

Synopsis
Caspase-2 is critical for genotoxic stress induced apoptosis and is activated by formation of the PIDDosome, an oligomeric caspase-2 activating complex. The PIDDosome comprises three protein components, PIDD, RAIDD and caspase-2. RAIDD contains both a death domain (DD) and a caspase recruitment domain (CARD). It acts as the bridge to recruit PIDD using the DD: DD interaction and to recruit caspase-2 via the CARD: CARD interaction. Here we report biochemical characterization and in vitro reconstitution of the core interactions in the PIDDosome. We show that RAIDD CARD and RAIDD DD interact with their binding partners, caspase-2 CARD and PIDD DD, respectively. However, full-length RAIDD fails to interact with either caspase-2 CARD or PIDD DD under a physiological buffer condition. We reveal that this lack of interaction of full-length RAIDD is not due to its tendency to aggregate under the physiological buffer condition, as decreasing full-length RAIDD aggregation using high salt or high pH is not able to promote complex formation. Instead, full-length RAIDD forms both binary and ternary complexes under a low salt condition. Successful in vitro reconstitution of the ternary complex provides a basis for further structural studies of the PIDDosome.
Keywords: Apoptosis, Inflammation, Caspase-2, RAIDD, PIDD, PIDDosome
Introduction
The development and homeostasis of multi-cellular organisms are dependent on a delicate balance of cell proliferation and cell death [1-4]. Failure to control cell death or apoptosis leads to serious diseases that threaten the existence of the organism [1, 5, 6]. Apoptosis proceeds through characteristic morphological changes that are dependent on the activities of caspases, a family of cysteine proteases that cleave specifically after aspartic acid residues [7-9]. Caspases are synthesized as single-chain zymogens and require a highly regulated process for their activation. Fully activated caspases are dimeric with two large subunits and two small subunits. Both cleavage and dimerization are important for the integrity of the caspase active sites and therefore required for caspase activation [10, 11].
Based on their roles in apoptosis, caspases are divided into two classes, initiator caspases such as caspase-2, -8, -9, -10, and effector caspases such as caspase-3 and -7 [7]. Initiator caspases are mostly monomeric in their pro-forms and are activated and auto-processed upon induced dimerization by recruitment to oligomeric signaling complexes. In contrast, effector caspases are constitutive dimers and activated upon cleavage by initiator caspases. The well-known oligomeric signaling complexes for the activation of initiator caspases are the death-inducing signaling complex (DISC) for caspase-8 activation [12], the apoptosome for caspase-9 activation [13], the inflammasome for caspase-1 activation [14, 15], and the PIDDosome for caspase-2 activation [16, 17]. Despite the important role of caspase activating complexes in apoptotic and inflammatory signaling pathways, the assembly mechanisms are not well characterized [4].
Accumulating data have shown that caspase-2 play an important role in genotoxic stress induced apoptosis by acting upstream of mitochondria at the mitochondrial apoptotic pathway [18, 19]. PIDDosome, the caspase-2 activating complex, is composed of three proteins, p53-induced protein with a death domain (PIDD), RIP-associated Ich-1/Ced-3 homologous protein with a death domain (RAIDD) and caspase-2 [4, 16, 20] (Figure 1(a)). Interestingly, it has been reported that activation of caspase-2 can also happen in the absence of PIDDosome formation [21]. In addition, elimination of PIDD or RAIDD did not significantly influence caspase-2 activation, suggesting that alternative modes of caspase-2 activation may exist [22]. RAIDD is an adaptor protein that contains both an N-terminal caspase recruitment domain (CARD) and a C-terminal death domain (DD) [23-25]. Caspase-2 possesses an N-terminal CARD prodomain while PIDD contains a C-terminal DD [26-29]. Both CARD and DD belong to the DD superfamily, which also includes the death effector domain (DED) and the Pyrin domain (PYD) [4, 30]. Collectively, the DD superfamily is one of the largest and most studied domain superfamilies, which contains more than 100 protein members and is involved in both cell death and inflammation. Many of these proteins participate in formation of large molecular machineries for the activation of signaling enzymes such as caspases and kinases. The DD superfamily is characterized by a common structural fold with six anti-parallel α-helices.
Figure 1. RAIDD CARD and RAIDD DD interact with caspase-2 CARD and PIDD DD respectively.
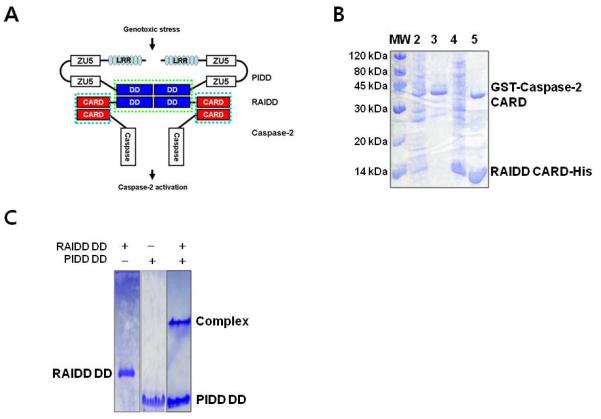
A. Schematic model of PIDDosome assembly. RAIDD that contains both CARD at N-terminus and DD at C-terminus works as adaptor between Caspase-2 and PIDD. RAIDD interacts to Caspase-2 via CARD-CARD interaction and PIDD via DD-DD interaction. B. Co-expression and His-tag pull-down of GST-caspase-2 CARD by His-tagged RAIDD CARD. Lane1: Marker; Lane2: Supernatant of the cell lysate; Lane3: Pellets of the cell lysate; Lane4: Flow through after incubation with Ni-NTA beads; Lane5: Imidazole eluted fraction. C. Native-PAGE for RAIDD DD (Lane 1), PIDD DD (Lane 2), and the mixed sample (Lane 3). Complex band and excess PIDD DD are appeared.
