

Abstract
Cu/O2 intermediates in biological, homogeneous, and heterogeneous catalysts exhibit unique spectral features that reflect novel geometric and electronic structures that make significant contributions to reactivity. This review considers how the respective intermediate electronic structures overcome the spin forbidden nature of O2 binding, activate O2 for electrophillic aromatic attack and H-atom abstraction, catalyze the 4 e- reduction of O2 to H2O, and discusses the role of exchange coupling between Cu ions in determining reactivity.
Our focus has been on the use of spectroscopic methods to elucidate active sites in catalysis. In the area of Cu/O2 chemistry, this has mostly involved studies on metalloenzymes, however these have led to parallel studies in Cu coordination chemistry and now to studies on Cu sites in zeolites. There are five main topics in Cu/O2 biological, homogeneous and heterogeneous reactivity that will be the scope of this overview. First is the spin-forbidden, reversible binding of dioxygen by the coupled binuclear Cu site in hemocyanin. Next, we will consider O2 activation by coupled binuclear copper sites for electrophilic attack on phenolic substrates in tyrosinase and related model complexes. We will then consider H-atom abstraction from relatively weak C-H bonds (~85 kcal/mol) by the “non-coupled” binuclear Cu enzymes and how the difference in magnetic “exchange” coupling can control reactivity. We will then move to the four e- reduction of O2 to H2O by the multi-copper oxidases at a trinuclear Cu cluster, a structural motif originally defined to be present by MCD spectroscopy.1, 2 Finally, we will focus on O2 activation for H-atom abstraction from the strong C-H bond of methane (~105 kcal/mol) which in biology is accomplished by methane monooxygenases (MMO) but can now be achieved in the active sites of zeolites (CuZSM-5). The copper-oxygen intermediates in these systems have unique spectroscopic features that we have shown to reflect novel geometric and electronic structures that make key contributions to reactivity.
I. Reversible O2 Binding: Coupled binuclear Cu Sites
Hemocyanin (Hc) functions as an extracellular oxygen transport protein in arthropods and mollusks.3 Deoxy-Hc contains 2 Cu(I) ions that reversibly bind O2 to form the binuclear cupric site in oxy-Hc. Thus, 2e− are transferred to O2 reducing it to the peroxide level. As will be discussed below, oxy-Hc has unique spectral features, and to understand these we first consider “normal” peroxide-Cu(II) bonding.4
O2 is a triplet that has two unpaired electrons in the doubly degenerate π* orbitals. Reduction of O2 to peroxide leads to a fully occupied π* HOMO. As shown in Fig. 1A, upon binding O22− end-on to a Cu(II), one π* orbital is stabilized due to σ bonding with the d9 Cu(II) half occupied d orbital, which is in turn destabilized. This leads to the characteristic EPR spectrum of an unpaired e− in the Cu d based orbital (here dx2− y2) shown in Fig. 1A. This bonding also produces a characteristic O22− charge transfer (CT) transition at ~500 nm with a molar extinction coefficient (ε) of ~ 5000 M−1cm−1. The intensity of this charge transfer transition quantifies the strength of the peroxide donor interaction with the Cu(II). Tuning a laser into this CT transition leads to resonance enhancement of an O-O stretch at 803 cm−1 (Fig. 1A bottom inset) in its Raman spectrum, a feature that is characteristic of peroxo bound end-on to a single Cu(II).
Fig. 1.

Molecular orbital energy diagrams for mononuclear copper peroxo (A) and binuclear copper peroxo end-on species (B) with their corresponding spectroscopic features given below.
We next consider the additional factors involved in bridging the peroxide between two Cu(II) (Fig. 1B).5 Since the two Cu(II) ions are related by symmetry, we take symmetric and anti-symmetric combinations of their d orbitals which then bond to the peroxide. From Fig. 1B, the O22− π* HOMO is involved in a σ bonding interaction with the symmetric combination of d orbitals that splits the two Cu based MOs in energy. This splitting is large enough to overcome electron-electron repulsion resulting in a spin paired, singlet ground state. This splitting is the origin of antiferomagnetic coupling, hence the nomenclature “coupled” binuclear Cu(II) sites for proteins with two bridged Cu(II) ions; in Fig. 1B, the bridging peroxide orbital serves as the superexchange pathway for the anti-ferromagnetic coupling. This singlet ground state, of course, has no EPR signal which is the characteristic spectroscopic feature of the coupled binuclear Cu active sites. The O22− donor bonding again produces a CT transition, but now with twice the intensity (ε ~10000 M−1cm−1) due to the bridging peroxo having donor interactions with two Cu(II) ions. Tuning a laser into this CT transition produces an O-O stretch of 830 cm−1 (Fig. 1B bottom insert), higher then the mononuclear Cu(II)-O22− case shown in Fig. 1A. The stronger σ donor interaction of the bridging peroxo removes more electron density from the antibonding O22− π* orbital, resulting in a stronger O-O bond.
In Fig. 2B, we compare the normal spectral features of an O22− bound end-on to a single Cu(II) to those of oxy-Hc. Oxy-Hc has O22− bound in a side-on (μ-η2:η2) bridged binuclear cupric structure (Fig. 2A). Its CT transition is four times as intense as in the mononuclear-Cu(II)-peroxo complex and is 10,000 cm−1 higher in energy. Its resonance Raman spectrum6, 7 shows that the O-O stretch has now decreased to 750 cm−1 (insert). Given that it contains a coupled binuclear Cu(II) site, it exhibits no EPR signal. SQUID magnetic susceptibility8 indicates it is anti-ferromagnetic coupled with a singlet/triplet splitting > 600 cm−1.
Fig. 2.

The active site structure (A) and the unique spectral features of oxy-Hc (red) are compared to those of normal copper (blue) in (B). A schematic molecular orbital diagram and the corresponding HOMO and LUMO orbital contours determined from SCF-Xα-SW calculations illustrates the importance of σ* backbonding in oxy-Hc (C). A comparison between peroxo and disulfido Cu(II) cores indicated that limited backbonding into σ* was responsible for the low νO-O observed in oxy-Hc.
The novel electronic structure description we defined for this side-on bridged Cu2(O2) structure elucidated its unique spectral features, Fig. 2C. Again, we take symmetric and anti-symmetric contributions of the valence d orbitals on each copper. The symmetric contribution undergoes a strong bonding interaction with the O22− orbital. From the contour of the corresponding antibonding LUMO, the side-on peroxide now has four donor interactions with the two Cu(II), leading to the higher energy and higher intensity (ε ~ 20000 M−1cm−1) of its Cu(II) CT transition. This O22− donor interaction takes even more electron density out of the orbital (relative to the end-on bridged peroxide in Fig. 1B). Thus, the O-O stretch would be expected to increase in energy, however, it decreases to ~750 cm−1. This results from an additional bonding interaction that only occurs in the side-on peroxide bridged structure. The peroxide has a higher energy unoccupied σ* orbital which now interacts with the occupied copper based HOMO. This backbonding interaction9, 10 shifts electron density from the Cu(II)2 into the O22− σ* orbital, Fig. 2C. From a comparative study of peroxo and disulfido model complexes11, electron density in the σ* orbital is very efficient at weakening the O-O bond (Fig. 2D) leading to the low νO-O. Further, the large LUMO/HOMO splitting leads to the strongly coupled antiferromagnet singlet ground state where the dominant contribution to this coupling is the bonding interaction of the Cu(II)’s with the O22− orbital. This electronic structure provides the basis for overcoming the spin-forbidden nature of O2 binding to Hc.
Triplet O2 binds to singlet deoxy-Hc to form oxy-Hc which has a singlet ground-state (vide supra). The reaction coordinate for O2 binding is given in Fig. 3A.12 Starting from Oxy-Hc, as peroxide is raised from the molecular plane, the structure first butterflies, then goes to an asymmetric end-on/side-on bridged structure, then to an end-on/end-on bridged structure, and finally O2 is lost. These structures are a result of maximizing the metal-ligand overlap along the reaction coordinate. Fig. 3B presents the transfer of charge along this reaction coordinate. The peroxide gets less negative and the Cu’s get less positive at intermediate distances of O2 from the Cu molecular plane indicating CT from the peroxo to the two Cu(II)’s. Importantly, the charge on the two Cu’s (red and blue) change at the same rate, even in the asymmetrically bridged structure (dashed lines). This indicates a simultaneous two electron transfer for O2 binding to this coupled binuclear Cu active site.
Fig. 3.

