

Abstract
Although seasonal influenza vaccines play a valuable role in reducing the spread of the virus at the population level, ongoing viral evolution to evade immune responses remains problematic. No current vaccines are likely to elicit enduring protection in the face of emerging and re-emerging influenza viruses that rapidly undergoing antigenic drift. Eliciting broadly cross-neutralizing antibody responses against influenza virus is a crucial goal for seasonal and pandemic influenza vaccine preparation. Recent three-dimensional structure information obtained from crystallization of influenza antigens in complex with neutralizing antibodies (nAbs) have provided a framework for interpreting antibody-based viral neutralization that should aid in the design of vaccine immunogens. Here, we will review current knowledge of the structure-based mechanisms contributing to the neutralization and neutralization escape of influenza viruses. We will also explore the potential for this structure-based approach to overcome the challenge of obtaining the highly desired “universal” influenza vaccine.
Introduction
As global travel increases, so too does the transmission of pathogens. Currently, no biothreat is more palpable than that of pandemic influenza outbreaks. The global effort to control influenza through vaccination has expanded continually since the pandemic of 1918–1919, which was responsible for an estimated 50 million to 100 million deaths worldwide [1]. Nearly a century later, many still wonder not if but when influenza might again seriously threaten public health on such a global scale. The most recent influenza pandemic of 2009 proved to not be as severe as initially feared, but the emergence and rapid worldwide dissemination of the virus prompted health providers, policy makers, and researchers alike to more critically re-evaluate the adequacy of our current ability to deal with outbreaks. Despite the successes of prophylactic vaccination strategies that have been implemented to reduce disease burden in the last several decades, seasonal influenza epidemics are still responsible for substantial morbidity and mortality, resulting in the deaths of between 250,000 and 500,000 people every year [2] [3] [4].
Influenza viruses are classified into three subtypes: A, B and C as defined by the antigenicities of the nucleocapsid (NP) and matrix (M) proteins [5]. Influenza A and B are responsible for epidemics of seasonal flu, with influenza A being associated with more severe clinical disease in humans. Influenza A viruses are further divided into subtypes based on differences in two viral surface-expressed proteins: hemagglutinin (HA) which initiates virus entry into cells by binding to sialic acid on glycoconjugates of host membrane proteins, and neuraminidase (NA) which enables release of virions bound to the surface of producer cells by enzymatically cleaving sialic acid of neighboring glycojugates [4] [5]. There are 16 antigenically different HA subtypes and 9 antigenically distinct NA subtypes which in combination define all known subtypes of influenza A viruses. Three of these viral subtypes have caused pandemics in recent history: H1N1 in 1918 (and 2009), H2N2 in 1957 and H3N2 in 1968. With such diversity and potential for recombination between the different virus strains, the continuing challenge to the vaccine effort is to provide antigens that effectively elicit potent neutralizing antibodies (nAbs) that give broad strain protection against any future seasonal or pandemic influenza outbreak.
While the influenza surface HA glycoprotein is the antigenic target of vaccine-induced nAbs, the virus is evolutionarily capable of rapidly changing vulnerable epitopes within this protein in order to avoid detection and elimination by the immune system. Therefore, it is crucial to understand at the molecular level how this virus successfully gains entry into the host and, more importantly, how this first step in the infectious life cycle can be interrupted by nAbs. In this chapter, we provide an overview of our present understanding of the structural basis of influenza neutralization, focusing on the three-dimensional structure, function, and evolution of HA and nAb responses to this protein. We will describe the structural properties, based on the three-dimensional structures of an nAb-HA complex, of the receptor-binding and hydrophobic fusion machinery sites that are located in the globular head and stem regions, respectively. We will also describe the antigenic evolution of HA, mechanisms of neutralization escape as well as recent advances in structure-based vaccine strategies. Detailed structure based analysis of neutralization is necessary to increase our understanding of how the ever-changing influenza virus survives detection and elimination by the immune system. Implementation of vaccine approaches that can prevent infection or clinical disease progression worldwide is the ultimate goal of these efforts.
Antibody-mediated neutralization of viral infectivity
There are several mechanisms by which antibodies can inhibit influenza, and they can do so at different steps in the early viral life cycle. Antibodies against HA can neutralize the virus by directly blocking the initial virus attachment to target cells by binding to sites surrounding the receptor-binding pocket on the membrane-distal surface of HA, thereby interfering with virus receptor interaction (Fig. 1a). Subsequent to the initial attachment, receptor-bound viruses are taken into cells by endocytosis. The low pH environment of the endosome causes major conformational changes in the HA ectodomain, which activates fusion of the virus with the endosomal membrane and the eventual release of the uncoated viral ribonucleoprotein (RNP) complex into the cytoplasm. Anti-HA antibodies can also interfere with these conformational changes and/or the requisite interactions between the viral and endosomal membranes required for fusion (Fig. 1b) [6] [7] [7–8]. Thus, inhibition of the essential initial steps of viral infection steps can effectively interrupt transmission.
Figure 1.

Mechanisms of antibody-mediated neutralization of the influenza virus. (a) Antibodies can block influenza HA1 glycoprotein binding to sialic acid residues of receptor proteins on host cells. (b) Antibodies specific to the HA2 glycoprotein of the virus can inhibit its low-pH triggered fusion activity in the endosome at the post-binding/pre-fusion stage, which inhibits replication of the virus. (c) Antibodies to surface neuraminidase can prevent the release of influenza virions from the infected cell surface.
From the cytoplasm, the viral RNP is transported into the nucleus where transcription and replication of viral RNA occur. New viral RNP and protein complexes are subsequently assembled into viral particles which bud at the plasma membrane, a process that requires the enzymatic activity of NA to release the virions [9] [10]. NA-specific antibodies have been shown to restrict viral replication by preventing release of progeny from the infected cells, thereby shortening the severity and duration of illness [11] [12] [13] [14] [15], but they cannot prevent influenza virus infection. Therefore, we will focus in this review on describing mechanisms of virus neutralization by antibodies directed towards HA, the principal target of vaccine-induced protective immune responses.
