

Abstract
Peroxiredoxin 6 (Prdx6) exerts its protective role through peroxidase activity against H2O2 and phospholipid hydroperoxides. We hypothesized that targeted disruption of Prdx6 would lead to enhanced susceptibility to cigarette smoke (CS)-mediated lung inflammation and/or emphysema in mouse lung. Prdx6 null (Prdx6−/−) mice exposed to acute CS showed no significant increase of inflammatory cell influx or any alterations in lung levels of pro inflammatory cytokines compared to wild-type (WT) mice. Lung levels of antioxidant enzymes were significantly increased in acute CS-exposed Prdx6−/− compared to WT mice. Overexpressing (Prdx6+/+) mice exposed to acute CS showed significant decrease in lung antioxidant enzymes associated with increased inflammatory response compared to CS-exposed WT mice or air-exposed Prdx6−/− mice. However, chronic 6 months of CS exposure resulted in increased lung inflammatory response, mean linear intercept (Lm), and alteration in lung mechanical properties in Prdx6−/− when compared to WT mice exposed to CS. These data show that targeted disruption of Prdx6 does not lead to increased lung inflammatory response but is associated with increased antioxidants, suggesting a critical role of lung Prdx6 and several compensatory mechanisms during acute CS-induced adaptive response, whereas this protection is lost in chronic CS exposure leading to emphysema.
Keywords: antioxidants, cigarette smoke, COPD, inflammation, oxidants, Prdx6
Cigarette smoke (CS) is the major etiological agent in the pathogenesis of chronic obstructive pulmonary disease (COPD). CS consists of over 4700 chemical compounds and each puff contains 1014 to 1016 free radicals, including reactive aldehydes, quinones, and benzo(a)pyrene [1, 2]. CS induces oxidative stress, leading to lipid peroxidation thereby generating lipid hydroperoxides associated with inflammatory response and apoptosis [3]. Reduced glutathione (GSH), an important cellular thiol antioxidant, plays a key role in maintaining the redox balance and integrity in epithelial lining. GSH levels are decreased in response to CS exposure in lungs of rodents and in patients with COPD, highlighting the role for oxidative stress in the pathogenesis of COPD [2].
Peroxiredoxins (Prdxs), a family of thiol-specific antioxidant proteins, exert their protective role through peroxidase activity against hydrogen peroxide, peroxynitrite, and phospholipid hydroperoxides [4]. The family of Prdxs contains 6 isoforms, all of them have been detected in mammalian cells [5] and in the human lung [6, 7]. These proteins are classified into 3 subgroups: 2-cysteine (2-Cys), atypical 2-Cys, and 1-Cys based on the number and position of cysteine residues that participate in enzyme catalysis [8, 9]. 2-Cys Prdx1 to Prdx4 contain 2 conserved cysteines; atypical 2-Cys Prdx5 also utilizes thioredoxin as an electron donor [8]. Prdx6 is the only member of Prdxs that utilizes GSH as an electron donor to catalyze the reduction of peroxides [10]. Prdx6 has the ability to reduce phospholipid hydroperoxides, thereby being capable of repairing membrane damage caused by oxidative stress [11, 12]. Alteration in the Prdxs, particularly Prdx6, would lead to altered redox balance and hence ensue inflammatory response in the lung.
Prdx6 is highly expressed in the lungs [5, 13, 14], and detected in alveolar epithelial type II cells, bronchial Clara cells, and alveolar macrophages [14–17]. Prdx6 can protect against oxidative stress–induced membrane lipid peroxidation and apoptosis and regulate cellular signaling [16]. Thus it is possible that Prdx6 is an important protective enzyme that counteracts oxidant stress in the lung. Until now, there is no report describing the role of Prdx6 in CS-mediated lung inflammation using in vivo mouse model. Hence, in order to understand the potential role of Prdx6 in acute and chronic CS-mediated lung inflammation and injury in vivo, we hypothesized that deficiency of Prdx6 leads to susceptibility, whereas overexpression of Prdx6 protects against CS-induced lung oxidant stress and inflammation. In the present study, mice lacking and overexpressing Prdx6 were used to understand the role of Prdx6 in response to acute and chronic CS-induced lung inflammatory, injurious, or protective responses.
MATERIALS AND METHODS
Materials
Unless otherwise stated, all biochemical reagents used in this study were purchased from Sigma (St. Louis, MO). Antibodies for Western blotting such as anti-Prdx1 (LF-PA0095), anti-Prdx2 (LF-PA0007), anti-Prdx3 (LF-PA0030), anti-Prdx4 (LF-PA0009), anti-Prdx5 (LF-PA0010), anti-Prdx6 (LF-PA0011), anti-Prdx6 SO3 (LF-PA0005), and anti-Prdx-SO3 (LF-PA0004) were purchased from Lab Frontier (Seoul, Korea). Antibodies against sulfiredoxin 1 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) (sc-51211), anti-catalase (C 0979) from Sigma, anti-Cu/ZnSOD (SOD-101) and anti-MnSOD (06–984) from Assay Designs (Ann Arbor, MI) and Upstate (Lake Placid, NY), respectively.