The structures along the reaction coordinate of triplet dioxygen binding to deoxy-Hc is shown in 3A. The Mulliken charges of the atoms along the reaction coordinate are shown in 3B (triangles represent singlet states, circles represent triplet states, and dashed lines for asymmetric coordination). The potential energy surface of the triplet and the spin projected singlet as well as a schematic representation of the reaction that illustrates the mechanism for overcoming the spin-forbiddeness are shown in 3C. Adapted with permission from reference 12, © 2001 American Chemical Society.
The contribution of this coupling in overcoming the spin forbidden nature of this reaction is given by the potential energy surface in Fig. 3C. From the left, in the side on bridged structure the two 1/2 occupied Cu d orbitals overlap the orbital providing the antiferromagnetic coupling that strongly stabilizes the singlet ground state. Butterflying this structure along the reaction coordinate leads to each Cu(II)’s overlap with a different peroxide π* orbital; these orthogonal magnetic orbitals stabilize the triplet ground state (the red PES in Fig. 3C). From here, two electrons of the same spin can be transferred, one from each π* orbital to each Cu, and triplet O2 is released. Thus, along the reaction coordinate O2 binding leads to the redistribution of the spins to the remote copper centers, eliminating the two electron exchange stabilization of triplet O2. The strong O22− superexchange pathway, formed along the reaction coordinate, then stabilizes the singlet. Finally, note that the energy of O2 binding by the coupled binuclear Cu site is exothermic by ~ 4 kcal/mol.
II. O2 Activation for Electrophilic Aromatic Substitution: Coupled Binuclear Cu Sites
The enzyme tyrosinase (Tyr) binds O2 and activates it for the hydroxylation and oxidation of phenol to quinone. Our early spectroscopic studies on oxy-Tyr showed it has very similar unique spectral features to oxy-Hc.13 As shown in Fig. 4A, oxy-Tyr displays the same intense (ε ~ 20000 M−1cm−1) 350 nm peroxide to Cu CT transition in its absorption spectrum, (and equivalent weaker, characteristic transitions at lower energy, shown in the expanded scale in Fig. 4A) and the same low frequency intra-peroxide O-O stretching feature (~750 cm−1).14 Based on these spectral signatures, it is apparent that oxy-Tyr has the same geometric and electronic structure as oxy-Hc; a side on-peroxo bridged binuclear Cu(II) site.
Fig. 4.

The spectral features of oxy-Hc and oxy-Ty show in Fig 4A indicate that the two enzymes contain structurally equivalent active sites. Yet, the reaction of both with azide (4B left) shows differential substrate access between the two proteins. Addition of the competitive inhibitor mimosine displaces peroxide only in Tyr and results in a ligand to metal CT transition indicating substrate binds directly to the Cu. The unique EPR spectra of half-met Ty (N. crassa) with mimosine bound to the copper active site shows some dz2 character is mixed into the dx2y2 ground state indicating a trigonal bipyrimidal distortion at the active site. Reproduced with permission from reference 3, © 1996 American Chemical Society.
The major difference between these two enzymes is access to their coupled binuclear Cu sites.15 From Fig. 4B, the rate of the associative displacement of peroxide by N3− from the oxy sites varied by four orders of magnitude with oxy-Tyr having the most accessible active site. When the substrate analogue mimosine (shown in Fig. 4B middle) was added, it could only displace peroxide from the oxy-Tyr site. This resulted in a new absorption feature, a mimosine to Cu(II) CT transition.16 Since CT intensity requires orbital overlap, this was the first experiment that demonstrated that the substrate directly binds to the Cu center in Tyr. We could further probe this binding using a derivative of Tyr we prepared where the site was half oxidized and thus EPR active, the “half-met” state (Fig. 4B right).15,16 Addition of mimosine to this half-met derivative (of N. crassa Tyr) resulted in the unusual EPR spectrum (i.e. dz2 mixing) in Fig. 4B right indicating that substrate binding produces a trigonal bipyramidal distortion of the Cu site.16 These results lead us to the now generally invoked mechanism of Tyr, based on the side-on peroxy bridged structure of oxy Hc in Fig. 5 (note, the axial histidines are eliminated for clarity).15 In this mechanism, phenolate substrate binds directly to one Cu, the substrate undergoes a trigonal bipyramidal distortion leading to orthohydroxylation of the ring. The resulting bidentate coordinated bridging catecholate is then oxidized to the quinone, leaving a bicuprous center that reacts with O2 to continue the catalytic cycle.
Fig. 5.

Consensus mechanism of Tyr adapted from reference 15 to the side-on bridged structure (note axial His ligand not included for clarity).
The timely issue now is whether the side-on peroxy species directly reacts with the aromatic ring or is first converted to a bis μ-oxo structure before hydroxylation (Fig. 5 right). This issue derives from the chemistry of Tolman and co-workers where in some models, they found that the side-on peroxo species can be in an equilibrium with a bis μ-oxo species.17 We have studied the electronic structure of this bis μ-oxo Cu2 core in model complexes and from the Cu K edge XAS spectra in Fig. 6 Bottom,18 the 8979 eV Cu(II) pre edge feature in the side-on peroxide structure shifts up in energy by 1.9 eV in the bis μ-oxo structure demonstrating that it is oxidized to 2Cu(III). Thus in going from the side-on peroxo bridged structure to the bis μ-oxo species, the O22− σ* orbital that is involved in backbonding with the metal d based HOMO of the side-on bridged structure (Fig. 2C), comes down in energy upon O-O elongation and oxidizes the Cu based HOMO leading to the bis oxo-bridged Cu(III) electronic structure (Fig 6A right).19 This produces a new oxo to Cu(III) CT transition at lower energy than the CT transition of the side-on peroxo bridged species (400 nm vs 350 nm in Fig. 6B). Excitation into this transition produces a very different resonance Raman spectrum with an intense peak at 600 cm−1 associated with the symmetric stretch of the Cu2O2 core.17 The side-on peroxo to bis μ-oxo correlation19 in Fig. 6A predicts that there are two frontier MOs (i.e. Cu based LUMO’s with significant oxygen character) available for electrophilic attack on an aromatic ring (Fig 6C); the symmetric combination of Cu d containing the character for the side on peroxo and the antisymmetric combination of Cu d containing the σ* character of the bis μ-oxo structure. In functional model complexes, we have compared these two frontier MO’s to evaluated the reactivity of both structures.
Fig. 6.

A) Frontier molecular orbital correlation of Cu(II)2(O2) ⇌ Cu(III)2(O)2 (μ-peroxo (blue) ⇌ μ-oxo (green)) equilibrium based on B) the unique spectral signatures present in XAS, UV-Vis, and rR spectra. C) The LUMO in both structures as possible frontier molecular orbitals for electrophillic aromatic attack.
In collaborative efforts with Karlin and co-workers, we have investigated the pyridine coordinated xylyl bridged complex in Fig. 7A.20 This complex reacts with O2 to hydroxylate the ring of the tether,21, 22 and the oxy intermediate could be trapped at cryogenic temperatures. Resonance Raman data in Fig. 7A show no 600 cm−1 stretch indicating that no bis μ-oxo Cu(III)2 species is present, but does show the 750 cm−1 stretch of a side-on μ-peroxo Cu(II)2 species.20 This disappears as the C-O stretch of the hydroxylated product appears. Thus, the side-on peroxo bridged Cu(II)2 species is capable of direct electrophilic attack. With Stack and coworkers, we have studied in the ethylenediamine ligated oxy-species 3 in Fig. 7B.23, 24 The resonance Raman spectrum shows a υO-O = 730 cm−1 indicating that it is a side-on peroxy Cu(II)2 species. Upon addition of phenolate, the 730 cm−1 feature is eliminated and an ~600 cm−1 vibration grows in showing that coordination of phenolate to the Cu converts the side-on μ-peroxo to a bis μ-oxo Cu(III)2 species. This then goes on to react to produce the carbonyl stretch of the oxygenated product. These models studies show that both the bis μ-oxo and side-on μ-peroxo species are capable of direct electrophilic aromatic substitution.
Fig. 7.