Hemagglutinin is synthesized as a homo-trimeric precursor, HA0, which is assembled in the endoplasmic reticulum and is transferred to the plasma membrane via the Golgi complex [16] [17]. To be functional, HA0 must be cleaved by host cellular proteases into two disulfide-linked subunits, HA1 and HA2. The HA1 is highly divergent due to antigenic drift (discussed below), while the HA2 chain is relatively conserved [18]. The HA1 chain forms primarily the membrane-distal receptor binding domain that is responsible for adsorption of virus to the cell surface and also has a vestigial esterase domain containing a basic patch of four exposed histidines which contribute to virulence by enhancing membrane fusion (Fig. 2a) [19]. HA2 constitutes the core of the membrane fusion machinery around the hydrophobic fusion peptide at the amino-terminus of HA2. This cleavage event causes the partial burial of the fusion peptide into the charged cavity in the membrane-proximal stem region of HA2 [20–21].
Figure 2.

Crystal structure of the influenza virus HA protein and structural characterization of its rearrangement. (a) Overview of a trimeric HA0 precursor, shown as a ribbon diagram. One monomer is colored in yellow (HA1 subunit) and red (HA2 subunit) illustrating the locations of subdomains. The two other monomers that make up the trimer are shown in blue and green. (b) Ribbon diagram of a monomeric, cleaved form of the HA. Featured subdomains are highlighted with different colors: helix A (green), loop B (orange), helix C (pink), helix CD (blue), helix D (purple), β-sheet EF (cyan), helix G (black) and fusion peptide (red). (c) Stereodiagram of the HA2 subunit in the pH induced fusion conformation, the subdomains of which correspond to that of an intact HA monomer shown in b and are indicated by the same color. (d) The stereodiagram of HA2 in panel c is shown with the residues forming the hydrophobic core marked with red spheres. In (c) and (d), the N-terminal residues 1–37 of HA2 which includes the fusion peptide (residues 1–23) were removed from during the original preparation of the protein for crystal structure [32] and is illustrated in red within the circle. Figures were prepared using Chimera [82].
Cleavage of HA0 correlates with virus infectivity and pathogenicity, since it is essential for HA to undergo an acidic pH induced interaction that results in viral and endosomal membrane fusion [22–23] [24–26] [27–28]. The target cleavage site within HA0 requires host proteases that can vary among the different influenza viruses. For the three virus subtypes (H1, H2, and H3) that have caused pandemics in the past century, the HA0 precursor was shown to be cleaved at the sequence Q/E-X-R by trypsin-like proteases in bronchiolar epithelium Clara cells [29]. In contrast, the two avian subtypes (H5 and H7) contain polybasic sequences at the cleavage site [26, 30–31] and are cleaved intracellularly at the furin recognition sequence (R-XR/K-R) by subtilisin-like enzymes [26] [24] [25]. After HA0 cleavage, the membrane fusion process triggered by the low pH in the endosome results in an irreversible conformational change of HA. In this fusion process, a dramatic re-folding of HA2 causes the fusion peptide to move more than 100 Å to the end of the extended coiled-coil core (Fig. 2b & c) [32–33]. This dramatic transformation includes repacking of the central helix C with helix A which form an N-terminal cap at the central core, and conversion of the connecting segment between C and D (CD) to form a loop relocating helix D to folds back together with the small anti-parallel coiled coil in an inverted orientation [32] [34] (Fig. 2b & c). These multiple subdomain movements form a novel hydrophobic core that seemingly stabilizes the post-fusion conformation (Fig. 2d) [32].
Antigenic variation and evolution in influenza viruses
Unlike other viruses for which vaccines have been developed, influenza undergoes rapid and unpredictable antigenic variation of its surface expressed HA. This ever-evolving antigenic variation of HA is the key to the virus's ability to evade immune surveillance, therefore causing annual epidemics and periodic pandemics. Continuous host immune pressure after infection in the face of influenza's low fidelity RNA polymerase can lead to antigenic drift, i.e. gradual accumulation of point mutations and changes in the antigenicity of viral surface proteins [35]. In addition, as a result of having a segmented genome, shuffling of the virus' eight gene segments can occur if two different subtypes of influenza A virus infect the same cell. Such genetic reassortment can lead to a so-called antigenic shift, which is a major change in antigenicity that enables a virus strain to jump from one animal species to another, including humans. For example, phylogenic studies have shown that the 1957 pandemic H2N2 virus came from the circulating H1N1 virus, which acquired the H2 and N2 genes from avian specific strains [36]. Similarly, the 1968 pandemic H3N2 virus possibly came from the circulating H2N2 virus which acquired its H3 gene from avian viruses [37]. An avian influenza virus may also directly adapt after an initial zoonotic infection by undergoing significant genetic mutations to be efficiently transmitted from human to human. Aside from the cross-species route of transmission, still another plausible explanation for increased rates of influenza infections is for a human strain such as the H3N2 virus that has undergone antigenic drift to acquire certain internal gene segments of the pathogenic avian H5N1 virus (such as NS1 or PB2) which is associated with higher virulence. These newly emerging viruses tend to have atypical or more pathogenic infection cycles at the population level due to a lack of pre-existing immunity [38].
Vaccines have historically been the mainstay of infection control. However, due to rapid antigenic drift discussed above, it is difficult to predict the changes in HA-based vaccines that would be needed to protect against future epidemic strains. Consequently, the vaccine antigen that is updated biannually based on intensive global influenza surveillance [39] [40] is not always successful. Vaccine failure occurred in the 2003–2004 and 2007–2008 influenza seasons which caused increases in elderly and childhood deaths from influenza in the Northern Hemisphere [41] [42] [43]. A comparison of all available HA sequences of strains from 1968–2010 clearly shows that the variations occurred predominantly in the HA1 domain of the HA protein where the most dominant antigenic epitopes are located [44]. Regions of antigenic variations have been identified in H1 and H3 serotypes. For H3, the sites are designated A, B, C, D and E (Fig. 3a), while for H1, these sites are designated Sa, Sb, Ca and Cb (Fig. 3b) [45] [46]. Discovery of such extreme HA1-specfic amino acid variations occurring at positions corresponding to antigenic sites reveals that immununological selection by the host induced by vaccination or natural infection is a driving force for influenza virus evolution. For viruses of the H3 vaccine strains selected between 2005 and 2010, amino acid changes occurred in the HA1 domain at a rate of approximately 2.1%, per each new drift variant (Fig. 3a). The H1 vaccine strain followed a related pattern of variation with a rate of approximately 3% between 1999 and 2010 (Fig. 3b). An antigenic shift with roughly a 10-fold increase in amino acid variation was observed in the HA protein of an H1 strain from the 2009 to 2010 season that became the dominant pandemic influenza strain (Fig. 3b). Despite the variations in HA, the receptor binding region, including residues Tyr98, Trp153, Glu190, Leu194, and His183, has remained conserved during drift of the H1 and H3 subtypes. In the HA2 domain, only a total of 3 different amino acids (residues 32, 46, and 121) changed from 2005–2010 for H3 vaccine strains and 3 amino acids (residues 18, 89, and 303) changed from 1999–2010 for the H1 vaccine strains (Fig. 3). Interestingly, the highly conserved HA2 region is immunogenic, and antibodies reactive with the region have been demonstrated after vaccine-induced and natural infection in humans [47] [48]. However, it is unclear if these antibodies reach “protective” levels in vivo [49].