While RAIDD and PIDD interact with each other via their DDs, RAIDD and caspase-2 interact with each other via their CARDs [25, 26, 31]. Full length PIDD contains 910 residues with seven leucine rich repeats (LRRs), two ZU-5 domains and a C-terminal DD (Figure 1(a)). It is often auto-processed via an intein-like mechanism into shorter fragments of 51kD, 48kD and 37kD [16, 32, 33]. The cleavage sites have been mapped to S446 and S588 [32]. Cleavage at S446, which locates in between the two ZU-5 domains, generates a PIDD-N fragment of 48kD (residues 1-446) and a PIDD-C fragment of 51kD (residues 447-910). Further cleavage at S588, which locates in between the second ZU-5 domain and the C-terminal DD, generates a PIDD-CC fragment of 37kD (residues 589-910). Auto-cleavage of PIDD determines the outcome of the downstream signaling events. The initially formed PIDD-C fragment mediates the activation of NF-κB via the recruitment of RIP1 and NEMO and the subsequent formation of PIDD-CC causes caspase-2 activation and cell death [32]. Because PIDD-CC contains essentially the DD, the death-inducing PIDDosome is represented mostly by the DD: DD and the CARD: CARD interactions.
Our previous structural studies have shown that 5 PIDD DD and 7 RAIDD DD molecules cooperatively assemble into a large oligomeric structure and that the DD interaction between PIDD and RAIDD forms the core oligomerization platform of the PIDDosome [20]. To further elucidate how RAIDD, PIDD and caspase-2 assemble into the PIDDosome, we attempted to reconstitute the core interactions in the PIDDosome in vitro. Our results revealed that although RAIDD CARD and RAIDD DD interacted with caspase-2 CARD and PIDD DD, respectively, full-length RAIDD failed to interact with either caspase-2 CARD or PIDD DD at a physiological buffer condition. We characterized the aggregation property of full-length RAIDD and showed that high salt dramatically reduced RAIDD aggregation and precipitation. However, we further showed that reducing RAIDD aggregation did not promote complex formation. Instead, a low salt condition facilitated both RAIDD binary and ternary complex formation. These studies established the basis for future structural studies on the PIDDosome.
Materials and methods
Protein expression and purification
The cDNA of full length human RAIDD (1-199) was used as a template for PCR and plasmid vector pET26b (Novagen) was used to add a hexa-histidine tag at the carboxy-terminus of RAIDD (1-199) and RAIDD CARD (2-93) for affinity purification. PCR products of full length RAIDD and RAIDD CARD were digested with NdeI and XhoI (NEB) restriction enzymes and were ligated into pET26b. The cloning process for RAIDD DD (94-199) and PIDD DD (777-883) was introduced elsewhere [25]. The cDNA of mouse caspase-2 was used as a template for PCR and pGEX 4T-3 plasmid vector (Novagen) was used to add GST (Glutathione S-transferase) tag at the carboxy-terminus of Casapse-2 CARD (32-121). For sub-cloning, BamHI and XhoI restriction enzymes were used.
Recombinant RAIDD CARD, RAIDD DD and PIDD DD was expressed in E.coli BL21 (DE3) RILP and purified as previously described [25]. Full-length RAIDD and GST-tagged Caspase-2 CARD were expressed in BL21 (DE3) E.coli line. The purification process for full-length RAIDD was similar with the process used for RAIDD DD. Briefly, the expression was induced by 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) for overnight at 20°C. The bacteria were then collected, resuspended and lysed by sonication in 80 ml lysis buffer (20 mM Tris-HCl at pH 7.9, 500 mM NaCl, 10 mM imidazole, and 5 mM β-ME). The cell debris was removed by centrifugation at 16,000 rpm for 1 hour at 4°C. His-tagged target was purified by affinity chromatography using Ni-NTA beads (Qiagen) and gel-filtration chromatography using S-200 (GE healthcare) pre-equilibrated with buffer containing 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM DTT. GST fusion protein containing caspase-2 CARD was purified using glutathione-sepharose 4B affinity chromatography column. To obtain a ternary complex, we co-expressed RAIDD, PIDD DD and caspase-2 CARD.