The reaction pathway of Cu2(μ-peroxo) (A) vs Cu(III)2(μ-oxo)2 (B) with aromatic substrates as determined by resonance Raman spectroscopy. Both electrophiles proceed via similar reaction pathways, and thus should be considered in the Tyr mechanism.
To better understand the mechanism, we have computationally evaluated both reaction coordinates. The computationally determined reaction coordinates are shown at the bottom of Fig. 7A and 7B.24 For the bis μ-oxo complex (Fig. 7B bottom), the ortho carbon reacts with the proximal oxo to form an sp3 carbon on the ring, predicting the inverse KIE that is observed experimentally.23 Two electrons from the ring reduce the two Cu(III), and finally the C-O bond is fully formed upon deprotonation of the ring. For the side-on peroxo structure in Fig. 7A (bottom),25 again an sp3 C is formed upon peroxo attack on the ring and the C-O bond is fully formed upon loss of the proton. However, for the side on peroxy structure the two electrons from the ring flow directly into the σ* orbital of the peroxide to cleave the O-O bond. Thus, no bis μ-oxo Cu(III)2 is formed along the reaction coordinate, and both the side-on peroxo and bis μ-oxo structures are equivalently effective in electrophilic attack on an aromatic ring. It now remains to be determined whether the side on peroxo Cu(II)2 species of oxy Tyr directly attacks the substrate or if it is converted by substrate coordination to a bis μ-oxo Cu(III)2 prior to electrophilic aromatic substitution.
III. O2 Activation for H-atom Abstraction: Non-coupled Binuclear Cu sites
Dopamine β monooxygenase (DβM) and peptidylglycine α-hydroxylating monooxygenase (PHM) both activate O2 to abstract an H atom from the activated C-H bonds in the relevant substrates26 (Fig. 8). Both contain non-coupled binuclear Cu active sites in that the two Cu’s involved in reactivity are > 11 Å apart in the crystal structure of PHM and show no spectroscopic feature indicative of magnetic coupling.26 The CuM center (having a coordinated methionine) is the catalytic site while CuH (all His ligands) supplies the extra electron required for the reaction (Fig. 8)27, 28. Initially, it was proposed that O2 was reduced by two electrons to produce a hydroperoxo intermediate at the CuM site which went on to perform the H-atom abstraction.29 Thus we studied a tris-pyrazolyl-borate Cu(II)-OOH model complex prepared by Fujisawa et al which exhibited a υO-O at 843 cm−1 (Fig. 9A, right)30. We computationally modeled this end-on bound hydroperoxo into the CuM site based on spectroscopic data on the resting and ligand bound PHM sites.31 This led to the FMO shown in Fig. 9A that contained little character on the distal oxygen (2%) and was thus not activated for H atom abstraction. We then considered the alternative; a one electron reduction of O2 by CuM to produce a superoxide intermediate.31 Fujisawa et al. had prepared a side-on bound O2-Cu tris-pyrazolyl-borate complex, which from its resonance Raman spectrum (Fig. 9B, exhibiting a υO-O at 1043 cm−1) is a superoxide and from SQUID magnetic susceptibility has a singlet ground state.32 We modeled this superoxide at the CuM site and found an FMO (Fig. 9B) that is non-polarized and covalently delocalized over the Cu and the orbital.31 This FMO has ~ 32% character on each oxygen and thus is far more activated for H atom abstraction.
Fig. 8.

Left: The regiospecific and stereospecific C-H bond hydroxylation reactions catalyzed by DβM and PHM. Right: Structures of oxidized CuIIM and CuIIH for PHM. The two sites are separated by 11 Å and show no magnetic interaction, thus are non-coupled.
Fig. 9.

A. Energy optimized geometry of oxidized putative hydroperoxo intermediate CuIIM-OOH, [CuIIM(Met)(His)2(H2O)(OOH)]+, relevant bond lengths (Å) are indicated. Color codes: Cu, green; N, blue; C, gray; O, red; S, yellow; H, light blue. Surface contour plot of frontier molecular orbital (LUMO) of the CuIIM-OOH model. Orbital decompositions (%) are given. rR spectra of the CuII-OOH tris-pyrazolyl-borate model complex in the υO-O region excited at 568.2 nm.30 B. Energy optimized geometry of putative side-on CuIIM-superoxo species, [CuIIM(Met)(His)2 (O2−)]+. Surface contour plot of frontier molecular orbital (LUMO) of the side-on CuIIM-superoxo. rR spectra of the CuII-superoxo model complex in the υO-O region excited at 482.5 nm. Reproduced with permission from references 31, 38, © 2004 American Chemical Society and © 2004 the National Academy of Sciences of the United States of America.
A crystal structure33 subsequently appeared for an intermediate in PHM which showed O2 end-on bound to CuM, and a structurally defined end-on O2 bound Cu model complex [TMG3trenCuO2]+34 became available which has a triplet ground state35 (Fig. 10A). From electronic structure calculations36 (Fig. 10B and 11A), this triplet has two half occupied orthonormal magnetic orbitals, one being the Cu d orbital involved in antibonding interaction with as in the side-on superoxo structure but the other is the second superoxide π* orbital, labeled . Thus in going from the side-on bound structure, which has a covalent, delocalized, fully unoccupied d- LUMO, to the end-on bound superoxo, the splitting has decreased, resulting in the high spin ground state (Fig. 11A). To experimentally probe this splitting we turned to resonance Raman spectroscopy. The end-on bound superoxide exhibits a υO-O of 1120 cm−1, which was used to generate an excitation profile by scanning the laser through the absorption spectrum of this complex.36 From Fig. 11B, two peaks are observed. One is intra-ligand transition of (the first time this has been assigned for any bound superoxide complex), and the higher energy feature is the to Cu CT transition. The splitting between these transitions (9500 cm−1) reflects the energy splitting of the two half occupied orbitals, which is less, by a factor of ~2, than the electron-electron repulsion required to spin pair. Importantly, for the end-on bound superoxo-Cu(II), this leads to a new FMO, the low energy which is dominantly localized on the superoxide and thus has ~50% character on the distal oxygen (Fig. 12). This FMO would be predicted to be highly activated for H-atom abstraction.
Fig. 10.

A. Crystal structures of the precatalytic PHM-O2 complex36 and of [TMG3trenCuO2]+ 34. B. The two triplet frontier molecular orbitals of [TMG3trenCuO2]+.
Fig. 11.

A. Schematic of bonding in end-on superoxo [TMG3trenCuO2]+ (left) and side on superoxo HB(3-R-5-iPrpz)3CuO2 (right). B. Low temperature (5K) absorption spectrum of [TMG3trenCuO2]+ (black) and rR profiles for υ(Cu-O) (435 cm−1, blue) and υ(O-O) (1120 cm−1, red) (77K). Insets: rR spectra of [TMG3trenCuO2]+ with 16O2 in the regions of υ(Cu-O) and υ(O-O). Adapted from reference 36. Assignments of peaks in the profiles given at bottom.
Fig. 12.

The frontier molecular orbital of a CuIIM-superoxo modeled in PHM.
We then extended these FMO results to evaluate the reaction coordinates for H-atom abstraction by 2 electron reduced hydroperoxo and by 1 electron reduced superoxide-Cu(II)M intermediates.31, 37 From Fig. 13 left, 2 electron reduction of O2 to form a Cu(II)M-OOH species gives an activation barrier of > 40 kcal/mol and should not be active in H-atom abstraction. Alternatively, 1 electron reduction to produce a triplet end-on Cu(II)-O2− complex (Fig. 13 right) has a calculated activation barrier of ~ 19 kcal/mol, and a calculated activation enthalpy in reasonable agreement with experiment.37 Thus the one electron reduced superoxide species is a viable intermediate in the non-coupled binuclear copper enzymes.
Fig. 13.

