutations–correlation with incidence data and application to screening. Hum Mutat. 2002;19(6):575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 4.Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, Retuerto AIA, Imielinski M, Hadley D, Bradfield JP, Kim C, Gidaya NB, Lindquist I, Hutman T, Sigman M, Kustanovich V, Lajonchere CM, Singleton A, Kim J, Wassink TH, McMahon WM, Owley T, Sweeney JA, Coon H, Nurnberger JI, Li M, Cantor RM, Minshew NJ, Sutcliffe JS, Cook EH, Dawson G, Buxbaum JD, Grant SFA, Schellenberg GD, Geschwind DH, Hakonarson H. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009;5(6):e1000536. doi: 10.1371/journal.pgen.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan CS, Song JS. Ccctc-binding factor confines the distal action of estrogen receptor. Cancer Research. 2008;68(21):9041–9049. doi: 10.1158/0008-5472.CAN-08-2632. [DOI] [PubMed] [Google Scholar]
- 6.Clark AG, Boerwinkle E, Hixson J, Sing CF. Determinants of the success of whole-genome association testing. Genome Res. 2005;15(11):1463–1467. doi: 10.1101/gr.4244005. [DOI] [PubMed] [Google Scholar]
- 7.Consortium EP, Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H, Giresi PG, Goldy J, Hawrylycz M, Haydock A, Humbert R, James KD, Johnson BE, Johnson EM, Frum TT, Rosenzweig ER, Karnani N, Lee K, Lefebvre GC, Navas PA, Neri F, Parker SCJ, Sabo PJ, Sandstrom R, Shafer A, Vetrie D, Weaver M, Wilcox S, Yu M, Collins FS, Dekker J, Lieb JD, Tullius TD, Crawford GE, Sunyaev S, Noble WS, Dunham I, Denoeud F, Reymond A, Kapranov P, Rozowsky J, Zheng D, Castelo R, Frankish A, Harrow J, Ghosh S, Sandelin A, Hofacker IL, Baertsch R, Keefe D, Dike S, Cheng J, Hirsch HA, Sekinger EA, Lagarde J, Abril JF, Shahab A, Flamm C, Fried C, Hackermüller J, Hertel J, Lindemeyer M, Missal K, Tanzer A, Washietl S, Korbel J, Emanuelsson O, Pedersen JS, Holroyd N, Taylor R, Swarbreck D, Matthews N, Dickson MC, Thomas DJ, Weirauch MT, Gilbert J, Drenkow J, Bell I, Zhao X, Srinivasan KG, Sung W-K, Ooi HS, Chiu KP, Foissac S, Alioto T, Brent M, Pachter L, Tress ML, Valencia A, Choo SW, Choo CY, Ucla C, Manzano C, Wyss C, Cheung E, Clark TG, Brown JB, Ganesh M, Patel S, Tammana H, Chrast J, Henrichsen CN, Kai C, Kawai J, Nagalakshmi U, Wu J, Lian Z, Lian J, Newburger P, Zhang X, Bickel P, Mattick JS, Carninci P, Hayashizaki Y, Weissman S, Hubbard T, Myers RM, Rogers J, Stadler PF, Lowe TM, Wei C-L, Ruan Y, Struhl K, Gerstein M, Antonarakis SE, Fu Y, Green ED, Karaöz U, Siepel A, Taylor J, Liefer LA, Wetterstrand KA, Good PJ, Feingold EA, Guyer MS, Cooper GM, Asimenos G, Dewey CN, Hou M, Nikolaev S, Montoya-Burgos JI, Löytynoja A, Whelan S, Pardi F, Massingham T, Huang H, Zhang NR, Holmes I, Mullikin JC, Ureta-Vidal A, Paten B, Seringhaus M, Church D, Rosenbloom K, Kent WJ, Stone EA, Batzoglou S, Goldman N, Hardison RC, Haussler D, Miller W, Sidow A, Trinklein ND, Zhang ZD, Barrera L, Stuart R, King DC, Ameur A, Enroth S, Bieda MC, Kim J, Bhinge AA, Jiang N, Liu J, Yao F, Vega VB, Lee CWH, Ng P, Shahab A, Yang A, Moqtaderi Z, Zhu Z, Xu X, Squazzo S, Oberley MJ, Inman D, Singer MA, Richmond TA, Munn KJ, Rada-Iglesias A, Wallerman O, Komorowski J, Fowler JC, Couttet P, Bruce AW, Dovey OM, Ellis PD, Langford CF, Nix DA, Euskirchen G, Hartman S, Urban AE, Kraus P, Calcar SV, Heintzman N, Kim TH, Wang K, Qu C, Hon G, Luna R, Glass CK, Rosenfeld MG, Aldred SF, Cooper SJ, Halees A, Lin JM, Shulha HP, Zhang X, Xu M, Haidar JNS, Yu Y, Ruan Y, Iyer VR, Green RD, Wadelius C, Farnham PJ, Ren B, Harte RA, Hinrichs AS, Trumbower H, Clawson H, Hillman-Jackson J, Zweig AS, Smith K, Thakkapallayil A, Barber G, Kuhn RM, Karolchik D, Armengol L, Bird CP, de Bakker PIW, Kern AD, Lopez-Bigas N, Martin JD, Stranger BE, Woodroffe A, Davydov E, Dimas A, Eyras E, Hallgŕmsdóttir IB, Huppert J, Zody MC, Abecasis GR, Estivill X, Bouffard GG, Guan X, Hansen NF, Idol JR, Maduro VVB, Maskeri B, McDowell JC, Park M, Thomas PJ, Young AC, Blakesley RW, Muzny DM, Sodergren E, Wheeler DA, Worley KC, Jiang H, Weinstock GM, Gibbs RA, Graves T, Fulton R, Mardis ER, Wilson RK, Clamp M, Cuff J, Gnerre S, Jaffe DB, Chang JL, Lindblad-Toh K, Lander ES, Koriabine M, Nefedov M, Osoegawa K, Yoshinaga Y, Zhu B, de Jong PJ Program, N. C. S., of Medicine Human Genome Sequencing Center, B. C., Center, W. U. G. S., Institute, B., Institute, C. H. O. R. Identification and analysis of functional elements in 1 genome by the encode pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Consortium WTCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cordell HJ. Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet. 2002;11(20):2463–2468. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- 10.Cordell HJ. Genome-wide association studies: Detecting gene-gene interactions that underlie human diseases. Nat Rev Genet. 2009 doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295(5558):1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 12.Dostie J, Dekker J. Mapping networks of physical interactions between genomic elements using 5c technology. Nat Protoc. 2007;2(4):988–1002. doi: 10.1038/nprot.2007.116. [DOI] [PubMed] [Google Scholar]
- 13.Eleftherohorinou H, Wright V, Hoggart C, Hartikainen A-L, Jarvelin M-R, Balding D, Coin L, Levin M. Pathway analysis of gwas provides new insights into genetic susceptibility to 3 inflammatory diseases. PLoS ONE. 2009;4(11):e8068. doi: 10.1371/journal.pone.0008068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emily M, Mailund T, Hein J, Schauser L, Schierup MH. Using biological networks to search for interacting loci in genome-wide association studies. Eur J Hum Genet. 2009;17(10):1231–1240. doi: 10.1038/ejhg.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fisher R. The correlation between relatives on the supposition of Mendelian inheritance. Transactions of the Royal Society of Edinburgh. 1918;52(2):399–433. [Google Scholar]
- 16.Gertz J, Gerke JP, Cohen BA. Epistasis in a quantitative trait captured by a molecular model of transcription factor interactions. Theor Popul Biol. 2009 doi: 10.1016/j.tpb.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PMA, Chiavacci R, Annaiah K, Thomas K, Hou C, Glaberson W, Flory J, Otieno F, Garris M, Soorya L, Klei L, Piven J, Meyer KJ, Anagnostou E, Sakurai T, Game RM, Rudd DS, Zurawiecki D, McDougle CJ, Davis LK, Miller J, Posey DJ, Michaels S, Kolevzon A, Silverman JM, Bernier R, Levy SE, Schultz RT, Dawson G, Owley T, McMahon WM, Wassink TH, Sweeney JA, Nurnberger JI, Coon H, Sutcliffe JS, Minshew NJ, Grant SFA, Bucan M, Cook EH, Buxbaum JD, Devlin B, Schellenberg GD, Hakonarson H. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459(7246):569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gobbi MD, Viprakasit V, Hughes JR, Fisher C, Buckle VJ, Ayyub H, Gibbons RJ, Vernimmen D, Yoshinaga Y, de Jong P, Cheng J-F, Rubin EM, Wood WG, Bowden D, Higgs DR. A regulatory snp causes a human genetic disease by creating a new transcriptional promoter. Science. 2006;312(5777):1215–1217. doi: 10.1126/science.1126431. [DOI] [PubMed] [Google Scholar]
- 19.Haiman CA, Marchand LL, Yamamato J, Stram DO, Sheng X, Kolonel LN, Wu AH, Reich D, Henderson BE. A common genetic risk factor for colorectal and prostate cancer. Nat Genet. 2007;39(8):954–956. doi: 10.1038/ng2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459(7243):108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Idaghdour Y, Czika W, Shianna KV, Lee SH, Visscher PM, Martin HC, Miclaus K, Jadallah SJ, Goldstein DB, Wolfinger RD, Gibson G. Geographical genomics of human leukocyte gene expression variation in southern morocco. Nat Genet. 2010;42(1):62–67. doi: 10.1038/ng.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia L, Landan G, Pomerantz M, Jaschek R, Herman P, Reich D, Yan C, Khalid O, Kantoff P, Oh W, Manak JR, Berman BP, Henderson BE, Frenkel B, Haiman CA, Freedman M, Tanay A, Coetzee GA. Functional enhancers at the gene-poor 8q24 cancer-linked locus. PLoS Genet. 2009;5(8):e1000597. doi: 10.1371/journal.pgen.1000597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson AD, O’Donnell CJ. An open access database of genome-wide association results. BMC Med Genet. 2009;10:6. doi: 10.1186/1471-2350-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein ctcf-binding sites in the human genome. Cell. 2007;128(6):1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436(7052):876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-tcf complex in apc−/− colon carcinoma. Science. 1997;275(5307):1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 27.Kotti S, Bickeboller H, Clerget-Darpoux F. Strategy for detecting susceptibility genes with weak or no marginal effect. Hum Hered. 2007;63(2):85–92. doi: 10.1159/000099180. [DOI] [PubMed] [Google Scholar]