Pull-down assay
Co-expression system was used to perform pull-down assay. RAIDD and RAIDD CARD in pET 26b vector were co-transformed with caspase-2 CARD in pGEX4T-3 vector separately into BL21(DE3) E.coli competent cells. Expression was induced with 0.5 mM IPTG overnight at 20°C. The cells were collected and lysed by sonication in lysis buffer (20 mM Tris buffer at pH 7.9, 500 mM NaCl, 10 mM imidazole). The lysate was then removed by centrifugation and the supernatant fractions were applied to gravity-flow column (Bio-rad) packed with Ni-NTA affinity beads (Qiagen). The unbound bacterial proteins and overexpressed caspase-2 CARD fused to GST were removed from the column using washing buffer (20 mM Tris-HCl at pH 7.9, 500 mM NaCl, 60 mM imidazole, 10 % glycerol). Stable complex was detected by SDS-PAGE.
Native PAGE shift assay
Protein interaction between RAIDD and PIDD DD and between RAIDD DD and PIDD DD was monitored by Native (non-denaturing) PAGE on a PhastSystem (GE Healthcare) with pre-made 8-25% acrylamide gradient gels (GE Healthcare). Separately purified proteins were pre-incubated at room temperature for 1 hour before loading on the gel. Coomassie Brilliant Blue was used for staining and detection a shifted band.
Gel-filtration chromatography
For gel filtration analysis to detect complex formation, purified RAIDD was mixed with a molar excess of PIDD DD and applied to a gel-filtration column (Superdex 200 HR 10/30, GE healthcare) which was pre-equilibrated with a solution 20 mM Tris-HCl pH 8.0, 50 mM NaCl and 1 mM DTT. The fractions were collected and subjected to SDS-PAGE. Similar methods were used to detect ternary complex.
Solubility assay
The general strategy of solubility assay was based on the method introduced by Bondos and Bicknell 2006.
In Brief, purified RAIDD from gel-filtration chromatography in 20 mM Tris-HCl, 150 mM NaCl, 1 mM DTT buffer was incubated with various different concentration of NaCl for salt dependent test, different pH for pH dependent test, different temperature for temperature dependent test and different time for time dependent test. Among around 400 ul solution of each sample, 300 ul was used for turbidity assay. Turbidity of each sample was directly measured by the optical density at 600 nm at spectrophotometer (Backman). The other 100 ul samples were used for aggregation assay. % of aggregation was detected by both SDS-PAGE and Bradford assay (Bio-Rad, Richmond, CA). For this experiment, each sample that contains RAIDD precipitants at each condition tested was centrifugated at 10,000 g at 4°C for 20 min. Precaution was taken to avoid foam formation throughout all processes. RAIDD remaining in the supernatant after centrifugation was defined as soluble RAIDD at certain condition. Decreased RAIDD by aggregate that was removed by centrifugation was detected by SDS-PAGE. All the samples are boiled 10 min at 90°C for SDS-PAGE. Coomassie brilliant blue was used for staining and detection of bands appeared on the acylamide gels. Decreased RAIDD also quantitatively measured by calculating protein concentration.
Results
Purified RAIDD CARD and RAIDD DD interact with caspase-2 CARD and PIDD DD respectively
As a first step to elucidate the molecular basis of PIDDosome assembly (Figure. 1A), we attempted to express and purify PIDD DD, RAIDD DD, RAIDD CARD and caspase-2 CARD. While PIDD DD, RAIDD DD, and RAIDD CARD expressed well in E. coli as C-terminal His-tag fusion proteins and were purified with Ni affinity chromatography, caspase-2 CARD did not express with a C-terminal His-tag. By exchanging the affinity tag from a C-terminal His-tag to an N-terminal GST-tag, we could obtain caspase-2 CARD expression. However unfortunately, the GST-caspase-2 CARD was not soluble.
To overcome the insolubility of the GST-tag fused caspase-2 CARD, we co-expressed the fusion protein with its binding partner RAIDD CARD in E. coli. Co-expression is often important for obtaining soluble protein complexes when the component proteins are insoluble without their binding partners [34]. Although GST-caspase-2 CARD was still detected in the insoluble fraction, most of the expressed protein was present in the soluble fraction. With this information, we performed His-tag pull-down assay with the co-expression sample. The bacterial cell pellets were resuspended, homogenized and fractionated into the soluble supernatant fraction and the insoluble fraction. The soluble supernatant fraction that contained both His-tagged RAIDD CARD and GST-fused caspase-2 CARD was incubated with Ni-NTA beads. After extensive washing, the beads were eluted using imidazole and the eluted fraction was examined using Coomassie Brilliant Blue stained SDS-PAGE. In agreement to previous cellular experiments, Figure 1B clearly showed that GST-fused caspase-2 CARD co-eluted with His-tagged RAIDD CARD, indicating that capase-2 CARD interacts with RAIDD CARD in vitro.
For the interaction between His-tagged PIDD DD and RAIDD DD, we have shown previously that PIDD DD and RAIDD DD co-migrate on gel filtration chromatography as a ~150 kD complex [20]. This result was also confirmed using native PAGE (Figure 1C). Unbound left over PIDD DD was always detected. This maybe because of the aggregation tendency of PIDD, not all PIDD can be recruited into the RAIDD/PIDD complex.