Figure 3.
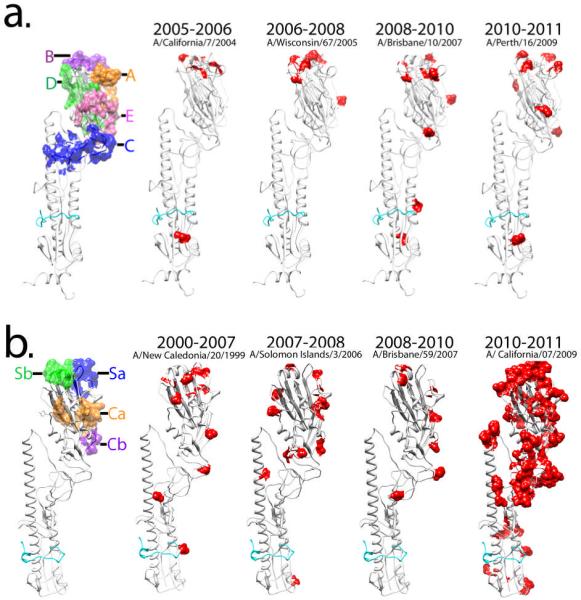
Antigenic variations in the HA protein of human influenza A vaccine strains. (a) Amino acid variations of H3 vaccine strains from 2005–2010 are compared synchronologically and represented on the HA monomer with a solvent accessible surface area. (b) Amino acid changes of H1 vaccine strains are compared from 1999 to 2010, and red colored residues on each HA structure indicate variations from the previous year's vaccine. Striking changes appeared in the amino acids in the HA of H1 strains from 2009 to 2010 which triggered the 2009–2010 pandemic H1N1. Figures were prepared using Chimera [82].
Mechanism of antibody-mediated neutralization of viral infection
The susceptibility of the influenza virus to antibody-mediated neutralization depends, in part, on the strength of an antibody binding to virus surface expressed HA; the higher the affinity of antibody for HA the greater the neutralization of virus infection [50–52] [53]. However, the preeminent determinant of virus neutralization is the nature of the viral epitope. Comparison of neutralization rate constants (Kneut.) and affinities (Kdissoc.) of five monoclonal antibodies (mAbs) with specificity for different epitopes on the HA1 of influenza virus revealed that neutralization is not directly proportional to antibody affinity. Instead, properties unique to the epitope are a primary factor in determining the efficiency of neutralization, suggesting that vaccine strategies that are focused on preferential induction of antibodies to the most potent neutralization sites or antiviral agents made directed towards these epitopes are more likely to have the most durable effects on neutralization and to prevent neutralization escape [52].
A large body of evidence has revealed that many antibodies efficiently neutralize influenza by occupying the receptor binding site [53–57]. Although the receptor binding pocket in the membrane distal HA1 subunit of HA is highly conserved, a vast majority of nAbs that target HA1 are strain-specific and lack broad cross-neutralizing activity, which is due primarily to the antibodies binding to hypervariable loops and regions surrounding the receptor binding site or they bind to epitopes on HA1 that only partially overlap with the receptor binding region and hypervariable regions [53–57]. For example, the structural comparison of nAbs complexed with the HA1 domain of the influenza virus showed that an antibody (HC19) which has an epitope overlapping with the receptor binding site has a higher neutralization efficiency than another antibody (HC45) that binds at a distance from this site (Fig. 4a) [55]. Dramatic conformational changes were observed in the HC19 complex, particularly in the distal tip of the H3 complementarity-determining region (CDR), where the side chains of Tyr102 and Asp103 shifted up to 10 Å compared to the unliganded structure of HC19. These changes allow the H3 CDR loop to interact with the HA receptor binding site (Fig. 4b) [54] [58]. The HC45 complex, on the other hand, involved only two local changes: one with a 2.1 Å movement of Asp63 carboxylate oxygen in HA due to a salt bridge formation with the HC45 Fab residue Arg94, and the second change in the HA structure resulted in a 1.5 Å movement of a residue Lys92 that prevented steric hindrance with the HC45 Fab [55] [59]. Such antibody-induced conformational changes could also enhance avidity of antibodies and therefore contribute to the neutralization efficiency.
Figure 4.

Stereo view of neutralizing antibodies and HA complexes. (a) Interactions of four antibodies with HA illustrate the locations of their neutralizing epitopes. HC19 binds to an epitope that overlaps with the receptor binding site. HC45 interacts with HA1 residues at the base of the β-sheet structure of the vestigial esterase E region. F10 and CR6261 contact residues on the HA2 hydrophobic membrane proximal stem region. (b) Ribbon diagram of HC19-HA interface shows that residues Tyr102 and Asp103 on the CDR-H3 loop (blue) of the HC19 heavy chain contribute to direct contact with residues Ser136, Asn137, Thr155 and Leu194 in the receptor binding region (peach shaded circle). (c) Close-up view of superimposed F10 and CR6261 nAbs in complex with HA depicting the exclusive binding with the heavy chain of the antibodies. The CDR chains of F10 are in darker shades corresponding to that of CR6261 in lighter shades of the same color (purple, CDR-H1; CDR-H2, green; CDR-H3, blue). The fusion peptide chain of HA is shown in red. The key interaction residue is a phenylalanine on the tip of CDR2-H2 which inserts into the hydrophobic pocket formed by the fusion peptide chain. (d) Overlapping structural representation of H3 (blue) and H5 (orange) subtype HAs shows the HA group-specific differences at residues His111 of HA2 unique to group 1 and His17 of HA1 unique to group 2. The group 2-specific His17 causes a change in orientation (90° turn) of the Trp21 side chain on the C-terminal end of the fusion peptide (cyan). This difference forms the basis for group-specific epitopes and group-wide broad-spectrum neutralization by antibodies that target the HA2 membrane proximal stem region. Figures were prepared using Chimera [82].