Animals
Prdx6 knockout mice (Prdx6−/−), which had targeted disruption of Prdx6 gene [14, 18–20], Prdx6 transgenic (Prdx6+/+) mice overexpressing Prdx6 gene [21–23], and wild-type (WT) mice of genetic background C57BL/6J (Jackson Laboratory, Bar Harbor, ME) were used. Mice were bred and maintained under specific pathogen–free condition in the vivarium facility at the University of Rochester. Targeted disruption and overexpression of Prdx6 gene was verified by genotyping and only homozygous knockout and transgenic overexpressing mice were used along with wild-type mice in this study. Wild-type, Prdx6−/− and Prdx6+/+ mice were 8 to 10 weeks of age at the beginning of experiments (air/CS exposure). All experimental protocols were approved by the University Committee on Animal Research at the University of Rochester.
Cigarette Smoke Exposure
Mice were exposed to acute (3 days) CS using Baumgartner-Jaeger CSM2082i cigarette smoking machine (CH Technologies, Westwood, NJ) and subchronic (2 months) and chronic (4 or 6 months) using a smoking machine (Model TE-10; Teague Enterprises) in the Inhalation Core Facility at the University of Rochester [24]. For acute CS exposure, mice were placed in individual compartments of a wire cage, which was placed inside a closed plastic box connected to the smoke source. The smoke was generated from 3R4F research cigarettes containing 10.9 mg of total particulate matter (TPM), 9.4 mg of tar, and 0.726 mg of nicotine, and carbon monoxide 11.9 mg per cigarette (University of Kentucky, Lexington, KY). Mice received two 1-hour exposures per day, 1 hour apart, according to the Federal Trade Commission protocol (1 puff/min of 2-second duration and 35 mL volume) for 3 days (acute exposure). Mainstream cigarette smoke was diluted along with filtered air and directed into the exposure chamber. Monitoring of CS exposure (TPM per cubic meter of air) was done in real time using a MicroDust Pro-aerosol monitor (Casella CEL, Bedford, UK) and verified daily by gravimetric sampling immediately after the exposure was completed. By adjusting the number of cigarettes used to produce smoke and the flow rate of the dilution air, the concentration of smoke was set at a nominal value (~300 mg/m3 TPM). Similarly, the control mice were exposed to filtered air for the same duration of time and the carbon monoxide concentration (290 to 300 ppm) in the chamber was monitored [24].
For subchronic and chronic CS exposures, mice received a total of 5 hours of exposures per day, 5 days a week for 2 months, 4 months, and 6 months, respectively. Each lighted cigarette was puffed for 2 seconds and once every minute for a total of 8 puffs, with the flow rate of 1.05 L/min, a standard puff of 35 cm3. The smoke machine (Model TE-10; Teague Enterprises) was adjusted to produce a mixture of side stream smoke (89%) and mainstream smoke (11%) by smoldering 5 cigarettes at one time and the smoke chamber atmosphere was monitored for total suspended particulates (90 mg/m3) and carbon monoxide (350 ppm) [25, 26].
Tissue Harvest and Differential Cell Count in Bronchoalveolar Lavage (BAL) Fluid
Mice were injected intraperitoneally with 100 mg/kg body weight of pentobarbiturate (Abbott laboratories, Abbott Park, IL) and then sacrificed by exsanguination. The lungs were lavaged 3 times with 0.6 mL of saline via a cannula inserted into the trachea. The aliquots were combined, centrifuged, and the BAL inflammatory cell pellet was resuspended in saline. The total cell number was determined with a hemocytometer, and cytospin slides (Thermo Shandon, Pittsburgh, PA) were prepared using 50,000 cells per slide. Differential cell counts (~500 cells/slide) were performed on cytospin-prepared slides stained with Diff-Quik (Dade Behring, Newark, DE).
Cytokine Analysis
The level of proinflammatory mediators, such as monocyte chemotatic protein (MCP)-1 and the chemokine keratinocyte chemoattractant (KC), in lung homogenates were measured by enzyme-linked immunosorbent assay (ELISA) using respective dual-antibody kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. The results were expressed in the samples as pg/mg protein.
Protein Extraction From Lung Tissues
One lobe of the lung tissue (~50 mg) was homogenized (Pro 200 homogenizer, at maximum speed, 5th gear for 40 seconds) in 0.5 mL of ice-cold RIPA buffer containing complete protease inhibitor cocktail (Sigma) [24]. The tissue homogenate was then incubated on ice for 45 minutes to allow total cell lysis. The homogenate was then centrifuged at 13,000 × g for 5 minutes at 4°C to separate the protein fraction from the cell/tissue debris. The supernatant containing protein was aliquoted and stored at −80°C for Western blotting.