Full-length RAIDD failed to interact with either caspase-2 CARD or PIDD DD
To reconstitute the PIDDosome in vitro with purified protein components, we successfully purified full-length RAIDD in E. coli (Figure 2A). Since full-length RAIDD, which contains CARD and DD for homotypic interactions, expected to interact with caspase-2 CARD and PIDD DD, we tested the dimeric interactions between full-length RAIDD and caspase-2 CARD and between full-length RAIDD and PIDD DD. We used a co-expression system followed by a pull-down assay to investigate the interaction between full-length RAIDD and caspase-2 CARD. To investigate full-length RAIDD and PIDD DD interaction, we used gel-filtration chromatography. Using a co-expression system for investigating the interaction between full-length RAIDD and caspase-2 CARD is critical because GST-tagged caspase-2 CARD was insoluble when expressed alone. In contrast, the PIDD DD itself was soluble and easily purified. Based on these biochemical analyses, we found that neither caspase-2 CARD nor PIDD DD formed a complex with full-length RAIDD in a physiological buffer condition (Figure 2B, 2C).
Figure 2. Full-length RAIDD failed to interact with neither Caspase-2 CARD nor PIDD DD.
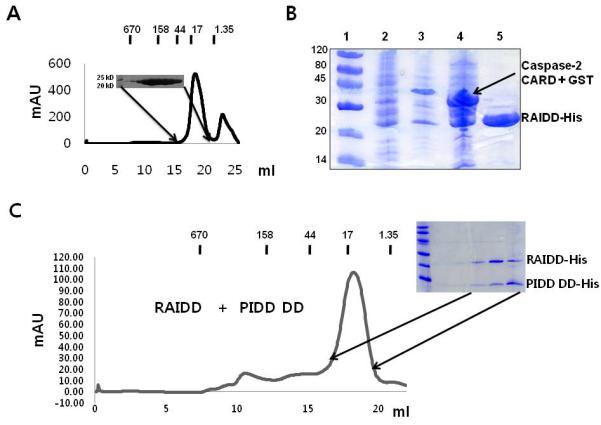
A. Gel filtration chromatogram and fractions of full-length RAIDD. B. Co-expression and His-tag pull-down of GST-caspase-2 CARD by His-tagged full-length RAIDD. Lane1: Marker; Lane2: Supernatant of the cell lysate; Lane3: Pellet of the cell lysate; Lane4: Flow through after incubation with Ni-NTA beads; Lane5: Imidazole eluted fraction. Flow-through contains unbounded GST-caspase-2 CARD. Only RAIDD is present at the eluted fraction. C. Gel-filtration profile of the mixture of full-length RAIDD and PIDD DD. SDS-PAGE of the peak fraction is shown.
Purified RAIDD is prone to aggregation at a physiological salt and pH condition
During the purification steps, we noticed that full-length RAIDD had a tendency to precipitate, as shown by visible turbidity, after elution from the gel filtration chromatography in a standard buffer condition that we routinely use. The buffer contained 20 mM Tris-HCl pH 8.0, 150 mM NaCl, and 1 mM DTT and mimics the physiological salt and pH condition. We wondered whether the solubility of full-length RAIDD is sensitive to buffer conditions and whether this property is related to its inability to interact with PIDD DD or casapse-2 CARD.
To investigate this property of full-length RAIDD more accurately, we conducted a solubility assay introduced by Bondos and Bicknell [35]. Based on this method, protein aggregation was measured by turbidity and sedimentation, which generally give mutually confirmatory results. We first tested time-dependent aggregation of full-length RAIDD at two different temperatures, 4°C and room temperature (R.T.). Turbidity, which increases with RAIDD precipitation, was detected by optical density (OD) at 600 nM using a spectrophotometer. The degree of aggregation was assayed by centrifuging down the precipitates followed by SDS-PAGE and the Bradford assay. As expected, turbidity of the RAIDD solution markedly increased as a function of time at both temperatures (Figure 3A). Measurement of loss of protein concentration after centrifugation in comparison with the initial RAIDD concentration showed the same trend of aggregation (Figure 3B). The precipitation and aggregation of RAIDD were also affected by temperature, with 4 °C producing the least amount of precipitation, 20 °C producing much more severe precipitation and 37 °C producing the most amount of precipitation (Figure 3C, 3D).
Figure 3. Full-length RAIDD is prone to aggregation in a temperature-dependent manner.