Less well-known but even more potent nAbs are the fusion-inhibiting antibodies that interact with the membrane proximal HA2 portion of the HA. The mAb C179 was first identified as an broadly neutralizing antibody (BnAb) with activity against H1, H2, and H5 viruses by blocking membrane fusion [60] [61] [62]. Recently, through human antibody phage display library selected against trimeric HA of H5N1 strain, several BnAbs were identified which neutralize multiple influenza subtypes, including H1, H2, H5, H6, H8, and H9 [63] [7]. Additional human antibodies that display broad neutralization against H1 and H5 have been identified in survivors to H5N1 infection [64]. BnAbs that bind viruses belonging to several HA subtypes (H1, H2, H5, H6, and H9) were also identified from some individuals vaccinated with the seasonal influenza vaccine.
An unprecedented yet common finding of these BnAb studies in the preferential, although not exclusive use of germline VH1-69 segment in forming critical interactions with their target. Co-crystal structures of HA with two VH1-69 encoded BnAbs, CR6261 [8] and F10 [7], that were determined at atomic resolution using X-ray crystallography, demonstrated binding to a common epitope in a highly conserved pocket in the HA2 membrane-proximal stem region of HA. Interestingly, VH1-69 is the only VH gene with two hydrophobic residues, 53 and 54 (Kabat numbering), with residue 54 encoding exclusively a phenylalanine at the tip of its CDR-H2 loop [65], which allows the antibody to make several crucial interactions with the conserved hydrophobic pocket in the HA2 membrane-proximal stem region. Both the CR6261 and F10 phage-derived antibodies bind to a highly conserved helical region in the membrane proximal stem, and their binding to the epitopes are exclusively through the heavy chain with no contribution of the epitope binding through the antibody light chain (Fig. 4c). The epitopes of these nAbs were shown to encompass the entire hydrophobic pocket adjacent to helix A, which contains the buried fusion peptide (Fig. 4c).
Phylogenic analysis of the HA gene subdivides influenza strains into 4 different clades within 2 phylogenic groups; group 1, which contains 10 of the 16 subtypes, including the H1 clade (H1, H2, H5, H6, H11, H13, and H16) and H9 clade (H8, H9, and H12), and group 2 includes the H3 clade (H3, H4, and H14) and H7 clade (H7, H10, and H15) [66–67]. The highly conserved hydrophobic pocket on HA is group-specific which explains the ability of these BnAbs to recognize all influenza group 1 viruses, including the experimentally tested H1, H2, H5, H6, H8, and H9 [8] [7]. Comparison of crystal structures of HA, three from group 1 (H1, H5, and H9) and two from group 2 (H3 and H7) [67–70], with the fusion-inhibiting epitopes recognized by antibodies F10 and CR6261 revealed that the group-specific differences are in the location of buried residues, particularly His111 on the HA2 subunit unique to group 1 and His17 on the HA1 subunit unique to group 2 [8] [7]. These unique histidine residues have been proposed to be the critical components for pH-induced conformation changes, indicating that the structural differences causes the side chain of Trp21 of HA2 to rotate at a 90 degree angle in group 2 subtypes, which could contribute to group-specific mechanisms for initiating the fusion process (Fig. 4d) [71].
Viral escape from neutralizing antibodies
Ongoing influenza virus evolution to evade immune responses remains problematic, and therefore understanding the mechanisms that govern neutralization escape is a prerequisite for developing effective prophylactic strategies. Recent studies indicate the unique role of immune pressure, mediated predominantly by humoral immunity, in shaping viral evolution within and among hosts infected by influenza A virus. It is generally assumed that influenza virus evasion of neutralization by antibodies is accompanied by single or multiple mutations in the viral HA structure. Bizebard et al. [54] reported the crystallographic structure of a complex of HA and an antibody that allowed viral escape from neutralization. Comparison of an antibody footprint (600–1000 Å2) with the surface area of the receptor-binding site (315 Å2) indicated that the HA-antibody interface contained many residues not essential for receptor-binding function, which might permit the virus to circumvent antibody neutralization in conjunction with mutations at hypervariable residues located adjacent to the receptor binding site [54]. The structural basis for escape has been further demonstrated by comparison of mutated HA residues at the antibody HC19-HA interface that either rendered escape from the neutralizing antibody or failed to do so [54] [72]. In this study, all recovered escape mutants contained mutations in epitopes where a solvent accessible surface area (A.s.a.) of the antibody-HA complex is less than 5 Å2 [54]. The replacement of wild-type residues (Thr155, Ser157, and Gly158) with the much bulkier mutant residues (Tyr155, Leu157, and Glu158) could potentially cause steric hindrance in the antibody-HA complex, as it cannot accommodate the bulkier amino acid without a significant structural rearrangement [54] (Fig. 5a). Additionally, the steric hindrance could cause reduced affinity of the antibody to its epitopes, which may contribute to antibody-mediated neutalization escape. However, steric hindrance cannot fully explain the mechanism of neutralization of other HA variants, particularly with a mutation of the residue at position 131 (Ile replacing Thr) from the same HC19-HA complex. The location of the complex provides 16 Å2 of A.s.a. where the side chain of the mutant residue Ile should be free enough to avoid steric hindrance. Fleury et al. [73] provided a structural based explanation of escape with this particular mutant by comparing the structures of mutant HA-Fab and wild-type complexes. The differences in the structures indicated that, although HC19 can still bind specifically to mutant HA, its binding is less favorable due to a distorted conformation of the mutant created from a side chain rotation at residue 131 together with loop movement adjacent to the mutated residue (Fig. 5c). Consequently, this distorted HA lowers binding affinity of the HC19 antibody to the epitope and permits neutralization escape by this antibody.
Figure 5.