Western Blot Analysis
Proteins (20 μg) from lung tissue homogenates, were separated on a 7.5% or 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel, transferred onto nitrocellulose membranes (Amersham, Arlington Heights, IL), and blocked using 5% bovine serum albumin (BSA) for 1 hour at room temperature. The membranes were then probed with a specific primary antibody (1:1000 dilution in 5% BSA in phosphate-buffered saline [PBS] containing 0.1% Tween 20) at 4°C for overnight. After three 10-minute washing steps, the membrane was probed with suitable secondary anti-rabbit, or anti-mouse, or anti-goat antibody (1:10,000 dilution in 5% BSA) linked to horseradish peroxidase for 1 hour, and detected using the enhanced chemiluminescence method (Perkin Elmer, Waltham, MA). Equal loading of the gel was determined by quantitation of protein as well as by reprobing the same membranes for β-actin. The levels of the overoxidation/hyperoxidation forms of Prdxs (Prdx SO(2,3) and Prdx6 SO(2,3)) and other antioxidant enzymes: catalase (CAT), copper/zinc superoxide dismutase (Cu/ZnSOD), and manganese superoxide dismutase (MnSOD) were determined from the lung homogenates of mice after (3 days) exposure to CS.
Hematoxylin and Eosin (H&E) Staining and Mean Linear Intercept (Lm) Analysis
Mice lungs (which had not been lavaged) after acute, subchronic, and chronic exposures, were inflated by 1% low-melting agarose at a pressure of 25 cm H2O, and then fixed with neutral buffered formalin. H&E-stained paraffin-embedded tissue sections (4 μm) were used to evaluate the alveolar size from the mean linear intercept (Lm) of the alveolar airspace. In each sample, 10 random fields at a magnification of ×200 using cross-lines were used to calculate Lm as described previously [27–29].
Protein Assay
Protein level in lung samples were measured by bicinchoninic acid (BCA) colorimetric assay (Thermo Scientific, Rockford, IL) using BSA as a standard.
Statistical Analysis
Data were presented as mean ± SEM. Statistical analysis of significance was calculated using 1-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for multigroup comparisons using StatView software. P < .05 is considered significant whereas P > .05 is considered non significant.
RESULTS
Inflammatory Cell Influx into the Lung of WT, Prdx6−/−, and Prdx6+/+ Mice in Response to Acute and Chronic Air and CS Exposures
In order to determine the role of Prdx6 in acute and chronic CS-mediated lung inflammation, WT, Prdx6−/−, and Prdx6+/+ mice were exposed from 3 days to 6 months CS, and the inflammatory cell in-flux into bronchoalveolar lavage (BAL) fluid was assessed by Diff-Quik staining. Wild-type mice showed increased neutrophil influx (P < .001) into BAL fluid at 3 days of CS exposure (TPM: 300 mg/m3). Surprisingly, when Prdx6−/− mice were exposed to CS, they showed a decrease (P < .001), and overexpressing Prdx6+/+ mice showed an increase, in neutrophil influx when compared to CS-exposed WT mice (P < .05) and to Prdx6−/− mice (P < .001) (Figure 1A). The number of total cells (data not shown) or macrophages were not altered significantly in air- and CS-exposed WT or in Prdx6−/− mice after acute CS exposure, whereas air- and CS-exposed overexpressing Prdx6+/+ mice showed significant decrease in macrophages (P < .001 and P < .01, respectively) compared to air- and CS- exposed WT mice (Figure 1B).
FIGURE 1.

Neutrophils, macrophages, and total cell influx into BAL fluid of WT, Prdx6−/−, and Prdx6+/+ mice after acute and chronic CS exposures. Mice were exposed to CS at 300 mg/m3 total particulate matter (TPM) for 3 days and 90 to 100 mg/m3 TPM for 6 months and sacrificed 24 hours after the last exposure. At least 500 cells in the BAL fluid were counted using hemocytometer in a blinded manner to determine the number of neutrophils (A) and macrophages (B, D) and total cells (C) on cytospin slides stained with Diff-Quik. Values are mean ± SEM (n = 4–5 mice per group). ***P < .001, significant compared to air-exposed WT mice. #P < .05, significant compared to CS-exposed WT mice. ###P < .001, significant compared to CS-exposed WT and Prdx6+/+ mice. **P < .01, significant compared to air-exposed Prdx6+/+ mice, CS-exposed WT, and Prdx6−/− mice. +++P < .001, significant compared to air-exposed WT and Prdx6−/− mice.
The total number of cells and macrophages increased (P < .05) in WT mice in response to chronic 6 months of CS exposure, whereas no change was observed in the Prdx6−/− mice (TPM: 90 mg/m3) (Figures 1C, D). Because subchronic CS exposure of Prdx6+/+ mice produced only mild, if any, changes in inflammatory cell influx, no long-term CS exposures were conducted in these mice. Overall, these results suggest that Prdx6 differentially regulates lung inflammatory cell influx in response to acute and chronic CS exposures.