- 28.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones S, Marra M. Circos: an information aesthetic for comparative genomics. Genome research. 2009;19(9):1639. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lupien M, Brown M. Cistromics of hormone-dependent cancer. Endocr Relat Cancer. 2009;16(2):381–389. doi: 10.1677/ERC-09-0038. [DOI] [PubMed] [Google Scholar]
- 31.Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, Brown M. Foxa1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132(6):958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 33.McClellan J, King M-C. Genetic heterogeneity in human disease. Cell. 2010;141(2):210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 34.Michalski R. A theory and methodology of inductive learning. Machine Learning: an artificial intelligence approach. 1983;1:83–134. [Google Scholar]
- 35.Moore JH, Asselbergs FW, Williams SM. Bioinformatics challenges for genome-wide association studies. Bioinformatics. 2010 doi: 10.1093/bioinformatics/btp713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore JH, Williams SM. Epistasis and its implications for personal genetics. Am J Hum Genet. 2009;85(3):309–320. doi: 10.1016/j.ajhg.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Riddersträle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. Pgc-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 38.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-tcf signaling in colon cancer by mutations in beta-catenin or apc. Science. 1997;275(5307):1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 39.Orphanides G, Reinberg D. A unified theory of gene expression. Cell. 2002;108(4):439–451. doi: 10.1016/s0092-8674(02)00655-4. [DOI] [PubMed] [Google Scholar]
- 40.Pattin KA, Moore JH. Exploiting the proteome to improve the genome-wide genetic analysis of epistasis in common human diseases. Hum Genet. 2008;124(1):19–29. doi: 10.1007/s00439-008-0522-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pattin KA, Moore JH. Role for protein-protein interaction databases in human genetics. Expert review of proteomics. 2009;6(6):647–659. doi: 10.1586/epr.09.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phillips JE, Corces VG. Ctcf: master weaver of the genome. Cell. 2009;137(7):1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, Beckwith CA, Chan JA, Hills A, Davis M, Yao K, Kehoe SM, Lenz H-J, Haiman CA, Yan C, Henderson BE, Frenkel B, Barretina J, Bass A, Tabernero J, Baselga J, Regan MM, Manak JR, Shivdasani R, Coetzee GA, Freedman ML. The 8q24 cancer risk variant rs6983267 shows long-range interaction with myc in colorectal cancer. Nat Genet. 2009;41(8):882–884. doi: 10.1038/ng.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reif DM, Dudek SM, Shaffer CM, Wang J, Moore JH. Exploratory visual analysis of pharmacogenomic results. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing. 2005:296–307. [PubMed] [Google Scholar]
- 45.Ripperger T, Gadzicki D, Meindl A, Schlegelberger B. Breast cancer susceptibility: current knowledge and implications for genetic counselling. Eur J Hum Genet. 2009;17(6):722–731. doi: 10.1038/ejhg.2008.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ritchie MD, Hahn LW, Roodi N, Bailey LR, Dupont WD, Parl FF, Moore JH. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Hum Genet. 2001;69(1):138–147. doi: 10.1086/321276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee Y-H, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimäki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King M-C, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, Sharp PA. Divergent transcription from active promoters. Science. 