Crystal structures of an HA mutant that escapes recognition by a neutralizing antibody. (a) Mutant residues on a small solvent-accessible surface area (red and orange spheres) abrogate neutralization by the HC19 antibody that targets epitopes in the receptor binding region within the HA globular head. Other mutant residues with greater than 20 Å surface interface (blue spheres) do not contribute to escape neutralization. The receptor binding surface is colored in yellow. (b) The space-filling model of the wild-type HA and antibody complex shows a sufficient solvent accessible area for the carbonyl oxygen of Lys156 residue on HA1 to form a hydrogen bond with the hydroxyl of Thr131. (c) The substitution of Thr with a hydrophobic Ile at residue 131 of HA1 completely buries the carbonyl oxygen of the Lys156 and prevents formation of hydrogen bond between Lys156 and Ile131 in the mutant HA-antibody complex. Hence, the antibody-HA complex becomes energetically unfavorable, resulting in a 4000-fold reduction of affinity of the antibody for the mutant HA as compared to the wild-type. Figures were prepared using Chimera [82].
Most escape mutants from anti-HA nAbs have been found to contain mutations in the hypervariable region of the membrane-distal domain, mainly due to its accessibility and wide-spread immunodominant domains in the globular head. Conversely, escape mutants in the HA2 region are notably scarce. Three viral mutants escaped from a fusion-inhibiting mAb C179, specific to the epitope comprising HA1 and HA2, have been generated for the purpose of identifying its epitopes [61] [62] [74]. However, selection of HA mutants that can escape neutralization has been difficult due to lack of structural information of the antibody C179-HA complex. Recently, the H5N1 virus variant that showed partial escape from the broadly neutralizing antibody CR6261 specific to regions in the membrane-proximal stem of H1 and HA2 was produced after extensive cell passage in vitro [63]. The identified mutation, a replacement of a histidine with a leucine at residue 111, was located in the HA2 helix. In the pre-fusion state, wild-type His111 is buried by the HA2 Try21 of the fusion peptide in the hydrophobic pocket; by mutating histidine to a hydrophobic leucine at this position, the result is diminished activity of antibody neutralization; however, the replication competence of this virus escape mutant was not reported [63]. Recently, three independent studies attempted to generate escape mutants from human antibodies that target the conserved pocket in the HA2 stem region of H1 [49], H3 [75] or H5 [7] viruses but were unable to isolate escape mutants. Thus, generation of escape mutants in the presence of BnAbs that target the HA2 membrane-proximal stem region may be constrained by fitness requirements such that mutations of critical residues in the epitope are more likely to affect the survival of the virus.
Structure-based influenza vaccine design strategies
Current trivalent influenza vaccines induce subtype-dependent immunity against antigenically well-matched strains of human influenza A subtypes H1N1 and H3N2 and of influenza B virus. These bi-annually determined seasonal vaccines are designed to principally induce nAbs against HA and thus are standardized on the basis of the HA content [76]. Consequently, mismatch of the vaccine strain antigens to circulating viruses due to continuous antigenic drift can cause suboptimal vaccine efficacy. Furthermore, it is currently impossible to predict when the next pandemic influenza will occur or what the subtype of the virus will be. A vaccine that elicits BnAbs against multiple influenza A subtypes, a so-called universal influenza vaccine, could eliminate much of the guesswork associated with vaccine strain selection and protection from emerging pandemic viruses. However, obtaining a universal vaccine has been hampered by the difficulties of designing an effective immunogen that can induce immune response to circumvent antigenic drift and neutralization escape of the virus.
Graves et al. [77] engineered an influenza virus lacking the HA1 subunit to exam differences of antigenicity and cross-reactivity between unmasked and wild-type HA2 which is topped by a large HA1 globular head. Based on the structural analysis of HA1 and HA2 interface, the disulfide bond joining the two HA subunits could be removed by acid treatment and dithiothreitol (DTT) reduction. HA2-specific antibodies generated by the subviral particles lacking the HA1 subunit was shown to partially cross-react with the HA2 of different subtypes while antibodies raised from the wild-type virus could not [77]. A strategy to create “designer” broadly protective immunogens was experimentally implemented with the intention of refocusing the immune system to avoid generating antibodies to undesired epitopes either by deletion of hypervariable epitopes on the HA globular head region or by masking the strain-specific antigenic sites with glycans [78] [77] [79]. These immune-refocused HA mutants induced neutralizing antibodies that produced significantly higher anti-viral activity against heterologous influenza viruses [77].
From decades of intensive influenza virus surveillance for vaccine strain selection and our knowledge of HA structure, we now understand that the hypervariable, immunodominant epitope regions are not functionally important but exclusively serve as decoys for the virus to avoid immune clearance (Fig. 6). The recently identified anti-HA BnAbs were shown to target the hydrophobic stem region of HA2 and inhibit the fusion process, strongly indicating that a broadly protective vaccine should be developed to refocus HA-specific immune responses away from those redundant epitopes of HA1 into more broadly heterosubtypic, conserved, and functionally important epitopes. Studies from two research groups demonstrated that a “headless” HA construct containing the conserved HA stalk domain and lacking the globular head could defy immunodominant, decoy responses generated by the virus and direct antibody responses to focus on more protective targets [80]. To further gain insight into the antigenic profile of this “headless” HA immunogen and structural requirements for BnAb binding, further studies are warranted to determine whether these HA proteins maintain a conformation similar to that of the wild-type HA to provide authentic conformational or discontinuous epitopes, whether they mirror the cleaved or uncleaved pre-fusion HA forms, or if similar BnAbs can be elicited that also block conformational changes and membrane fusion.
Figure 6.
Structure-based strategies for design of influenza vaccine immunogens. Current trivalent influenza vaccines are effective against only vaccine strains and closely related subtype-matched strains, generating many non-protective and/or type-restricted Abs against immunodominant epitopes of HA acting as an immunogenic decoy as shown in the left panel. In the middle panel, the HA immunogen shown is designed to elicit immune responses to target critical epitopes that can confer broad-spectrum neutralization. The engineered HA incorporates glycan moieties at selective positions in order to selectively divert and direct Ab responses to those critical epitopes on the receptor binding region and HA2 stalk domain (right upper panel). In the right bottom panel, an HA immunogen lacking the globular head region would ideally eliminate most of the immunodominant decoy epitopes and expose the highly conserved HA2 stalk domain for induction of broadly nAbs. Figures were prepared using Chimera [82].