Summary of the 2-e− (left, blue) and 1-e− (right, red for H atom abstraction, green for subsequent steps) reaction pathways in PHM. For clarity, His and Met ligands are omitted in the structures. Only species that are essential to the reactions are indicated on the scheme. Free energies are referenced to the initial reactions, which are set to zero. The proton and H2O ligand in steps v and a and the H2O ligand in step iii are from the solvent.
The differences between the coupled and non-coupled binuclear copper proteins led us to evaluate the impact exchange coupling had on determining the individual reaction mechanisms.38 The exchange coupling (J) can be related to the electronic coupling matrix element for electron transfer , where U is the repulsion of two electrons on the same Cu.39, 40 For the coupled-binuclear copper sites, the exchange coupling is large, simultaneous two electron reduction of O2 is observed (Fig. 3) and this generates an FMO active in electrophilic aromatic attack. For the non-coupled binuclear copper Cu enzymes, the exchange interaction is unmeasurably weak, thus HDA is very small. This would allow initial one electron reduction of O2 at CuM generating a Cu(II)-O2− intermediate with an FMO active for H-atom abstraction. Then, as shown on the right of Fig. 13, a high potential intermediate can be generated at a later stage in the reaction (proposed to be a cupryl species), which can drive the transfer of the 2nd electron from CuH to complete the reaction even with a low electronic coupling between the Cu’s.
At this point, spectroscopic data on the O2/Cu intermediate in PHM are required to define the relevance of these model studies and calculations to the non-coupled binuclear Cu enzymes.
IV. O2 Reduction to H2O: Trinuclear Cu Cluster Sites
The multicopper oxidases (MCOs) couple four 1 electron oxidations of substrate to the 4 electron reduction of O2 to H2O, often with a low overpotential.3 From Fig. 14A, electrons enter at the T1 (or blue Cu) site and are rapidly transferred over 13 Å through a Cys-His pathway to a trinuclear Cu Cluster where O2 is reduced to H2O.41, 42 The trinuclear Cu cluster (TNC)1, 2 is comprised of a T3 pair of coppers and a single T2 Cu center. The T3 pair is held in the protein by 3His per Cu and the two Cu’s are bridged by hydroxide leading to an antiferromagnetically coupled singlet ground state. The T2 Cu is 3.5 Å from the T3 pair; it is coordinated by one His above and one below the trigonal plane and has a hydroxide ligand oriented external to the cluster. The T2 has a normal Cu(II) EPR signal. We have studied the resting trinuclear Cu site in some detail both experimentally and computationally and find open coordination positions on all three Cu’s oriented towards the center of the cluster, ready to bridge oxygen intermediates (Fig 14B).43 It is interesting that this 4+ site stays coordinatively unsaturated in H2O solvent; this results from four anionic carboxylates within 10 Å of the cluster, which destabilize H2O from binding as hydroxide or oxide in the center of the cluster and tune the redox properties of the site.
Fig. 14.

The stucture of the MCO active site (A) with arrows indicating the flow of substrates, electrons (e−) and O2, and the electronic structure of the resting TNC (B). Reproduced with permission from reference 42, © 2008 RSC Publishing.
While the T3 site has 3 His per Cu and is antiferomagnetically coupled, it has a very different O2 reactivity relative to the coupled binuclear Cu sites in hemocyanin and tyrosinase.3 We first defined this difference using XAS at the Cu K-edge. From Fig. 15A, reduced Cu has a characteristic peak at 8984 eV, while oxidized Cu does not.44 In fact Fig. 15A shows deoxy Hc reacting with O2 to form oxy-Hc with 2 Cu(I) oxidized to 2 Cu(II). For the multicopper oxidases, we can prepare a T2 depleted form,45 which only contains the reduced T3 center (and the reduced T1 Cu).46 As shown in Fig. 15B, the reduced T3 does not react with O2 in the absence of the T2 Cu.3 From the potential energy surface in Fig. 16A, O2 binding to the T3 center is uphill by ~ 6 kcal/mol (and with an additional barrier) while for hemocyanin in Fig. 3C O2 binding is downhill by ~ 4 kcal/mol.47 Much of this 10 kcal/mol difference in O2 binding energy reflects the relative energies of the reduced sites. From Fig. 16B, the deoxy-Hc site has two Cu(I)s held tightly together at a distance of ~ 4 Å, while in the reduced T3 site the two coppers are allowed to relax to a distance of ~ 6 Å. For deoxy-Hc, this short Cu(I)-Cu(I) distance leads to significant electostatic repulsion in the low dielectric environment of the protein, which destabilizes the reduced site, drives O2 binding and allows its cooperative regulation by changing Cu(I)-Cu(I) distance. This large different in O2 reactivity between the coupled binuclear and T3 Cu sites reflects their very different coordination in the protein. From Fig 17, in hemocyanin the two coppers are coordinated by His ligands on α-helixes held together by salt bridges, while for the T3 site the Cu’s are coordinated by H-X-H motifs on loops from β-sheets.47
Fig. 15.

Cu K-edge X-ray absorption spectra of deoxy Hc (A) and fully reduced T2D laccase (B) and their reactions with O2. Reproduced with permission from reference 3, © 1996 The American Chemical Society.
Fig. 16.

(A) Energy of O2 binding to the T3 site with S = 0 in red and S = 1 in blue. (B) Potential energy surfaces for deoxy-Hc and deoxy-T3 sites as a function of Cu-Cu distance (dashed lines show the energy of electrostatic interactions between the two Cu(I) centers). Reproduced with permission from reference 47, © 2009 the National Academy of Sciences of the United States of America.
Fig. 17.

Comparison of the more and less constrained structures of deoxy-Hc (PDB code 1JS8) and deoxy-T3, respectively (PDB code 1GYC). Reproduced with permission from reference 47, © 2009 the National Academy of Sciences of the United States of America.
The holo-multicopper oxidases do, of course, react with O2 to reduce it to H2O with a bimolecular rate constant of 1.7 × 106 M−1 s−1 (Fig 18)48, 49. This produces the native intermediate (NI). We also characterized a derivative of the MCOs where the T1 is depleted (T1D)50, 51 or replaced by the redox inactive Hg(II) ion52, 53. This contains an active trinuclear cluster that, when reduced, reacts with O2 to form what we have shown to be a two electron reduced peroxide intermediate (PI)54. The rate of PI formation, within error, is the same as for the native enzyme and thus is kinetically competent to be a precursor to NI.55 We have in fact found that PI decays to NI. The rate of this is very fast in the native enzyme intermediate but is slowed down by >106 in the T1D forms, as the electron from the T1 center is not available to drive the reduction of PI.56 Below, we evaluate the unique spectral features of PI and NI to determine their geometric and electronic structures, which can be used to explore the reductive cleavage of the O-O bond.
Fig. 18.

Schematic and rates of the O2 reactivity of both native and T1D/T1Hg forms of the MCOs and the cleavage of the O-O bond. Reproduced with permission from reference 42, © 2008 RSC Publishing.
For PI, a variety of our past studies have shown that two of the three coppers reduce O2 to peroxide which binds in the center of the cluster.42 Importantly from Fig 19, PI has a very different peroxide to Cu(II) CT spectrum from oxy-Hc, in having less intensity in the ~350 nm region and a number of relatively intense CT transitions at lower energy. This indicates a different geometric and electronic structure for PI relative to oxy-Hc. However, when O2 is placed in the center of the reduced trinuclear cluster and the site is geometry optimized it goes to a side-on peroxo bridging the two T3 Cu’s, which as presented above is not energetically favorable.57 Alternatively, when the structure is optimized including an anionic Asp at the T2/T3B edge, the structure that is most favorable has the peroxide bound side-on to T3B and end-on bridged to T3A and the T2 Cu (Fig. 20A). Addition of D94 to the calculated structure was based on mutation studies where its replacement with a neutral Ala or Asn turns off the O2 reduction of the reduced TNC while substituting D94 with Glu recovers this reactivity.58, 59
Fig. 19.

Peroxide to Cu(II) charge transfer absorption spectra of PI (red) and oxy-Hc (black). Reproduced with permission from reference 42, © 2008 RSC Publishing.
Fig. 20.