Levels of Proinflammatory Mediators in Lungs of WT, Prdx6−/−, and Prdx6+/+ Mice in Response to Acute Air and CS Exposures
In order to confirm the pro inflammatory response in these mice, the levels of pro inflammatory mediators (MCP-1 and KC) were measured in lung homogenates of acute air- and CS-exposed WT, Prdx6−/−, and Prdx6+/+ mice. CS-exposed WT mice showed MCP-1 increase (P < .001) when compared to air-exposed WT. CS-exposed Prdx6−/− mice showed MCP-1 decrease (P < .001) and overexpressing Prdx6+/+ mice showed an increase (P < .001) when compared to corresponding air-exposed mice (Figure 2A). CS-exposed WT mice showed an increase in KC levels (P < .01) when compared to air-exposed WT mice. The levels of KC were lower (P < .001) in CS-exposed Prdx6−/− mice compared to CS-exposed WT mice. However, the levels of KC did not differ in lungs of air- and CS-exposed Prdx6−/−mice. Surprisingly, the levels of KC were lower (P < .001) in CS-exposed overexpressing Prdx6+/+ mice compared to CS-exposed WT mice (Figure 2B). Similarly, overexpressing Prdx6+/+ mice exposed to CS showed decrease in KC levels (P < .05) compared to air-exposed Prdx6+/+ mice (Figure 2B). Chronic CS exposure led to KC increase (P < .05) in WT mice compared to air-exposed WT mice, whereas Prdx6−/− mice exposed to CS revealed a pattern of KC increase when compared to the corresponding air-exposed mice (Figure 2C). Because CS exposure to Prdx6+/+ mice produced only mild changes in inflammatory cell influx into the lung after sub-chronic exposure, no further cytokine analyses were conducted on these mice.
FIGURE 2.

The levels of lung pro inflammatory mediators in WT, Prdx6−/−, and Prdx6+/+ mice after acute and chronic CS exposures. The levels of pro inflammatory mediators, MCP-1 and KC after acute (3 days) (A and B) and chronic (6 months) (C) CS exposures. MCP-1 and KC were measured by ELISA in lung homogenates of air- or CS-exposed mice. Values are mean ± SEM (n = 4 mice per group). *P < .05, **P < .01, and ***P < .001, significant compared to corresponding air-exposed WT mice. +++P < .001 significant compared to CS-exposed WT mice.
Chronic CS Exposure Led to Airspace Enlargement and Emphysema in Prdx6−/−Mouse Lung
Mean linear intercept (Lm) is commonly used as an index for characterizing airspace enlargement (emphysema) and the severity associated with structural destruction in the lungs. Increase in Lm is directly related to an increase in airspace sizes [30]. To assess the role of Prdx6 in chronic CS-mediated air-way enlargement/emphysema, the Lm was assessed in Prdx6−/− mice exposed to air or CS for 6 months. Lungs from the Prdx6−/− mice appeared to have greater Lm than the WT mice, in general the alveolar size being quantitatively greater (P < .05) both in air- and CS-exposed Prdx6−/− mice at 6 months when compared to air-exposed WT mice (Figure 3A and B). Increased airspace enlargement in chronic air-exposed Prdx6−/− mice may be due to developmental abnormalities such as airspace formation and maintenance as well as lung morphogenesis in Prdx6-deficient mice, which requires future studies. CS-exposed Prdx6−/− mice showed no change in Lm after 6 months of CS exposure when compared to WT mice exposed to CS.
FIGURE 3.
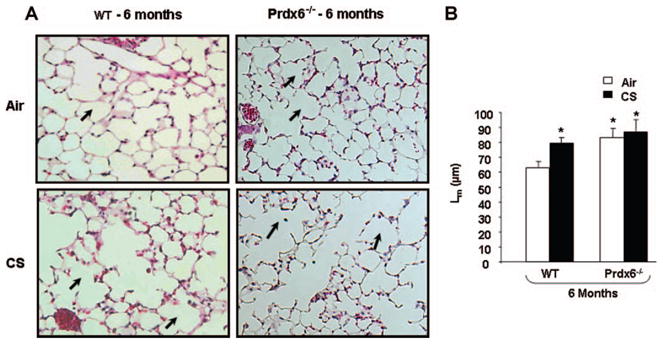
Airspace enlargement in lungs of WT and Prdx6−/− mice exposed to chronic CS. Representative figure of hematoxylin and eosin (H&E) stained lung sections from chronic (6 month) air- and CS-exposed WT and Prdx6−/− mice (A). Mean linear intercept (Lm) was calculated using H&E stained slides (B, original magnification: ×200). No significant change in Lm between the air- and CS-exposed Prdx6−/− mice. WT exposed to chronic CS shows an increase in Lm when compared to the air-exposed WT mice. Dark arrow indicates alveolar airspace enlargement. Values are mean ± SEM (n = 5 mice per group). *P < .05, significant compared to air-exposed WT mice.