Concluding remarks
Antigen and antibody interactions involve intimate complementary contact, which are susceptible to environmental change. The high resolution three-dimensional structure of antigen-antibody complexes, particularly with the recently identified anti-HA BnAbs, has contributed greatly to our understanding of how this conserved neutralization epitope is recognized and how we might induce heterosubtypic immunity to influenza infection. Moreover, the structure-based studies of the antibody-HA complexes has facilitated solving the conundrum of antigenic variation of HA in association with the annual recurrence of influenza epidemics/pandemics and immunity in man. It is our feeling that this new structural information can lead to the rational design of immunogens that can confer cross-protective immunity against multiple influenza subtypes and prevent neutralization escape or if it occurs it will be at significant cost to viral fitness. Continued implementation and refinement of novel prevention and treatment strategies can only serve to enhance our current understanding of the structural basis of how RNA viruses rapidly undergo neutralization escape. This is turn will lead to more robust immunization strategies, both passive and active, that can interrupt immune-mediated virus evolution. It is vital to understand the process of viral evolution under selective immune pressure and to seek answers from the structurally-informed perspective of this phenomenon. Although, it is clear that many challenges remain, approaches that recapitulate the structural characteristic of conserved neutralization epitopes and heterosubtypic antibodies, together with the use of new immunization strategies [81], should aid in the development of a truly universal vaccine for influenza.
ACKNOWLEDMENTS
This work was supported by grants from the National Insituttes of Health: U01-AI074518-01 to W.A.M. and 1K01AI073861 to T.H. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).
References
- 1.Johnson NP, Mueller J. Updating the accounts: global mortality of the 1918–1920 “Spanish” influenza pandemic. Bull Hist Med. 2002;76:105–115. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- 2.Fireman B, et al. Influenza vaccination and mortality: differentiating vaccine effects from bias. Am J Epidemiol. 2009;170:650–656. doi: 10.1093/aje/kwp173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nichol KL. Influenza vaccination in the elderly: impact on hospitalisation and mortality. Drugs Aging. 2005;22:495–515. doi: 10.2165/00002512-200522060-00004. [DOI] [PubMed] [Google Scholar]
- 4.Ginsberg J, et al. Detecting influenza epidemics using search engine query data. Nature. 2009;457:1012–1014. doi: 10.1038/nature07634. [DOI] [PubMed] [Google Scholar]
- 5.Kilbourne ED. Influenza. Plenum Publishing; New York: 1987. pp. 164–165. [Google Scholar]
- 6.Barbey-Martin C, et al. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology. 2002;294:70–74. doi: 10.1006/viro.2001.1320. [DOI] [PubMed] [Google Scholar]
- 7.Sui J, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 2009;16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ekiert DC, et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang IC, et al. Influenza A virus neuraminidase limits viral superinfection. J Virol. 2008;82:4834–4843. doi: 10.1128/JVI.00079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins PJ, et al. Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants. Nature. 2008;453:1258–1261. doi: 10.1038/nature06956. [DOI] [PubMed] [Google Scholar]
- 11.Powers DC, Kilbourne ED, Johansson BE. Neuraminidase-specific antibody responses to inactivated influenza virus vaccine in young and elderly adults. Clin Diagn Lab Immunol. 1996;3:511–516. doi: 10.1128/cdli.3.5.511-516.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kilbourne ED, et al. Antiviral activity of antiserum specific for an influenza virus neuraminidase. J Virol. 1968;2:281–288. doi: 10.1128/jvi.2.4.281-288.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy BR, Kasel JA, Chanock RM. Association of serum anti-neuraminidase antibody with resistance to influenza in man. N Engl J Med. 1972;286:1329–1332. doi: 10.1056/NEJM197206222862502. [DOI] [PubMed] [Google Scholar]
- 14.Couch RB, et al. Induction of partial immunity to influenza by a neuraminidase-specific vaccine. J Infect Dis. 1974;129:411–420. doi: 10.1093/infdis/129.4.411. [DOI] [PubMed] [Google Scholar]
- 15.Webster RG, Reay PA, Laver WG. Protection against lethal influenza with neuraminidase. Virology. 1988;164:230–237. doi: 10.1016/0042-6822(88)90640-x. [DOI] [PubMed] [Google Scholar]
- 16.Copeland CS, et al. Assembly of influenza hemagglutinin trimers and its role in intracellular transport. J Cell Biol. 1986;103:1179–1191. doi: 10.1083/jcb.103.4.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gething MJ, et al. Studies on the mechanism of membrane fusion: site-specific mutagenesis of the hemagglutinin of influenza virus. J Cell Biol. 1986;102:11–23. doi: 10.1083/jcb.102.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vareckova E, et al. Inhibition of fusion activity of influenza A haemagglutinin mediated by HA2-specific monoclonal antibodies. Arch Virol. 2003;148:469–486. doi: 10.1007/s00705-002-0932-1. [DOI] [PubMed] [Google Scholar]
- 19.Stevens J, et al. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science. 2004;303:1866–1870. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- 20.Ha Y, et al. H5 avian and H9 swine influenza virus haemagglutinin structures: possible origin of influenza subtypes. EMBO J. 2002;21:865–875. doi: 10.1093/emboj/21.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 22.Klenk HD, et al. Activation of influenza A viruses by trypsin treatment. Virology. 1975;68:426–439. doi: 10.1016/0042-6822(75)90284-6. [DOI] [PubMed] [Google Scholar]
- 23.Lazarowitz SG, Choppin PW. Enhancement of the infectivity of influenza A and B viruses by proteolytic cleavage of the hemagglutinin polypeptide. Virology. 1975;68:440–454. doi: 10.1016/0042-6822(75)90285-8. [DOI] [PubMed] [Google Scholar]