(A) Calculated geometric structures of PI without and with D94. The PI structure without D94 (left) has both T3 Cu’s oxidized and the T2 Cu reduced while in the structure with D94 (right), the T3B and T2 are oxidized and the T3A is reduced. (B) Contours of the α- (based on the T2 dx2−y2) and the β- (based on the T3B dx2–y2) LUMOs of PI + D94. Reproduced with permission from reference 57, © 2008 American Chemical Society.
We have studied the electronic structure of PI in some detail and find that the T3B and T2 Cu are oxidized (T3A reduced) and both have half-occupied d-orbitals that overlap the orbital of the peroxide (Fig. 20B) leading to the antiferromagnetic coupling we observe experimentally (from SQUID magnetic susceptibility studies of PI).54 From Fig. 20A right, the site is clearly asymmetric in its O2 reactivity, and this is clearly demonstrated by the fact that substitution of the His126 ligand on the T3CuA with an amide still allows for the formation of PI, while substitution of H483 on the T3CuB with an amide turns off the O2 reactivity.60 This asymmetry derives from a combination of D94 lowering the potentials of the T3CuB and T2 coppers for electron transfer and the fact that one of the His on the T3CuA is δ coordinated, which leads to a trigonal planar structure for that Cu stabilizing its reduced state. This trinuclear cluster asymmetry is important in its irreversible binding of O2 as peroxide and its cleavage to form H2O.
NI is produced by the reaction of the holoenzyme with O248, 49. The unique spectral features of NI are shown in Fig. 2161. It has two CT transitions at 365 and 318 nm and an absorption band at ~16,000 cm−1 (600 nm), the last corresponding to the oxidation of the T1Cu (Fig. 21A). Therefore, at least on more e− (from T1) has been added to PI to generate NI. The redox states of the Cu’s were again determined by XAS at the Cu K-edge (Fig. 21 B). No 8984 eV feature of reduced Cu is present in NI, thus its trinuclear Cu cluster is fully oxidized (and dioxygen is fully reduced). This is also the case for the resting enzyme, but the trinuclear Cu cluster in NI has very different spectral features from the resting trinuclear cluster. From the EPR spectrum of the trinuclear cluster in Fig. 21D, the resting site has a normal EPR signal associated with the T2 Cu(II), while NI exhibits a broad signal with g-values below 2.0, which is very unusual for Cu(II). We obtained major insight into NI through magnetic circular dichroism (MCD) studies. The CT bands of NI show an intense derivative shaped feature in its MCD spectrum in Fig. 21A (called a pseudo-A term). We used these excited state MCD features to probe the unusual ground state of NI through variable temperature variable field studies. Fig. 21 C shows the saturation behavior of the MCD intensity with increasing magnetic field at low temperatures. This fits to a Brillouin function for an S = ½ identifying the spin state that is associated with the unusual EPR signal in Fig. 21 D. Key insight into the origin of the unique features of this ground state came from the temperature dependence of the MCD signal at high magnetic field. Normally, the MCD signal of a paramagnetic system should decrease in intensity with increasing temperature (as 1/T), the MCD analog of Curie Law behavior in magnetic susceptibility. However, as shown in Fig 21 E for NI, the MCD spectrum changes shape with increasing temperature. At 25,000 cm−1, for example, the intensity first decreases but then increases with increasing temperature. This demonstrates that a low-lying excited state is being Boltzmann populated with a different MCD spectrum than the ground state. From the fit to the temperature dependent MCD shown in Fig. 21F, this state is 150 cm−1 above the ground state. The presence of this very low-lying excited state allows assignment of the unique geometric and electronic structure of NI.
Fig. 21.

Stopped flow absorption spectra (top) and rapid freeze quench MCD spectra (A), Cu k-edge XAS spectra (B), variable temperature, variable field behavior of the MCD spectra with Brillouin function fits with different S values (C), low temperature X-band EPR (D), temperature dependence of the MCD spectra (E) and plot of temperature dependence of MCD intensity at 25,000 cm-1 of NI. Reproduced with permission from reference 61 and 42, © 2002 American Chemical Society and © 2008 RSC Publishing.
From the Fourier transformed EXAFS data in Fig. 22A, NI has a pair of Cu(II)’s bridged tightly together at a phase shift corrected distance of 3.3 Å.61 From the magnetostructural correlation in Fig. 22B, this corresponds to a pair of Cu(II)’s that are antiferromagnetically coupled with a singlet/ triplet splitting of ~520cm−1. Allowing for the presence of the third, non-interacting Cu(II) with S = 1/2 leads to an STot= 1/2 ground state and STot= 1/2, 3/2 degenerate excited state at 520cm−1 (Fig. 23 left). Introducing one bridging ligand to the third Cu(II) turns on an additional antiferromagnetic (AF) coupling which energy splits the excited doublet and quartet states, but neither is shifted below ~500cm−1(Fig. 23 middle). However, addition of a third bridge to the TNC (Fig. 23 right) leads to a spin-frustrated system (Fig. 24A). Antiferromagnetic coupling of Cu1 with Cu2 and Cu2 with Cu3 results in parallel alignment of the spins on Cu1 and Cu3, although their spins want to be antiferromagnetically coupled due to the third bridge. This results in a dramatic decrease in the STot = 1/2 excited state energy in Fig. 23 right. The observed 150cm−1 splitting, combined with other spectral data, led to the all exchanged coupled, therefore bridged, TNC in Fig. 24B. Combining these results with the analysis of the unique EPR g values (due to the antisymmetric exchange associated with spin frustration)62 and the sign of the pseudo A term in the MCD spectrum in Fig. 21A63, led to the geometric structure of NI in Fig. 24C. The reduction of peroxide leads to a μ3-oxo bridge in the center of the cluster plus the second oxygen of O-O cleavage generates a μ2 OH− between the T3 Cu(II)’s.64
Fig. 22.

Fourier Transformed EXAFS spectra of NI (A) and magnetostructural correlation of the Cu-Cu distance (and angle) with exchange coupling constants (J) for OH bridged Cu(II) model complexes (B). Reproduced with permission from reference 61, © 2002 American Chemical Society.
Fig. 23.

Energy splittings of doublet and quartet states of a Cu(II) trimer with 1 (left), 2 (middle), and 3 (right) bridging interactions. Reproduced with permission from reference 42, © 2008 RSC Publishing.
Fig. 24.

Orientation of electron spins in the spin-frustrated ground state of NI (A), experimental estimate of pairwise exchange parameters for NI (B) and the spectroscopically derived calculated geometric structure of NI where the μ3-oxo and μ2-OH derive from the 4 e− reduction of O2 (C).
Having geometric and electronic structures of PI and NI, we explored the 2e− reductive cleavage of the O-O bond both experimentally58 and computationally.57 Starting with PI at the left in Fig. 25A, where the T3B Cu and the T2 Cu are oxidized and the T3A Cu is reduced, addition of a single e− from the T1 Cu reduces the T2 Cu and generates PI + e in Fig. 25A, the starting point for a 2D PES for the reductive cleavage of the O-O bond. As shown in Fig. 25B, one dimension is O-O cleavage (directed out on the left), while the second dimension is proton donation from a carboxylate at the T3 Cu edge (E487), from our mutation experiments on Fet3p (directed out on the right).58 Two pathways are found for this cleavage. In path 2 in Fig. 25B, the proton is transferred first, and then the O-O bond is cleaved.57 This predicts an inverse KIE that we observe experimentally.56 The second path, 1, involves O-O cleavage before the proton is transferred. Importantly, both have low barriers (5–6 kcal/mol, from calculation and experiment) with little contribution from the proton. This demonstrates the effectiveness of the TNC in O-O cleavage due to its frontier MOs. From Fig. 25C, the triangular topology of the TNC allows both the reduced T3A Cu and the reduced T2 Cu to have good overlap with the σ* orbital of the peroxide for concerted transfer of an e− pair in the reductive cleavage of this bond. Furthermore, while the T3β Cu is oxidized it still acts as a Lewis acid, equivalent to a proton, in lowering the energy of the O22− σ* orbital assisting in facile O-O cleavage with a low barrier.
Fig. 25.

Calculated structures of PI, PI+e and NI (A). 2-D Potential energy surface of the reductive cleavage of the O-O bond (B). Schematic of triangular topology of the TNC and depiction of the orbitals relevant in O-O cleavage. Note that T2 and T3A are reduced and transfer an α and β pair of electrons into the peroxide σ* orbital, while T3B is oxidized and acts and a lewis acid in stabilizing the σ*.
In summary (Fig 26), we have found that: in non-coupled binuclear Cu sites, one Cu can transfer one e− to reduce O2 to superoxide with a π* FMO for H atom abstraction from activated C-H bonds;37, 38 the coupled binuclear Cu centers transfer two e− ‘s reducing O2 to peroxide with a low lying σ* FMO for electrophilic attack on aromatic rings;24 and for the MCO’s four Cu’s transfer four electrons in two concerted steps, two into the π* orbital and two into the σ* orbital with a low barrier for O-O cleavage due to the triangular topology of the TNC.42 Below we consider the nature of the O2 intermediate required for H-atom abstraction from the strong H-C bond of methane.
Fig. 26.

Summary of copper-dioxygen active sites in biology and their pertinent frontier molecular orbitals involved in reactivity.
V. O2 Activation for Methane Oxygenation
In nature methane is converted to methanol by the methane monooxygenase (MMO) enzymes.65–67 Soluble (s)MMO contains a binuclear non-heme Fe site coordinated by 2 His and 4 carboxylate ligands (Fig 27A).65 Two oxygen intermediates are observed,68, 69 P and Q, but are not well characterized by spectroscopy leading to much computational speculation. Particulate (p)MMO is now thought to contain a binuclear Cu reactive site with 3 His and one amine ligand (Fig. 27B).66, 70 No intermediate has been reported and little is know about its reaction mechanism. Alternatively, it has been shown in reference 71 that Cu-ZSM-5 (Fig. 27C), heated in He to reduce the Cu and activated with O2 at 200°C, produces a new species with an absorption band at 22,700 cm−1. When this is reacted with CH4 at low temperature, this absorption feature disappears and methanol is produced in a single turnover (Fig. 28B).72 We have used the temperature dependence of this absorption feature to obtain the activation energy for this reaction as 15.7 kcal/mol and find a deuterium isotope effect that raises the barrier by 2.1 kcal/mol (Fig. 28C), consistent with the kinetic isotope effect determined from product analysis. Thus we can correlate this 22,700 cm−1 absorption feature with the reactive site for CH4 to CH3OH conversion, and can use this absorption feature to probe this active site.
Fig. 27.