The mechanical properties of the lungs were assessed by using Scireq Flexivent apparatus (Montreal, Canada) as described elsewhere [27]. Lung compliance was increased in CS-exposed WT mice compared to air-exposed WT (0.037 ± 0.003 versus 0.030 ± 0.002 mL/cm H2O; P < .01, n = 4 mice per group). The basal level of lung compliance in Prdx6−/− mice exposed to air and CS was already higher than in WT mice (0.050 ± 0.003; 0.052 ± 0.003 mL/cm H2O; P < .001, n = 4 mice per group). The only significant difference in CS-exposed Prdx6−/− mouse was evident when this group was compared with air- or CS-exposed WT mice. The lung resistance was significantly lower in CS-exposed WT as compared to air-exposed WT (0.50 ± 0.042 versus 0.58 ± 0.049 cm H2O·s/mL; P < .05, n = 4 mice per group), but the resistance was further lowered and there was no change in lung resistance between the air- and CS-exposed Prdx6−/− mice (0.49 ± 0.010 versus 0.51 ± 0.26 cm H2O·s/mL, n = 4 mice per group).
Overall, these results confirm that WT mice have pro inflammatory response to acute CS exposure. Prdx6−/− mice exposed to acute CS showed adaptation but chronic CS exposure led to sustained lung inflammatory response associated with alterations in Lm, and lung mechanical properties.
CS-Induced Changes of Antioxidant Enzymes in Lungs of Mouse Exposed to Acute CS
The abundance of antioxidant enzymes such as CAT, Cu/ZnSOD, and MnSOD in WT, Prdx6−/−, and Prdx6+/+ mice exposed to air or CS for 3 days were determined using immunoblot analysis. WT mice exposed to CS did not show significant changes in the levels of these antioxidant enzymes. Instead, Prdx6−/− mice exposed to CS showed an increase (P < .01) in levels of CAT, Cu/ZnSOD, and MnSOD when compared to air-exposed Prdx6−/− or air- and CS-exposed WT mice (Figure 4A and B). Prdx6+/+ mice showed a decrease in levels of Cu/ZnSOD both in air- (P < .01) and CS- (P < .001) exposed mice lungs compared to air- and CS-exposed WT mice (Figure 4C and D). CS-exposed WT mice showed a decrease (P < .001) in CAT compared to air-exposed WT mice. CAT and MnSOD were not altered in air-and CS- exposed Prdx6+/+ mice. Similarly, the levels of other antioxidant enzymes such as glutathione peroxidase 1 (GPx1) and sulfiredoxin 1 (Srx1) were also significantly increased in CS-exposed WT, Prdx6−/−and Prdx6+/+ mice compared to air-exposed WT, Prdx6−/−, and Prdx6+/+ mice (data not shown).
FIGURE 4.

Lung levels of antioxidant enzymes CAT, Cu/ZnSOD, and MnSOD in WT and Prdx6−/− and Prdx6+/+ mice exposed to acute CS. Mice were exposed to CS at 300 mg/m3 TPM for 3 days and sacrificed 24 hours after the last exposure. Levels of CAT, Cu/ZnSOD, and MnSOD in lung tissue homogenates were analyzed by Western blotting and β-actin was used as a housekeeping control (A–D). Prdx6−/− mice exposed to CS showed significant increase in the levels of CAT, Cu/ZnSOD, and MnSOD. Western blots are representative of 3 independent experiments and values are mean ± SEM (n = 3–5 mice per group). *P < .05, ***P < .001, ++P < .01, significant compared to air-exposed WT mice. *P < .05, **P < .01, significant compared to corresponding air-exposed Prdx6−/− or Prdx6+/+ mice. ++P < .01, +++P < .001, significant compared to CS-exposed WT mice.
Further studies were conducted to assess total GSH levels in lung homogenates of mice exposed to CS for 3 days (i.e., acute exposure). Total GSH level did not alter between the acute air- or CS-exposed WT mice as measured by the method described previously [31] (Figure 5). However, the Prdx6−/− mice exposed to CS for 3 days showed an increase in total GSH when compared to air-exposed Prdx6−/− and air- and CS-exposed WT mice. Prdx6+/+ mice exposed to CS showed decreased GSH compared to CS-exposed Prdx6−/− mice (Figure 5).
FIGURE 5.

Lung levels of total GSH in WT, Prdx6−/−, and Prdx6+/+ mice exposed to acute CS. Mice were exposed to CS at 300 mg/m3 TPM for 3 days and sacrificed 24 hours after the last exposure. Levels of GSH were measured spectrophotometrically in lung homogenates. Values are mean ± SEM (n = 4 mice per group). **P < .01, significant compared to the air-exposed Prdx6−/− mice. ++P < .01, significant compared to CS-exposed WT mice. +P < .05, significant compared to CS-exposed Prdx6−/− mice.