2008;322(5909):1849–1851. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4c) Nat Genet. 2006;38(11):1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 50.Stern MC, Lin J, Figueroa JD, Kelsey KT, Kiltie AE, Yuan J-M, Matullo G, Fletcher T, Benhamou S, Taylor JA, Placidi D, Zhang Z-F, Steineck G, Rothman N, Kogevinas M, Silverman D, Malats N, Chanock S, Wu X, Karagas MR, Andrew AS, Nelson HH, Bishop DT, Sak SC, Choudhury A, Barrett JH, Elliot F, Corral R, Joshi AD, Gago-Dominguez M, Cortessis VK, Xiang Y-B, Gao Y-T, Vineis P, Sacerdote C, Guarrera S, Polidoro S, Allione A, Gurzau E, Koppova K, Kumar R, Rudnai P, Porru S, Carta A, Campagna M, Arici C, Park SSL, Garcia-Closas M of Bladder Cancer, I. C. Polymorphisms in dna repair genes, smoking, and bladder cancer risk: findings from the international consortium of bladder cancer. Cancer Research. 2009;69(17):6857–6864. doi: 10.1158/0008-5472.CAN-09-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W, Barclay E, Lubbe S, Martin L, Sellick G, Jaeger E, Hubner R, Wild R, Rowan A, Fielding S, Howarth K, Consortium C, Silver A, Atkin W, Muir K, Logan R, Kerr D, Johnstone E, Sieber O, Gray R, Thomas H, Peto J, Cazier J-B, Houlston R. A genome-wide association scan of tag snps identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007;39(8):984–988. doi: 10.1038/ng2085. [DOI] [PubMed] [Google Scholar]
- 53.Torkamani A, Topol EJ, Schork NJ. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics. 2008;92(5):265–272. doi: 10.1016/j.ygeno.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tuupanen S, Turunen M, Lehtonen R, Hallikas O, Vanharanta S, Kivioja T, Björklund M, Wei G, Yan J, Niittymäki I, Mecklin J-P, Järvinen H, Ristimäki A, Di-Bernardo M, East P, Carvajal-Carmona L, Houlston RS, Tomlinson I, Palin K, Ukkonen E, Karhu A, Taipale J, Aaltonen LA. The common colorectal cancer predisposition snp rs6983267 at chromosome 8q24 confers potential to enhanced wnt signaling. Nat Genet. 2009;41(8):885–890. doi: 10.1038/ng.406. [DOI] [PubMed] [Google Scholar]
- 55.Tyler AL, Asselbergs FW, Williams SM, Moore JH. Shadows of complexity: what biological networks reveal about epistasis and pleiotropy. Bioessays. 2009;31(2):220–227. doi: 10.1002/bies.200800022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.von Mering C, Jensen LJ, Kuhn M, Chaffron S, Doerks T, Krüger B, Snel B, Bork P. String 7-recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007;35:D358–D362. doi: 10.1093/nar/gkl825. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King M-C, Sebat J. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320(5875):539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 58.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh T-Y, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40(7):897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wright JB, Brown SJ, Cole MD. Upregulation of c-myc in cis through a large chromatin loop linked to a cancer risk-associated snp in colorectal cancer cells. Mol Cell Biol. 2010 doi: 10.1128/MCB.01384-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zanke BW, Greenwood CMT, Rangrej J, Kustra R, Tenesa A, Farrington SM, Prendergast J, Olschwang S, Chiang T, Crowdy E, Ferretti V, Laflamme P, Sundararajan S, Roumy S, Olivier J-F, Robidoux F, Sladek R, Montpetit A, Campbell P, Bezieau S, O’Shea AM, Zogopoulos G, Cotterchio M, Newcomb P, McLaughlin J, Younghusband B, Green R, Green J, Porteous MEM, Campbell H, Blanche H, Sahbatou M, Tubacher E, Bonaiti-Pellié C, Buecher B, Riboli E, Kury S, Chanock SJ, Potter J, Thomas G, Gallinger S, Hudson TJ, Dunlop MG. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet. 2007;39(8):989–994. doi: 10.1038/ng2089. [DOI] [PubMed] [Google Scholar]
- 61.Zhao Z, Tavoosidana G, Sjölinder M, Göondoör A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, Pant V, Tiwari V, Kurukuti S, Ohlsson R. Circular chromosome conformation capture (4c) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38(11):1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]