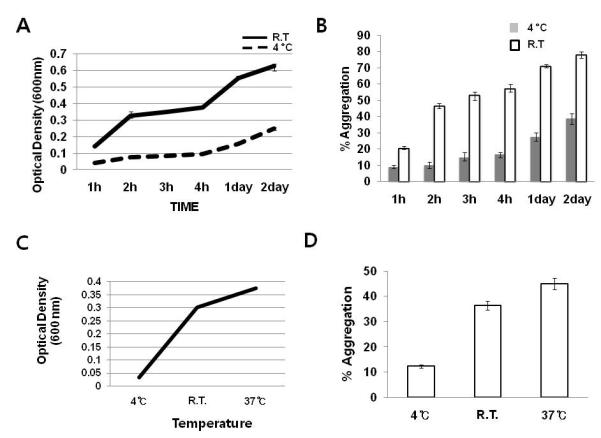
A. Solubility of full-length RAIDD as a function of time and temperature. All RAIDD samples were at 1 mg/ml concentration in 20 mM Tris-HCl pH 8.0 and 150 mM NaCl. Turbidity of each sample at the indicated incubation time and temperature was measured using the optical density at 600 nm. Values are means SD of n=3. B. Proportion of aggregated RAIDD. Upon incubation at the indicated time and temperature, each RAIDD sample was centrifuged and the loss of RAIDD concentration as a percentage of the initial concentration was denoted as the percentage of aggregation. Protein concentration was checked by the Bradford assay. Values are means SD of n=3. C. Solubility of full-length RAIDD as a function of temperature. Incubation time was 2 hours. Values are means SD of n=3. D. Proportion of aggregated RAIDD upon 2 hour incubation as a function of temperature. Values are means SD of n=3.
The solubility of RAIDD is sensitive to salt concentration
Although full-length RAIDD precipitated readily in the gel filtration buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl and 1 mM DTT), we realized that RAIDD did not precipitate after several days at 4°C in the Ni-NTA elution buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 250 mM Imidazole, and 5 mM β-ME). We hypothesized that the much higher NaCl concentration in the Ni-NTA elution buffer may increase the solubility of RAIDD. To evaluate this hypothesis, we tested the effect of NaCl concentration on the aggregation property of RAIDD. Increase of NaCl concentration gave a steady decrease in RAIDD aggregation as shown by both the OD600 assay and the centrifugation assay (Figure 4A, 4B). Since high concentrations of NaCl are required to prevent RAIDD aggregation, we examined whether NaCl could also solubilize pre-precipitated RAIDD aggregates. Our data clearly showed that 1.5 M of NaCl completely dissolved the aggregate, suggesting that the aggregation process of RAIDD is reversible (Figure 4C, 4D). “Salting out” effect was not observed at up to a concentration of 3M NaCl.
Figure 4. The solubility of RAIDD is sensitive to salt concentration.
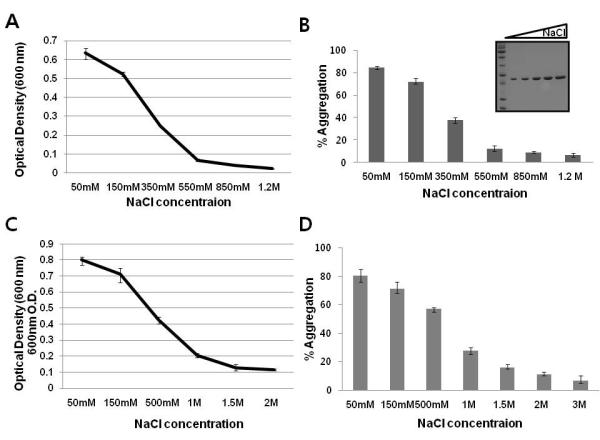
A. Solubility assay of salt concentration effect on RAIDD aggregation. RAIDD protein was prepared 1mg/ml concentration in buffer containing 20 mM Tris-HCl pH 8.0, 150 mM NaCl. Turbidity of each samples incubated with various different salt concentration for 3 days at 4°C, as measured by the optical density at 600 nm. Values are means SD of n=3. B. Proportion of aggregated full-length RAIDD following incubation with various NaCl concentration at 4°C for 3 days after centrifugation expressed as a percentage of the amount of initial concentration of RAIDD. Protein concentration of soluble portion of RAIDD in the solution was checked by Bradford assay. Values are means SD of n=3. The soluble portion of RAIDD in the solution was also loaded onto SDS-PAGE. Protein bands on a 15 % SDS-PAGE gel were detected by coomasie blue staining. The migration of molecular size marker is indicated. C. Solubility assay of salt effect on reversible RAIDD aggregation. Prepared RAIDD protein was incubated at R.T. for 12 hours and let protein aggregate. The aggregate was placed in the buffer containing various salt concentration indicated for a day and turbidity of each samples was measured by the optical density at 600 nm. Values are means SD of n=3. D. Proportion of reduced aggregate following incubation with various NaCl concentrations for a day expressed as a percentage of the amount of initial concentration of RAIDD. Protein concentration of solubilized portion of RAIDD in the solution was checked by Bradford assay. Values are means SD of n=3.
Complex formation of full-length RAIDD and PIDD DD at low salt
The salt dependence of full-length RAIDD solubility may have several implications in its interaction with PIDD DD. One possibility is that RAIDD has to de-aggregate at higher salt to interact with PIDD DD. Another possibility is that aggregation of RAIDD at lower salt is due to the lack of binding partners and that higher salt either changes RAIDD conformation or masks surface charge patches to prevent aggregation.