Immunohistochemistry of CS-exposed WT, Prdx6−/−, and Prdx6+/+ mouse lung showed an increase in abundance of Srx1 expression in bronchial epithelium and alveolar septum compared to air-exposed WT, Prdx6−/−, and Prdx6+/+ mice (data not shown). Similarly, Srx1 expression levels by immunoblot analysis revealed a significant increase in lung homogenates of WT, Prdx6−/−, and Prdx6+/+ mice in response to CS exposure (data not shown). The level of Nrf2 was not changed in lungs of Prdx6−/− mice as compared to WT mice exposed to CS, but it was decreased in CS-exposed Prdx6+/+ mice and CS-exposed WT mice (data not shown), suggesting that Nrf2, which is altered by CS [32], is not affected in Prdx6−/− mice, whereas the level was decreased in Prdx6+/+ mice compared to WT mice exposed to CS. This confirms that Srx1 (which is Nrf2 dependent) plays an important role in protection against acute CS-induced oxidative stress. Thus, the antioxidant enzymes could play a compensatory role against acute CS-induced oxidative stress in Prdx6−/− mice.
In order to understand the role of other 2-Cys peroxiredoxins in Prdx6−/− and Prdx6+/+ mice exposed to acute CS, the abundance of other 2-Cys peroxiredoxins (Prdx1 to Prdx5) and Prdx overoxidation (Prdx SO(2,3), and Prdx6 SO(2,3)) forms were measured using immunoblotting. Immunohistochemistry revealed that Prdx6 was expressed widely throughout the WT lung except in vascular structures and airway smooth muscle, high expression being detected in airway epithelium and alveolar septum. The distribution of Prdx6 in lung of WT mice was almost similar to that in transgenic mice as reported earlier [23]. Exposure to CS resulted in a slight decrease in Prdx6 expression in airway epithelium and alveolar septum of WT mice lungs when compared to air-exposed WT mice (data not shown). No major alterations in the levels of Prdx1 to Prdx3 and overoxidation forms (Prdx SO(2,3) and Prdx6 SO(2,3)) could be seen between the acute air- and CS-exposed WT mice as well as in Prdx6−/− and Prdx6+/+ mice (data not shown). Prdx4 was significantly increased in response to CS compared to corresponding air-exposed and CS-exposed WT mice. The basal level of Prdx5 was higher both in air- and CS-exposed Prdx6−/− mice compared to air- and CS-exposed WT mice (data not shown). Because Prdx6−/− mice lost this protective effect, as shown by the increased lung inflammatory and physiological/mechanical responses, no further measurements of antioxidants or other parameters in chronic CS exposures in Prdx6−/− or Prdx6+/+ mice were conducted.
DISCUSSION
Peroxiredoxins play a protective role in maintaining an appropriate redox balance under normal conditions and during oxidative stress [8, 33, 34]. Thus, different Prdxs are capable of detoxifying reactive oxygen species (ROS) and hydroperoxides in many cell compartments and tissues including the lung. In human COPD lung, the expression of Prdxs, including Prdx6 appears to be relatively constant, although Prdx6 has been detected in airway secretions/induced sputum supernatants in COPD [13]. In light of the presence of Prdxs in the lung, an organ that is the primary target for inhaled toxicants, we hypothesized that knockdown or ablation of Prdx6 leads to susceptibility to oxidative stress associated with CS, whereas overexpression of Prdx6 would protect against CS-induced lung inflammation. When these mice were exposed to acute CS for 3 days, Prdx6−/− mice surprisingly showed no significant increase in the inflammatory cell influx into the lungs (BAL fluid) when compared to WT mice. Acute CS exposure of the Prdx6+/+ mice caused a significant increase in the inflammatory cell influx similar to WT mice exposed to CS.
We and others have reported that CS induces inflammatory response in the lung by release of pro inflammatory mediators such as MCP-1 and KC, growth factors, and adhesion molecules that in turn can cause changes in lung damage and function [1, 24, 35]. In the present study, not only WT mice but also Prdx6+/+ mice exposed to acute CS resulted in a significant increase in the levels of MCP-1 and KC, leading to pro inflammatory response. Instead, air- and CS-exposed Prdx6−/− mice revealed reduced levels of these pro inflammatory cytokines (MCP-1 and KC), thereby probably protecting against acute CS-mediated inflammatory response. In general, WT and Prdx6+/+ mice presented significant proinflammatory response to acute CS exposure compared to the air- and CS-exposed Prdx6−/− mice.