Active site structures in methane oxidizing enzymes (A and B) and Cu-Zeolite (C).
Fig. 28.

Formation of the 22,700 cm−1 absorption feature in Cu-ZSM-5 (A) and its decay upon reaction with methane (B). Arrhenius plots for the reaction of the 22,700 cm−1 species with CH4 and CD4 (C). Laser tuning into the 22,700 cm−1 feature that leads to resonance enhancement of the Raman vibrations of the Cu active site.
In bioinorganic chemistry the challenge of spectroscopy is to study the few active site metal ions in a large protein matrix. In heterogeneous systems the challenge is to spectroscopically study the reactive site in the presence of a dominant amount of spectator, in this case Cu, ions. We could accomplish this at high resolution using resonance Raman spectroscopy. As shown in Fig. 28 D tuning a laser into the 22,700 cm−1 band leads to the resonance enhancement of Raman vibrations of the associated active site.72 The impressive resonance Raman spectrum obtained is shown in Fig. 29. Oxygen isotope sensitive peaks are observed at 456, 870 and 237 cm−1. Isotope insensitive, resonance enhanced lattice modes are observed at 514 and 540 cm−1. Based on the low energy and high intensity of the 22,700 cm−1 absorption, it was originally considered to be a bis-μ-oxo Cu2(III) species. However from Table I, which summarizes the resonance Raman data presented throughout this paper, no vibrational feature is observed in the resonance Raman spectrum of O2 activated Cu-ZSM-5 at 600 cm−1 and this possibility can be eliminated. Also the side-on peroxo bridged Cu2(II) isomer can be excluded as no ~750 cm−1 vibration is observed in the spectrum in Fig. 29. Another possibility was that one Cu could reduce O2 by one e− leading to a bound superoxide, which appears to be active in H atom abstraction from relatively weak C-H bonds (vide supra). From section III, these show νO-O in the 1040 (side-on) to 1120 cm−1 (end-on) region but, such a feature is absent in the resonance Raman spectrum in Fig. 29, eliminating O2 activation as a superoxide. Alternatively, the 18O2 isotope sensitive 870 cm−1 feature could be the O-O stretch of a non-side on bridged peroxide (i.e. no backbonding into σ*). From Table I, for the end-on bridged peroxo νO-O is at 832 cm−1 while for the hydroperoxide this vibration occurs between 843–892 cm−1 depending on its binding mode (terminal vs bridged). If the 870 cm−1 feature is an O-O stretch, when made with mixed isotope 16,18O2, it should shift to a frequency intermediate between those of the 16O2 and 18O2 intermediates at 870 and 830 cm−1 respectively (i.e. ~850 cm−1). The insert in Fig. 29 shows the resonance Raman spectrum obtained by generating the O2 activated Cu-ZSM-5 intermediate with the commercially available 1:2:1 mixture of 18O2:16,18O2:16O2. No intermediate frequency vibration is observed and the experimental data can quantitatively be simulated by the average of the pure 18O2 + 16O2 resonance Raman spectra. Thus there is no O-O bond, thereby eliminating all known and characterized Cu/O2 cores. The oxygen intermediate in Cu-ZSM-5 is a new type of species in inorganic chemistry.72
Fig. 29.

rR spectra (λex = 457.9 nm) of Cu-ZSM-5 + 16O2 (red) and 18O2 (blue). Inset 16,18O2 (green) data obtained with a 1:2:1 mixture of 16O2 : 16/18O2 : 18O2 and normalized sum of 16O2 + 18O2 spectra (black). Reproduced with permission from reference 72, © 2009 the National Academy of Sciences of the United States of America.
Table I.
Spectroscopically characterized mononuclear and binuclear Cu/O2 species.
| Cu/O2 species | rR vibrations (Δ18O2)/cm−1 |
|---|---|
| O2 activated Cu-ZSM-5 |
456 (8) 870 (40) |
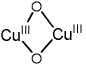 |
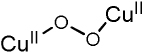
νCu-O=606 (23) |
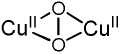 |
νCu-Cu=284 (0) νO-O=763 (40) |
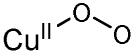 |
νCu-O=472 (20) νO-O=1121 (63) |
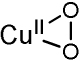 |
νCu-O=554 (20) νO-O=1043 (59) |
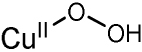 |
νCu-O=624 (17) νO-O=843 (44) |
 |
νCu-O=561 (26) νO-O=832 (44) |
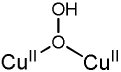 |
νCu-O=322 (10) νO-O=892 (52) |
The nature of this species could be defined by looking at an expanded energy region of the resonance Raman spectrum (Fig. 30). The intense isotope sensitive feature at 456 cm−1 must reflect a symmetric Cu-O stretch. The isotope sensitive weak 870 cm−1 feature can then be assigned as an antisymmetric stretch which would not be resonance Raman allowed. This is strongly supported by the fact that its 1st overtone at 1725 cm−1 is 6x more intense, consistent with the fact that the first overtone of an antisymmertic stretch is symmetric, hence resonance Raman allowed. Thus, this new species corresponds to a mono-μ-oxo bridged dicopper core with the 237 cm−1 isotope sensitive feature assigned as its bending mode. From Fig. 31 we can use the symmetric and antisymmetric vibrational frequencies (and their isotope shifts) to estimate the Cu-O-Cu angle, which is found to be 140°. From modeling this structure into the 10 member ring in the ZSM-5 channel (that is large enough for CH4 migration), this experimental angle fits well for oxygen bridging two coppers binding bidentate to oxygens of Al T sites separated by two Si T sites.
Fig. 30.

Extended rR spectra (λex = 457.9 nm) of Cu-ZSM-5 + 16O2 (red) and 18O2 (blue). Reproduced with permission from reference 72, © 2009 the National Academy of Sciences of the United States of America.
Fig. 31.

Estimation of the Cu-O-Cu angle based on normal coordinate analysis (including isotope perturbations) and the schematic of this structure inserted into the ZSM5 zeolite 10 member ring. Reproduced with permission from reference 72, © 2009 the National Academy of Sciences of the United States of America.
We then performed DFT calculations on a large model of this Cu2O core in the 10-ring of ZSM-5 (Fig. 32). We found that only CuII ions are supported by this core, as in calculations starting with CuIII ions, the holes delocalize into the zeolite lattice. The optimized CuII-O-CuII structure reproduces the experimental Cu-O-Cu angle; also from Table II there is good agreement between the calculated and experimental values of νs, νas and of the Cu-O-Cu bending modes and also with the lattice modes. Additionally, the TD-DFT predicts the oxo to CuII charge transfer transition as the assignment of the 22,700 cm−1 characteristic absorption feature of this site.
Fig. 32.

DFT calculated structural model of the Cu2O intermediate in ZSM-5. Reproduced with permission from reference 72, © 2009 the National Academy of Sciences of the United States of America.
Table II.
Experimental and calculated rR vibrations and charge transfer transitions for the Cu-ZSM Cu2O core.
| Exp. (Δ18O2) |
Calc. (Δ18O2) |
|
|---|---|---|
| Bend | 237(−3) | 253(−2) |
| νs | 456(−8) | 456(−5) |
| νas | 870(−40) | 852(−37) |
| Lattice | 514/540 | 556/568 |
| LMCT | 22,700 | 23,400 |
Having experimentally supported calculations on the geometric and electronic structure of this [Cu2O]2+ intermediate, we could then computationally evaluate its reactivity. From Fig. 33A and Table III, the low experimental activation energy for H-atom abstraction from CH4 and the measured isotope effect are reasonably reproduced by the calculations. There are two contributions to this low energy barrier for H-atom abstraction from methane. First as shown at the right of Fig 33A, the Cu2O-H bond of the first product is very strong (~90 kcal/mol) in part due to the delocalization of the abstracted e− of the H-atom over the two coppers. Second from Fig. 33B the frontier MO of the [Cu2O]2+ species becomes polarized as CH4 approaches at the transition state (TS) and goes from being mostly a hole on the metals to mostly a hole on the oxygen. Thus, this site gains significant CuI-oxyl character along the reaction coordinate (Fig. 33C), which is very efficient at H-atom abstraction, even from CH4. Finally from Fig. 34 we can insert this [Cu2O]2+ intermediate into the active site of pMMO and find the same spin polarization towards oxyl in the TS, thus low barrier for H-atom abstraction from CH4.73 However experimental data on oxygen intermediates in pMMO are required that can then be understood in terms of the large body of data now available on the electronic structure and reactivity of copper/oxygen species in inorganic chemistry.
Fig. 33.