- 24.Klenk HD, Garten W. Host cell proteases controlling virus pathogenicity. Trends Microbiol. 1994;2:39–43. doi: 10.1016/0966-842x(94)90123-6. [DOI] [PubMed] [Google Scholar]
- 25.Klenk HD, Rott R. The molecular biology of influenza virus pathogenicity. Adv Virus Res. 1988;34:247–281. doi: 10.1016/S0065-3527(08)60520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Webster RG, Rott R. Influenza virus A pathogenicity: the pivotal role of hemagglutinin. Cell. 1987;50:665–666. doi: 10.1016/0092-8674(87)90321-7. [DOI] [PubMed] [Google Scholar]
- 27.Godley L, et al. Introduction of intersubunit disulfide bonds in the membrane-distal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell. 1992;68:635–645. doi: 10.1016/0092-8674(92)90140-8. [DOI] [PubMed] [Google Scholar]
- 28.Maeda T, Ohnishi S. Activation of influenza virus by acidic media causes hemolysis and fusion of erythrocytes. FEBS Lett. 1980;122:283–287. doi: 10.1016/0014-5793(80)80457-1. [DOI] [PubMed] [Google Scholar]
- 29.Kido H, et al. Isolation and characterization of a novel trypsin-like protease found in rat bronchiolar epithelial Clara cells. A possible activator of the viral fusion glycoprotein. J Biol Chem. 1992;267:13573–13579. [PubMed] [Google Scholar]
- 30.Bosch FX, et al. Proteolytic cleavage of influenza virus hemagglutinins: primary structure of the connecting peptide between HA1 and HA2 determines proteolytic cleavability and pathogenicity of Avian influenza viruses. Virology. 1981;113:725–735. doi: 10.1016/0042-6822(81)90201-4. [DOI] [PubMed] [Google Scholar]
- 31.Perdue ML, et al. Virulence-associated sequence duplication at the hemagglutinin cleavage site of avian influenza viruses. Virus Res. 1997;49:173–186. doi: 10.1016/s0168-1702(97)01468-8. [DOI] [PubMed] [Google Scholar]
- 32.Bullough PA, et al. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 33.Carr CM, Kim PS. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- 34.Chen J, Skehel JJ, Wiley DC. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc Natl Acad Sci U S A. 1999;96:8967–8972. doi: 10.1073/pnas.96.16.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Treanor J. Influenza vaccine--outmaneuvering antigenic shift and drift. N Engl J Med. 2004;350:218–220. doi: 10.1056/NEJMp038238. [DOI] [PubMed] [Google Scholar]
- 36.Schafer JR, et al. Origin of the pandemic 1957 H2 influenza A virus and the persistence of its possible progenitors in the avian reservoir. Virology. 1993;194:781–788. doi: 10.1006/viro.1993.1319. [DOI] [PubMed] [Google Scholar]
- 37.Scholtissek C, et al. On the origin of the human influenza virus subtypes H2N2 and H3N2. Virology. 1978;87:13–20. doi: 10.1016/0042-6822(78)90153-8. [DOI] [PubMed] [Google Scholar]
- 38.Wood JM. Selection of influenza vaccine strains and developing pandemic vaccines. Vaccine. 2002;20(Suppl 5):B40–44. doi: 10.1016/s0264-410x(02)00509-1. [DOI] [PubMed] [Google Scholar]
- 39.Tosh PK, Jacobson RM, Poland GA. Influenza vaccines: from surveillance through production to protection. Mayo Clin Proc. 2010;85:257–273. doi: 10.4065/mcp.2009.0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly H, et al. Estimation of influenza vaccine effectiveness from routine surveillance data. PLoS One. 2009;4:e5079. doi: 10.1371/journal.pone.0005079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Jong JC, et al. Mismatch between the 1997/1998 influenza vaccine and the major epidemic A(H3N2) virus strain as the cause of an inadequate vaccine-induced antibody response to this strain in the elderly. J Med Virol. 2000;61:94–99. [PubMed] [Google Scholar]
- 42.Carrat F, Flahault A. Influenza vaccine: the challenge of antigenic drift. Vaccine. 2007;25:6852–6862. doi: 10.1016/j.vaccine.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 43.Louie JK, et al. Severe pediatric influenza in California, 2003–2005: implications for immunization recommendations. Pediatrics. 2006;117:e610–618. doi: 10.1542/peds.2005-1373. [DOI] [PubMed] [Google Scholar]
- 44.Wilson IA, Cox NJ. Structural basis of immune recognition of influenza virus hemagglutinin. Annu Rev Immunol. 1990;8:737–771. doi: 10.1146/annurev.iy.08.040190.003513. [DOI] [PubMed] [Google Scholar]
- 45.Caton AJ, et al. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype) Cell. 1982;31:417–427. doi: 10.1016/0092-8674(82)90135-0. [DOI] [PubMed] [Google Scholar]
- 46.Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature. 1981;289:373–378. doi: 10.1038/289373a0. [DOI] [PubMed] [Google Scholar]
- 47.Cox RJ, Brokstad KA. The postvaccination antibody response to influenza virus proteins. APMIS. 1999;107:289–296. doi: 10.1111/j.1699-0463.1999.tb01556.x. [DOI] [PubMed] [Google Scholar]
- 48.Styk B, Russ G, Polakova K. Antigenic glycopolypeptides HA1 and HA2 of influenza virus haemagglutinin. IV. Immunogenic properties of separated haemagglutinin glycopolypeptides. Acta Virol. 1979;23:9–20. [PubMed] [Google Scholar]
- 49.Corti D, et al. Heterosubtypic neutralizing antibodies are produced by individuals immunized with a seasonal influenza vaccine. J Clin Invest. 2010;120:1663–1673. doi: 10.1172/JCI41902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown LE, et al. An analysis of the properties of monoclonal antibodies directed to epitopes on influenza virus hemagglutinin. Arch Virol. 1990;114:1–26. doi: 10.1007/BF01311008. [DOI] [PubMed] [Google Scholar]
- 51.Jackson DC, et al. Enumeration of antigenic sites of influenza virus hemagglutinin. Infect Immun. 1982;37:912–918. doi: 10.1128/iai.37.3.912-918.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schofield DJ, Stephenson JR, Dimmock NJ. High and low efficiency neutralization epitopes on the haemagglutinin of type A influenza virus. J Gen Virol. 1997;78(Pt 10):2441–2446. doi: 10.1099/0022-1317-78-10-2441. [DOI] [PubMed] [Google Scholar]
- 53.Knossow M, et al. Mechanism of neutralization of influenza virus infectivity by antibodies. Virology. 2002;302:294–298. doi: 10.1006/viro.2002.1625. [DOI] [PubMed] [Google Scholar]