Prdx6 overexpressing transgenic mice have reduced cellular H2O2 levels [22] and increased defense against hyperoxia-induced lung injury [23]. Prdx6 antisense RNA–treated lung cells have increased susceptibility to oxidative stress and cell death [36], and Prdx6−/− mice exhibit increased susceptibility to paraquat-induced oxidative stress [18, 20]. Earlier study using Prdx6−/− mice exposed to hyperoxia have also shown that the levels of gene expression of several antioxidant enzymes (AOEs), such as CAT, GPx, and MnSOD, were unaltered, suggesting that Prdx6 cannot be compensated by other genes [18]. Contrasting observation was observed in this study perhaps due to the complex mixture of agents present in CS, which includes a number of different oxidants, free radicals, reactive aldehydes, and quinones. These compounds can cause overwhelming response due to increased aldehyde-mediated stress and oxidant burden in the lungs [1]. It is likely that oxidant/carbonyl burden can induce AOEs in the lung particularly in response to CS. Thus the results/protection obtained with the Prdx6−/− mice could be related to the compensatory elevation of other Prdxs and AOEs in the lungs.
To study the compensatory role of other Prdxs, the levels of several Prdxs were assessed in the lungs of Prdx6−/− mice exposed to CS. The expression levels of the other 2-Cys peroxiredoxins (Prdx1 to Prdx5) revealed virtually no change except for Prdx4, which was elevated. Also the basal level of Prdx5 was significantly higher in lung of air- and CS-exposed Prdx6−/−mice compared to WT and Prdx6+/+ mice (data not shown). It is therefore very likely that the increased protein levels of Prdx4 and Prdx5 in mouse lung can compensate other Prdxs and lead to enhanced antioxidant defense against acute CS-induced oxidative stress in the absence of Prdx6. These results are in line with another study where Prdx6−/− mice in a hepatic ischemia/reperfusion injury model revealed increased mitochondrial oxidative stress, which led to increased Prdx3 expression in mitochondria [37]. Interestingly, Prdx5 is present in high concentrations both in human lung [6] and in airway secretions [38, 39]. Expression of Prdx5 in human lung has been shown to increase during inflammation, and up-regulation of Prdx5 decrease DNA double-strand breaks and protein oxidation by cigarette smoke extract (CSE) and hydrogen peroxide [38, 39]. Overall, our results on increased Prdx4 and Prdx5 in lungs of Prdx6−/− mice suggest some of the Prdx isoforms play a compensatory response to acute CS exposure.
GSH and antioxidant enzymes in lung structural cells play a pivotal role against lung inflammatory reaction both intracellularly and extracellularly by offering significant protection against oxidants [1, 2]. CS-mediated decreases in GSH were observed in lungs of WT mice but the total GSH levels were constitutively increased in Prdx6−/− mice exposed to CS. This finding is in parallel with a study showing ethanol-mediated oxidative stress and liver damage in Prdx6−/− mice [40].
It is well known that nuclear factor (erythroid-derived 2)-like 2 (Nrf2) plays a protective role in oxidative stress–induced tissue injury via antioxidant response element (ARE)-mediated activation of antioxidant and phase II detoxifying enzymes, including glutamate cysteine ligase (GCL), the rate limiting enzyme in the GSH synthesis [41, 42]. Increased levels of intracellular oxidants have been shown to lead to the induction of other AOEs, such as SOD, GPx, or catalase, that can provide protection against oxidative damage in cells [1, 2]. A significant increase in the abundance of AOEs was also observed in the Prdx6−/− mice exposed to CS compared to the air-exposed Prdx6−/− mice. It is likely that oxidant burden induced by CS led to the activation of antioxidant defense thus playing a compensatory role in controlling the CS-mediated lung inflammation in Prdx6−/− mice. In contrast to Prdx6−/−, Prdx6+/+ mice had not been shown to be protective in several earlier studies [34, 40]. One explanation in those and the present studies is the possible modification of Prdx6 (hereby CS-induced 4-HNE aldehyde), which in turn causes loss of its activity [34, 40]. Overall, our studies revealed that alternative or compensatory regulatory mechanisms including GSH and several AOEs may play an important role in Prdx6−/− mice against acute CS-induced oxidative stress and lung inflammation.
Sulfiredoxin 1 (Srx1) catalyzes the reduction of sulfinic forms of Prdxs to sulfenic acid in presence of Mg2+, ATP, and thiol as electron donors, thus restoring the functional cysteine residues and activity of Prdxs [43–46]. An oxidative stress–inducible gene, Srx1 is regulated by Nrf2, which plays a protective role in antioxidant defense against acute CS-induced oxidative stress in mouse lung [42, 47, 48]. In the present study, CS-induced elevation of Srx1 expression and localization in bronchial epithelium and alveolar septum of Prdx6−/− mice exposed to CS suggests that Srx1 can be one of the key enzymes that contribute to protection against CS-mediated oxidative stress in Prdx6−/− mice. Prdx6 deficiency may lead to stabilization of Nrf2 by inducing Srx1 and reversing thiol modifications in Keap1 and Nrf2 proteins. Our earlier study has shown that Nrf2 levels are decreased in response to CS exposure [32], whereas Prdx6+/+ mice showed lower levels of AOEs, which is probably related to Nrf2 reduction in response to CS. Nrf2 plays a critical buffering role in altering the levels of Srx1, AOEs, and other phase II enzymes, which is in full agreement with the results obtained with the Prdx6−/− and Prdx6+/+ mice. Furthermore, the levels of AOEs, along with some isoforms of Prdxs (Prdx4 and Prdx5) and Srx1, are elevated and possibly induced coordinately to regulate acute CS-mediated lung inflammatory response in Prdx6−/− mice.