DFT calculated reactivity of CuII2O with methane (A), the LUMOs at the H-Atom abstraction TS; note that the top wavefunction is polarized towards the H-C bond of CH4, (B), and schematic of the Cu(I)-oxyl (C). Reproduced with permission from reference 72, © 2009 the National Academy of Sciences of the United States of America.
Table III.
Experimental and calculated activation energies for H-atom abstraction of methane.
| Exp. kcal/mol |
Calc. kcal/mol |
|
|---|---|---|
| Ea (CH4) | 15.7 | 18.5 |
| ΔEa (CD4) | 2.1 | 1.3 |
Fig. 34.

CuII2O core modeled in the active site of pMMO: H-Atom abstraction from methane at the transition state.
VI. Concluding Comments
While there has been major advances in our understanding of copper/dioxygen (bio)inorganic chemistry, there is still much to be achieved. For the coupled binuclear Cu proteins there is the major issue of whether the side-on bridged peroxide present in oxy-Tyr is directly active for electrophilic aromatic substitution or whether it converts to a bis-μ-oxo species upon substrate binding, and how the differences in the interactions of substrates with the coupled binuclear sites in different protein environments control the differences in reactivity i.e. cleaved oxy-Hc (where the site is thought to now be accessible to substrate), oxy-Tyr, and catechol oxidase (which also contains a coupled binuclear Cu site but can only oxidize catechol to quinone, not oxygenate the substrate). For the non-coupled binuclear Cu enzymes (DβM, PHM), the calculations now exceed the data and experimental definition of O2 intermediates in the enzyme is required to evaluate the mechanistic possibilities. For the multi-copper oxidases, it is clear that intermediate NI is the catalytically relevant fully oxidized form of the enzyme (rather then the crystallographically defined resting state),64 and one question is the mechanism of reduction of NI, which appears to be rate limiting in a number of multi-copper oxidases. There is also the goal of manipulating these enzymes to tune their properties and reactivities, as for example in biofuel cells. Finally, for the reduction of O2 to H2O, it is important to determine how the other class of enzymes that perform this reaction in biology, the cytochrome c oxidases, that contain a Cu-heme catalytic site, relate mechanistically to the MCO’s where the later are efficient in substrate oxidation while the former are required for proton pumping for ATP synthesis. For the MMO’s, CuZSM5 is an important breakthrough in understanding the nature of a site that can directly convert CH4 to CH3OH with a low barrier.72 One goal is now to make this system into a catalyic cycle (i.e. remove the methanol product for turnover), while another is to understand whether this [Cu2O]2+ species is, in fact, a model of the catalytic intermediate in pMMO. Also, in the field of Cu/O2 chemistry there are now an increasing number of sites in biology where the first function of the Cu after protein expression is to modify a nearby protein residue and create the catalytic active site. While the catalytic cycles of these enzymes are reasonably well understood, the nature of these cofactor biogenesis reactions is not. Finally, the five areas of reactivity considered in this review focus on O2 activation by reduced Cu(I) centers. There are, however, also an increasing number of reactions that use an oxidized, Cu(II) centers to activate substrate for reaction with O2. The field of copper/dioxygen (bio)inorganic chemistry has come far but there is still much to be understood.
Acknowledgements
E. I. Solomon wishes to thank past and present grad-students, post-docs, and collaborators referred to throughout this review for their extensive contributions to the science and the NIH for the long term financial support of these studies (DK031450 to E.I.S.).
References
- 1.Allendorf MD, Spira DJ, Solomon EI. Proc. Natl. Acad. Sci. U.S.A. 1985;82:3063–3067. doi: 10.1073/pnas.82.10.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spira-Solomon DJ, Allendorf MD, Solomon EI. J. Am. Chem. Soc. 1986;108:5318–5328. [Google Scholar]
- 3.Solomon EI, Sundaram U, Machonkin T. Chem Rev. 1996;96:2563–2606. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 4.Pate JE, Cruse RW, Karlin KD, Solomon EI. J Am Chem Soc. 1987;109:2624–2630. [Google Scholar]
- 5.Baldwin MJ, Root DE, Pate JE, Fujisawa K, Kitajima N, Solomon EI. J Am Chem Soc. 1992;114:10421–10431. [Google Scholar]
- 6.Freedman TB, Loehr JS, Loehr TM. J Am Chem Soc. 1976;98:2809–2815. doi: 10.1021/ja00426a023. [DOI] [PubMed] [Google Scholar]
- 7.Larrabee JA, Spiro TG, Ferris NS, Woodruff WH, Maltese WA, Kerr MS. J Am Chem Soc. 1977;99:1979–1980. doi: 10.1021/ja00448a053. [DOI] [PubMed] [Google Scholar]
- 8.Solomon EI, Dooley DM, Wang RH, Gray HB, Cerdonio M, Mogno F, Romani GL. J Am Chem Soc. 1976;98:1029–1031. doi: 10.1021/ja00420a035. [DOI] [PubMed] [Google Scholar]
- 9.Ross PK, Solomon EI. J Am Chem Soc. 1990;112:5871–5872. [Google Scholar]
- 10.Ross PK, Solomon EI. J Am Chem Soc. 1991;113:3246–3259. [Google Scholar]
- 11.Chen P, Fujisawa K, Helton ME, Karlin KD, Solomon EI. J Am Chem Soc. 2003;125:6394–6408. doi: 10.1021/ja0214678. [DOI] [PubMed] [Google Scholar]
- 12.Metz M, Solomon EI. J Am Chem Soc. 2001;123:4938–4950. doi: 10.1021/ja004166b. [DOI] [PubMed] [Google Scholar]
- 13.Himmelwright RS, Eickman NC, LuBien CD, Lerch K, Solomon EI. J Am Chem Soc. 1980;102:7339–7344. [Google Scholar]
- 14.Eickman NC, Solomon EI, Larrabee JA, Spiro TG, Lerch K. J Am Chem Soc. 1978;100:6529–6531. [Google Scholar]
- 15.Wilcox DE, Porras AG, Hwang YT, Lerch K, Winkler ME, Solomon EI. J. Am. Chem. Soc. 1985;107:4015–4027. [Google Scholar]
- 16.Winkler ME, Lerch K, Solomon EI. J Am Chem Soc. 1981;103:7001–7003. [Google Scholar]
- 17.Halfen JA, Mahapatra S, Wilkinson EC, Kaderli S, Young VG, Cramer CJ, Que L, Jr, Tolman WB. Science. 1996;271:1397–1400. doi: 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]
- 18.DuBois JL, Mukherjee P, Collier AM, Mayer JM, Solomon EI, Hedman B, Stack TDP, Hodgson KO. J Am Chem Soc. 1997;119:8578–8579. [Google Scholar]
- 19.Henson MJ, Mukherjee P, Root DE, Stack TDP, Solomon EI. J. Am. Chem. Soc. 1999;121:10332–10345. [Google Scholar]
- 20.Pidcock E, Obias HV, Zhang CX, Karlin KD, Solomon EI. J. Am. Chem. Soc. 1998;120:7841–7847. [Google Scholar]
- 21.Karlin KD, Nasir MS, Cohen BI, Cruse RW, Kaderli S, Zuberbuhler AD. J Am Chem Soc. 1994;116:1324–1336. [Google Scholar]
- 22.Karlin KD, Hayes JC, Gultneh Y, Cruse RW, McKown JW, Hutchison JP, Zubieta J. J. Am. Chem. Soc. 1984;106:2121–2128. [Google Scholar]
- 23.Mirica LM, Vance MA, Rudd DJ, Hedman B, Hodgson KO, Solomon EI, Stack TDP. Science. 2005;308:1890–1892. doi: 10.1126/science.1112081. [DOI] [PubMed] [Google Scholar]