To test the above possibilities, we investigated whether full-length RAIDD could form a stable complex with PIDD DD by modifying salt concentration. Although full-length RAIDD and PIDD DD were both monomeric and did not form a complex in solution containing 20 mM Tris-HCl pH 8.0, 150 mM NaCl and 1 mM DTT (Figure 2C), when purified RAIDD and PIDD DD were incubated at R. T. for 1 hour in the buffer containing 20 mM Tris-HCl pH 8.0, 50 mM NaCl and 1 mM DTT, a complex of RAIDD and PIDD DD was obtained as shown by gel filtration chromatography (Figure 5A, 5B) and native PAGE (Figure 5C). In contrast, a higher salt of 0.5 M did not produce a complex of RAIDD and PIDD DD (data not shown). The results suggested that the aggregation propensity of full-length RAIDD in lower salt is due to the absence of binding partners and that higher salt prevents aggregation but does not promote interaction.
Figure 5. Complex formation of full-length RAIDD and PIDD DD, binary PIDDosome complex, at low salt.
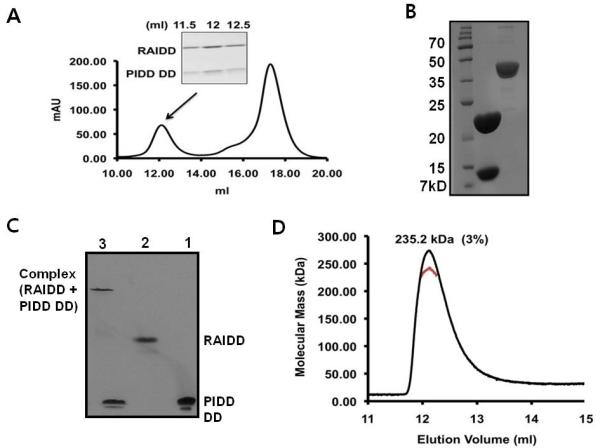
A. Separately purified both full-length RAIDD and PIDD DD are mixed together in the buffer containing 20 mM Tris-HCl pH 8.0, 50 mM NaCl, and 1 mM DTT and incubated R.T. for 1 hour. Gel-filtration profile showing formation of the complex between full-length RAIDD and PIDD DD. SDS-PAGE of gel filtration fractions showing formation of the complex. B. The final concentrated complex peak containing both RAIDD and PIDD showing on SDS-PAGE. The complex eluted at around 12 ml was collected and concentrated to 6 mg/ml: lane 1, final complex sample. Lane 2, Apo AI protein for comparison of concentration. C. Native-PAGE analysis of the interaction between full-length RAIDD and PIDD DD: lane1, PIDD DD only; lane 2, full-length RAIDD only; lane3, full-length RAIDD mixed with PIDD DD. D. Multi-angle light scattering (MALS) measurement of the RAIDD: PIDD DD complex peak, showing the molecular mass of the complex.
To obtain the accurate molecular mass and to elucidate the stoichiometry of the full-length RAIDD and PIDD DD complex, we used static multi-angle light scattering (MALS) in line with gel filtration chromatography. The calculated molecular masses of full-length RAIDD (residue 1-199) and human PIDD DD (residue 778-883) are 23.57 kDa and 13.38 kDa, respectively. MALS measurement gave a molecular mass of 235.2 kDa (3 % fitting error) for the complex (Figure 5D), suggesting that a 7 RAIDD: 5 PIDD DD complex was formed. The calculated molecular mass value for 7 full-length RAIDD and 5 PIDD DD is 232 kDa.
In vitro reconstitution of the core PIDDosome
To take the success of assembling the RAIDD: PIDD DD complex further, we performed ternary complex reconstitution of PIDD DD, RAIDD and caspase-2 CARD, the core interaction platform of the PIDDosome. We co-expressed RAIDD, PIDD DD and caspase-2 CARD. When the same condition of 20 mM Tris-HCl pH 8.0, 50 mM NaCl and 1 mM DTT was used for gel filtration chromatography, a peak centered at 9.5-10 ml in a Superdex 200 column was obtained (Figure 6A). Because the individual components would have elution peaks at around 16-18 ml, the much larger molecular complex containing all three proteins would correspond to the ternary complex of PIDD DD: RAIDD: caspase-2 CARD. The ternary complex was confirmed by Native-PAGE (Figure 6B)
Figure 6. In vitro reconstitution of ternary PIDDosome complex.
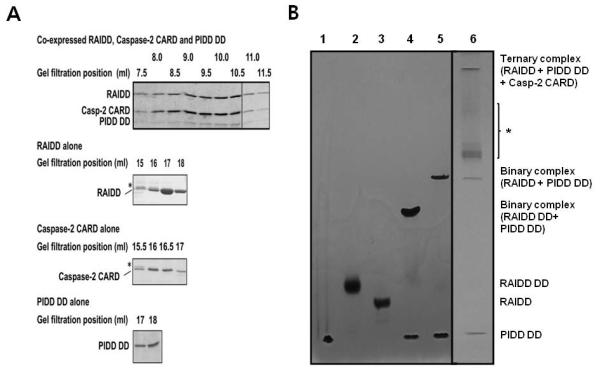
A. Co-expressed full-length RAIDD, PIDD DD and caspase-2 CARD were subjected to gel filtration at the 50 mM salt buffer, showing ternary complex formation. Elution positions of the individual components were also shown. B. Native-PAGE analysis of the interaction in the ternary complex: lane1, PIDD DD only; lane 2, RAIDD DD only; 3, full-length RAIDD only; lane4, RAIDD DD mixed with PIDD DD. lane 5, full-length RAIDD mixed with PIDD DD; 6, Ternary complex purified (including RAIDD, PIDD DD, and Caspase-2 CARD). * Possible ternary complex with less than full stoichiometric caspase-2 CARD.