- 54.Bizebard T, et al. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature. 1995;376:92–94. doi: 10.1038/376092a0. [DOI] [PubMed] [Google Scholar]
- 55.Fleury D, et al. A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nat Struct Biol. 1999;6:530–534. doi: 10.1038/9299. [DOI] [PubMed] [Google Scholar]
- 56.Taylor HP, Armstrong SJ, Dimmock NJ. Quantitative relationships between an influenza virus and neutralizing antibody. Virology. 1987;159:288–298. doi: 10.1016/0042-6822(87)90466-1. [DOI] [PubMed] [Google Scholar]
- 57.Taylor HP, Dimmock NJ. Competitive binding of neutralizing monoclonal and polyclonal IgG to the HA of influenza A virions in solution: only one IgG molecule is bound per HA trimer regardless of the specificity of the competitor. Virology. 1994;205:360–363. doi: 10.1006/viro.1994.1653. [DOI] [PubMed] [Google Scholar]
- 58.Bizebard T, et al. Refined three-dimensional structure of the Fab fragment of a murine IgGl,lambda antibody. Acta Crystallogr D Biol Crystallogr. 1994;50:768–777. doi: 10.1107/S0907444994001903. [DOI] [PubMed] [Google Scholar]
- 59.Fleury D, et al. Structural evidence for recognition of a single epitope by two distinct antibodies. Proteins. 2000;40:572–578. [PubMed] [Google Scholar]
- 60.Okuno Y, et al. Protection against the mouse-adapted A/FM/1/47 strain of influenza A virus in mice by a monoclonal antibody with cross-neutralizing activity among H1 and H2 strains. J Virol. 1994;68:517–520. doi: 10.1128/jvi.68.1.517-520.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okuno Y, et al. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J Virol. 1993;67:2552–2558. doi: 10.1128/jvi.67.5.2552-2558.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smirnov YA, et al. An epitope shared by the hemagglutinins of H1, H2, H5, and H6 subtypes of influenza A virus. Acta Virol. 1999;43:237–244. [PubMed] [Google Scholar]
- 63.Throsby M, et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One. 2008;3:e3942. doi: 10.1371/journal.pone.0003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kashyap AK, et al. Combinatorial antibody libraries from survivors of the Turkish H5N1 avian influenza outbreak reveal virus neutralization strategies. Proc Natl Acad Sci U S A. 2008;105:5986–5991. doi: 10.1073/pnas.0801367105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang CC, et al. Structural basis of tyrosine sulfation and VH-gene usage in antibodies that recognize the HIV type 1 coreceptor-binding site on gp120. Proc Natl Acad Sci U S A. 2004;101:2706–2711. doi: 10.1073/pnas.0308527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fouchier RA, et al. Characterization of a novel influenza A virus hemagglutinin subtype (H16) obtained from black-headed gulls. J Virol. 2005;79:2814–2822. doi: 10.1128/JVI.79.5.2814-2822.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Russell RJ, et al. H1 and H7 influenza haemagglutinin structures extend a structural classification of haemagglutinin subtypes. Virology. 2004;325:287–296. doi: 10.1016/j.virol.2004.04.040. [DOI] [PubMed] [Google Scholar]
- 68.Gamblin SJ, et al. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science. 2004;303:1838–1842. doi: 10.1126/science.1093155. [DOI] [PubMed] [Google Scholar]
- 69.Ha Y, et al. X-ray structure of the hemagglutinin of a potential H3 avian progenitor of the 1968 Hong Kong pandemic influenza virus. Virology. 2003;309:209–218. doi: 10.1016/s0042-6822(03)00068-0. [DOI] [PubMed] [Google Scholar]
- 70.Yamada S, et al. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature. 2006;444:378–382. doi: 10.1038/nature05264. [DOI] [PubMed] [Google Scholar]
- 71.Thoennes S, et al. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology. 2008;370:403–414. doi: 10.1016/j.virol.2007.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Knossow M, et al. Three-dimensional structure of an antigenic mutant of the influenza virus haemagglutinin. Nature. 1984;311:678–680. doi: 10.1038/311678a0. [DOI] [PubMed] [Google Scholar]
- 73.Fleury D, et al. Antigen distortion allows influenza virus to escape neutralization. Nat Struct Biol. 1998;5:119–123. doi: 10.1038/nsb0298-119. [DOI] [PubMed] [Google Scholar]
- 74.Tsuchiya E, et al. Antigenic structure of the haemagglutinin of human influenza A/H2N2 virus. J Gen Virol. 2001;82:2475–2484. doi: 10.1099/0022-1317-82-10-2475. [DOI] [PubMed] [Google Scholar]
- 75.Wang TT, et al. Broadly protective monoclonal antibodies against H3 influenza viruses following sequential immunization with different hemagglutinins. PLoS Pathog. 2010;6:e1000796. doi: 10.1371/journal.ppat.1000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gerdil C. The annual production cycle for influenza vaccine. Vaccine. 2003;21:1776–1779. doi: 10.1016/s0264-410x(03)00071-9. [DOI] [PubMed] [Google Scholar]
- 77.Graves PN, et al. Preparation of influenza virus subviral particles lacking the HA1 subunit of hemagglutinin: unmasking of cross-reactive HA2 determinants. Virology. 1983;126:106–116. doi: 10.1016/0042-6822(83)90465-8. [DOI] [PubMed] [Google Scholar]
- 78.Steel J, Lowen AC, Wang TT, Yondola M, Gao Qinshan., Haye Kester., Garcia-Sastre A, Palese P. Influenza Virus Vaccine Based on the Conserved Hemagglutinin Stalk Domain. mbio. 2010;1:1–9. doi: 10.1128/mBio.00018-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nara PL, Garrity R. Deceptive imprinting: a cosmopolitan strategy for complicating vaccination. Vaccine. 1998;16:1780–1787. doi: 10.1016/s0264-410x(98)00168-6. [DOI] [PubMed] [Google Scholar]
- 80.Sagawa H, et al. The immunological activity of a deletion mutant of influenza virus haemagglutinin lacking the globular region. J Gen Virol. 1996;77(Pt 7):1483–1487. doi: 10.1099/0022-1317-77-7-1483. [DOI] [PubMed] [Google Scholar]
- 81.Liu L, et al. Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell-mediated immunity. Nat Med. 2010;16:224–227. doi: 10.1038/nm.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]