- 24.Op't Holt BT, Vance MA, Mirica LM, Heppner DE, Stack TDP, Solomon EI. J Am Chem Soc. 2009;131:6421–6438. doi: 10.1021/ja807898h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qayyum MF, Sarangi R, Solomon EI. In preparation. [Google Scholar]
- 26.Klinman JP. Chem. Rev. 1996;96:2541–2562. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]
- 27.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Science. 1997;278:1300–1305. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- 28.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Nat Struct Biol. 1999;6:976–983. doi: 10.1038/13351. [DOI] [PubMed] [Google Scholar]
- 29.Tian G, Berry JA, Klinman JP. Biochemistry. 1994;33:226–234. doi: 10.1021/bi00167a030. [DOI] [PubMed] [Google Scholar]
- 30.Chen P, Fujisawa K, Solomon EI. J Am Chem Soc. 2000;122:10177–10193. [Google Scholar]
- 31.Chen P, Solomon EI. J Am Chem Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- 32.Chen P, Root DE, Campochiaro C, Fujisawa K, Solomon EI. J Am Chem Soc. 2003;125:466–474. doi: 10.1021/ja020969i. [DOI] [PubMed] [Google Scholar]
- 33.Prigge ST, Eipper BA, Mains RE, Amzel LM. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 34.Würtele C, Gaoutchenova E, Harms K, Holthausen MC, Sundermeyer J, Schindler S. Angew. Chem. Int. Ed. 2006;45:3867–3869. doi: 10.1002/anie.200600351. [DOI] [PubMed] [Google Scholar]
- 35.Lanci MP, Smirnov VV, Cramer CJ, Gauchenova EV, Sundermeyer J, Roth JP. J Am Chem Soc. 2007;129:14697–14709. doi: 10.1021/ja074620c. [DOI] [PubMed] [Google Scholar]
- 36.Woertink JS, Tian L, Maiti D, Lucas HR, Himes RA, Karlin KD, Neese F, Würtele C, Holthausen M, Bill E, Sundermeyer J, Schindler S, Solomon EI. Inorg Chem. doi: 10.1021/ic101138u. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian L, Woertink JS, Solomon EI. Unpublished results. [Google Scholar]
- 38.Chen P, Solomon EI. Proc Natl Acad Sci USA. 2004;101:13105–13110. doi: 10.1073/pnas.0402114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brunold TC, Gamelin DR, Solomon EI. J Am Chem Soc. 2000;122:8511–8523. [Google Scholar]
- 40.Tuczek F, Solomon EI. Coordin Chem Rev. 2001;219:1075–1112. [Google Scholar]
- 41.Solomon EI, Chen P, Metz M, Lee SK, Palmer AE. Angew. Chem. Int. Ed. 2001;40:4570–4590. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 42.Solomon EI, Augustine AJ, Yoon J. Dalton Trans. 2008;30:3921–3932. doi: 10.1039/b800799c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quintanar L, Yoon J, Aznar C, Palmer AE, Andersson K, Britt D, Solomon EI. J. Am. Chem. Soc. 2005;127:13832–13845. doi: 10.1021/ja0421405. [DOI] [PubMed] [Google Scholar]
- 44.Kau L-S, Spira-Solomon DJ, Penner-Hahn JE, Hodgson KO, Solomon EI. J Am Chem Soc. 1987;109:6433–6442. [Google Scholar]
- 45.Graziani MT, Morpurgo L, Rotilio G, Mondovi B. FEBS lett. 1976;70:87–90. doi: 10.1016/0014-5793(76)80732-6. [DOI] [PubMed] [Google Scholar]
- 46.LuBien CD, Winkler ME, Thamann TJ, Scott RA, Co MS, Hodgson KO, Solomon EI. J Am Chem Soc. 1981;103:7014–7016. [Google Scholar]
- 47.Yoon J, Fujii S, Solomon EI. Proc Natl Acad Sci USA. 2009;106:6585–6590. doi: 10.1073/pnas.0902127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andreasson LE, Branden R, Reinhammer B. Biochim. Biophys. Acta. 1976;438:370–379. doi: 10.1016/0005-2744(76)90254-0. [DOI] [PubMed] [Google Scholar]
- 49.Andreasson LE, Reinhammer B. Biochim. Biophys. Acta. 1976;445:579–597. doi: 10.1016/0005-2744(76)90112-1. [DOI] [PubMed] [Google Scholar]
- 50.Blackburn NJ, Ralle M, Hasset R, Kosman DJ. Biochemistry. 2000;39:2316–2324. doi: 10.1021/bi992334a. [DOI] [PubMed] [Google Scholar]
- 51.Palmer AE, Quintanar L, Severance S, Wang T-P, Kosman DJ, Solomon EI. Biochemistry. 2002;41:6438–6448. doi: 10.1021/bi011979j. [DOI] [PubMed] [Google Scholar]
- 52.Morie-Bebel MM, Morris MW, Menzie JL, McMillin DR. J. Am. Chem. Soc. 1984;106:3677–3678. [Google Scholar]
- 53.Cole JL, Clark PA, Solomon EI. J. Am. Chem. Soc. 1990;112:9534–9548. [Google Scholar]
- 54.Shin W, Sundaram UM, Cole JL, Zhang HH, Hedman B, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 1996;118:3202–3215. [Google Scholar]
- 55.Cole JL, Ballou DP, Solomon EI. J. Am. Chem. Soc. 1991;113:8544–8546. [Google Scholar]
- 56.Palmer AE, Lee SK, Solomon EI. J. Am. Chem. Soc. 2001;123:6591–6599. doi: 10.1021/ja010365z. [DOI] [PubMed] [Google Scholar]
- 57.Yoon J, Solomon EI. J. Am. Chem. Soc. 2007;129:13127–13136. doi: 10.1021/ja073947a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Augustine AJ, Quintanar L, Stoji CS, Kosman DJ, Solomon EI. J. Am. Chem. Soc. 2007;129:13118–13126. doi: 10.1021/ja073905m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quintanar L, Stoji CS, T.P. W, Kosman DJ, Solomon EI. Biochemistry. 2005:6081–6091. doi: 10.1021/bi047379c. [DOI] [PubMed] [Google Scholar]
- 60.Augustine AJ, Kjaergaard C, Qayyum M, Ziegler L, Kosman DJ, Hodgson KO, Hedman B, Solomon EI. J. Am. Chem. Soc. 2010;132:6057–6067. doi: 10.1021/ja909143d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee SK, George SD, Antholine WE, Hedman B, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 2002;124:6180–6193. doi: 10.1021/ja0114052. [DOI] [PubMed] [Google Scholar]
- 62.Yoon J, Mirica LM, Stack TDP, Solomon EI. J. Am. Chem. Soc. 2004;126:12586–12595. doi: 10.1021/ja046380w. [DOI] [PubMed] [Google Scholar]
- 63.Yoon J, Mirica LM, Stack TDP, Solomon EI. J. Am. Chem. Soc. 2005;127:13680–13693. doi: 10.1021/ja0525152. [DOI] [PubMed] [Google Scholar]
- 64.Yoon J, Liboiron BD, Sarangi R, Hodgson KO, Hedman B, Solomon EI. Proc. Natl. Acad. Sci. U.S.A. 2008;104:13609–13614. doi: 10.1073/pnas.0705137104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 66.Lieberman RL, Rosenzweig AC. Nature. 2005;434:177–182. doi: 10.1038/nature03311. [DOI] [PubMed] [Google Scholar]
- 67.Hakemian AS, Rosenzweig AC. Annu. Rev. Biochem. 2007;76:223–241. doi: 10.1146/annurev.biochem.76.061505.175355. [DOI] [PubMed] [Google Scholar]
- 68.Lee SK, Nesheim JC, Lipscomb JD. J. Biol. Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- 69.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 70.Balasubramanian R, Smith SM, Rawat S, Yatsunyk LA, Stemmler TL, Rosenzweig AC. Nature. 2010;465:115–119. doi: 10.1038/nature08992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Groothaert MH, van Bokhoven JA, Battiston AA, Weckhuysen BM, Schoonheydt RA. J. Am. Chem. Soc. 2003;125:7629–7640. doi: 10.1021/ja029684w. [DOI] [PubMed] [Google Scholar]
- 72.Woertink JS, Smeets PJ, Groothaert MH, Vance MA, Sels BF, Schoonheydt RA, Solomon EI. Proc Natl Acad Sci USA. 2009;106:18908–18913. doi: 10.1073/pnas.0910461106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hadt RG, Woertink JS, Smeets PJ, Solomon EI. In preperation. [Google Scholar]