Discussion
Structural studies for PIDDosome have been difficult. One problem is that full-length RAIDD did not readily interact with caspase-2 CARD or PIDD DD in an in vitro system at a physiological pH and salt concentration. However, purified RAIDD CARD and RAIDD DD can interact with caspase-2 CARD and PIDD DD respectively under the same conditions. Through a series of biochemical characterization studies, we found that the solubility of RAIDD and its interactions are sensitive to salt concentrations. At a high salt condition including a physiological salt concentration, RAIDD aggregates less, but could not interact with either PIDD DD or caspase-2 CARD. At an extreme low salt concentration, RAIDD tends to aggregate more but is able to interact with PIDD DD to form a binary complex and with both PIDD DD and caspase-2 CARD to form a ternary complex.
It appears that an extreme low salt likely changes the conformation of RAIDD to an open state to allow its interactions with the binding partners (Figure 7). The same open state also appears to be prone for aggregation. In contrast, high salt conditions likely promote a closed conformation of RAIDD that masks surface patches to prevent aggregation. It has been shown that spontaneous PIDDosome formation from endogeneous protein components can be induced when cells were broken open in a hypotonic buffer condition [16]. Physiologically, it is possible that genotoxic stress also causes a conformational change in RAIDD to allow PIDDosome formation and cell death induction. In vitro, a low salt condition somehow mimics this stress to promote assembly of the PIDDosome.
Figure 7. Model for regulation mechanism of PIDDosome formation.
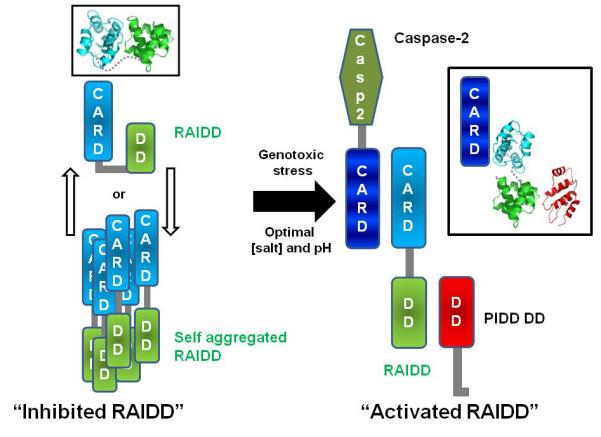
RAIDD may exist as equilibrium of the open and the closed states. Two states model (open or closed form) of RAIDD is shown at left side. Under low salt conditions or possibly genotoxic stress, RAIDD favors the open conformation to allow PIDDosome formation. Ribbon models are shown in black box. Ribbon model is generated by previously solved structures, RAIDD CARD (PDB ID: 3CRD_A), RAIDD DD (PDB ID: 2O71), PIDD DD (PDB ID: 2OF5).
Our model of conformational changes in RAIDD is reminiscent of that proposed for another adaptor protein, FADD, in the assembly of the caspase-activating complex DISC [36]. Similar to the case for RAIDD, FADD contains an N-terminal DED and a C-terminal DD. Full-length FADD failed to interact with either Fas DD or caspase-8 DED under a physiological buffer condition [36]. It appears then, that nature has evolved many regulatory elements including conformational changes to avoid accidental induction of cell death.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) of the Ministry of Education, Science and Technology (2010-0015311 to HHP) and by the National Institute of Health (RO1 AI076927 to HW)
References
- 1.Navratil JS, Ahearn JM. Apoptosis, clearance mechanisms, and the development of systemic lupus erythematosus. Curr Rheumatol Rep. 2001;3:191–198. doi: 10.1007/s11926-001-0018-1. [DOI] [PubMed] [Google Scholar]
- 2.Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival. Philos Trans R Soc Lond B Biol Sci. 1994;345:265–268. doi: 10.1098/rstb.1994.0104. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 4.Park HH, Lo YC, Lin SC, Wang L, Yang JK, Wu H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. 2007;25:561–586. doi: 10.1146/annurev.immunol.25.022106.141656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fisher DE. Pathways of apoptosis and the modulation of cell death in cancer. Hematol Oncol Clin North Am. 2001;15:931–956. ix. doi: 10.1016/s0889-8588(05)70258-6. [DOI] [PubMed] [Google Scholar]
- 6.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 7.Harvey NL, Kumar S. The role of caspases in apoptosis. Adv Biochem Eng Biotechnol. 1998;62:107–128. doi: 10.1007/BFb0102307. [DOI] [PubMed] [Google Scholar]
- 8.Salvesen GS. Caspases and apoptosis. Essays Biochem. 2002;38:9–19. doi: 10.1042/bse0380009. [DOI] [PubMed] [Google Scholar]
- 9.Park HH. Fifty C-terminal amino acid residues are necessary for the chaperone activity of DFF45 but not for the inhibition of DFF40. BMB Rep. 2009;42:713–718. doi: 10.5483/bmbrep.2009.42.11.713. [DOI] [PubMed] [Google Scholar]
- 10.Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15:725–731. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Pop C, Salvesen GS. Human caspases: Activation, specificity and regulation. J Biol Chem. 2009;284:21777–21781. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wajant H. The Fas signaling pathway: more than a paradigm. Science. 2002;296:1635–1636. doi: 10.1126/science.1071553. [DOI] [PubMed] [Google Scholar]
- 13.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 14.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 15.Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–574. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–846. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 17.Jang TH, Bae JY, Park OK, et al. Identification and analysis of dominant negative mutants of RAIDD and PIDD. Biochim Biophys Acta. 2010;1804:1557–1563. doi: 10.1016/j.bbapap.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 18.Lassus P, Opitz-Araya X, Lazebnik Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science. 2002;297:1352–1354. doi: 10.1126/science.1074721. [DOI] [PubMed] [Google Scholar]
- 19.Shin S, Lee Y, Kim W, Ko H, Choi H, Kim K. Caspase-2 primes cancer cells for TRAIL-mediated apoptosis by processing procaspase-8. EMBO J. 2005;24:3532–3542. doi: 10.1038/sj.emboj.7600827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park HH, Logette E, Raunser S, et al. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell. 2007;128:533–546. doi: 10.1016/j.cell.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manzl C, Krumschnabel G, Bock F, et al. Caspase-2 activation in the absence of PIDDosome formation. J Cell Biol. 2009;185:291–303. doi: 10.1083/jcb.200811105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olsson M, Vakifahmetoglu H, Abruzzo PM, Hogstrand K, Grandien A, Zhivotovsky B. DISC-mediated activation of caspase-2 in DNA damage-induced apoptosis. Oncogene. 2009;28:1949–1959. doi: 10.1038/onc.2009.36. [DOI] [PubMed] [Google Scholar]
- 23.Duan H, Dixit VM. RAIDD is a new ‘death’ adaptor molecule. Nature. 1997;385:86–89. doi: 10.1038/385086a0. [DOI] [PubMed] [Google Scholar]
- 24.Park HH, Wu H. Crystal structure of RAIDD death domain implicates potential mechanism of PIDDosome assembly. J Mol Biol. 2006;357:358–364. doi: 10.1016/j.jmb.2005.12.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park HH, Wu H. Crystallization and preliminary X-ray crystallographic studies of the oligomeric death-domain complex between PIDD and RAIDD. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:229–232. doi: 10.1107/S1744309107007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baptiste-Okoh N, Barsotti AM, Prives C. A role for caspase 2 and PIDD in the process of p53-mediated apoptosis. Proc Natl Acad Sci U S A. 2008;105:1937–1942. doi: 10.1073/pnas.0711800105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janssens S, Tinel A, Lippens S, Tschopp J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell. 2005;123:1079–1092. doi: 10.1016/j.cell.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 28.Lin Y, Ma W, Benchimol S. Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat Genet. 2000;26:122–127. doi: 10.1038/79102. [DOI] [PubMed] [Google Scholar]
- 29.Wu ZH, Mabb A, Miyamoto S. PIDD: a switch hitter. Cell. 2005;123:980–982. doi: 10.1016/j.cell.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 30.Reed JC, Doctor KS, Godzik A. The domains of apoptosis: a genomics perspective. Sci STKE. 2004;239:re9. doi: 10.1126/stke.2392004re9. [DOI] [PubMed] [Google Scholar]
- 31.Chou JJ, Matsuo H, Duan H, Wagner G. Solution structure of the RAIDD CARD and model for CARD/CARD interaction in caspase-2 and caspase-9 recruitment. Cell. 1998;94:171–180. doi: 10.1016/s0092-8674(00)81417-8. [DOI] [PubMed] [Google Scholar]
- 32.Tinel A, Janssens S, Lippens S, et al. Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and pro-survival NF-kappaB pathway. Embo J. 2006;26(1):197–208. doi: 10.1038/sj.emboj.7601473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pick R, Badura S, Bosser S, Zornig M. Upon intracellular processing, the C-terminal death domain-containing fragment of the p53-inducible PIDD/LRDD protein translocates to the nucleoli and interacts with nucleolin. Biochem Biophys Res Commun. 2006;349:1329–1338. doi: 10.1016/j.bbrc.2006.08.176. [DOI] [PubMed] [Google Scholar]
- 34.Dzivenu OK, Park HH, Wu H. General co-expression vectors for the overexpression of heterodimeric protein complexes in Escherichia coli. Protein Expr Purif. 2004;38:1–8. doi: 10.1016/j.pep.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 35.Bondos SE, Bicknell A. Detection and prevention of protein aggregation before, during, and after purification. Analytical Biochemistry. 2003;316:223–231. doi: 10.1016/s0003-2697(03)00059-9. [DOI] [PubMed] [Google Scholar]
- 36.Yang JK, Wang L, Zheng L, et al. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol Cell. 2005;20:939–949. doi: 10.1016/j.molcel.2005.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]