The compensatory response observed in acute CS-exposed Prdx6−/− mice during early time point was lost in chronic CS exposure, which was associated with increased inflammatory response and lung injury in these mice. Chronic exposure to CS in Prdx6−/− mice shows significant changes in the levels of pro inflammatory cytokines, increased alveolar destruction and airspace enlargement, increase in lung compliance, and decrease in lung resistance. Several earlier reports state that CS inhalation leads to oxidative stress, which further influences the pulmonary function as well [1]. As expected, chronic CS exposure caused a significant increase in lung compliance and decrease in lung resistance in CS-exposed WT mice compared to air-exposed WT mice. This is in support of previous studies showing alteration in lung mechanical properties in mice after chronic CS exposure [49]. We also found an increase in lung compliance in response to chronic CS exposure, which was consistent with airspace enlargement in mice.
In summary, these data show that WT, Prdx6−/−, and Prdx6+/+ mice display variable levels of susceptibility to acute and chronic CS exposures. CS-exposed WT mice, but not the Prdx6−/− mice, showed increased inflammatory cell influx into BAL fluid. Instead, Prdx6+/+ mice exposed to CS revealed increased inflammatory cell influx into the BAL fluid compared to Prdx6−/− mice. WT and Prdx6+/+ mice also had significant increase in pro inflammatory cytokines, MCP-1 and KC, in response to acute CS exposure, a response that could not be seen in the Prdx6−/− mice. The levels of total GSH, and AOEs CAT, Cu/ZnSOD, and MnSOD, were significantly increased only in the lungs of CS-exposed Prdx6−/−compared to WT mice, whereas GSH levels and Cu/ZnSOD were declined in Prdx6+/+ mice exposed to CS when compared to Prdx6−/− mice. Overall, targeted disruption of Prdx6 does not lead to increased lung inflammatory response but instead is associated with increased expression of AOEs and Srx1, suggesting a critical role of Prdx6 in maintenance of cellular redox balance during acute CS-mediated oxidative stress and inflammatory response in the lung, whereas this protection is lost in response to chronic CS exposure leading to emphysema. Results from this study provide a better insight in understanding the role of Prdx6 in CS-mediated chronic inflammatory diseases.
Acknowledgments
This study was supported by NIH–National Heart, Lung, and Blood Institute grant R01-HL085613, and National Institute of Environmental Health Sciences Center grant ES-01247. V.L.K. was supported by the funding of the Helsinki University Hospital (EVO) and by the Finnish Antituberculosis Association Foundation and Yrjö Jahnsson Foundation.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28:219–242. doi: 10.1183/09031936.06.00053805. [DOI] [PubMed] [Google Scholar]
- 2.Rahman I, MacNee W. Lung glutathione and oxidative stress: implications in cigarette smoke-induced airway disease. Am J Physiol. 1999;277:L1067–L1088. doi: 10.1152/ajplung.1999.277.6.L1067. [DOI] [PubMed] [Google Scholar]
- 3.Rahman I, van Schadewijk AA, Crowther AJ, Hiemstra PS, Stolk J, MacNee W, De Boer WI. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166:490–495. doi: 10.1164/rccm.2110101. [DOI] [PubMed] [Google Scholar]
- 4.Woo HA, Chae HZ, Hwang SC, Yang KS, Kang SW, Kim K, Rhee SG. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 5.Kang SW, Baines IC, Rhee SG. Characterization of a mammalian peroxiredoxin that contains one conserved cysteine. J Biol Chem. 1998;273:6303–6311. doi: 10.1074/jbc.273.11.6303. [DOI] [PubMed] [Google Scholar]
- 6.Kinnula VL, Lehtonen S, Kaarteenaho-Wiik R, Lakari E, Paakko P, Kang SW, Rhee SG, Soini Y. Cell specific expression of peroxiredoxins in human lung and pulmonary sarcoidosis. Thorax. 2002;57:157–164. doi: 10.1136/thorax.57.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehtonen ST, Markkanen PM, Peltoniemi M, Kang SW, Kinnula VL. Variable overoxidation of peroxiredoxins in human lung cells in severe oxidative stress. Am J Physiol Lung Cell Mol Physiol. 2005;288:L997–L1001. doi: 10.1152/ajplung.00432.2004. [DOI] [PubMed] [Google Scholar]
- 8.Rhee SG, Kang SW, Chang TS, Jeong W, Kim K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life. 2001;52:35–41. doi: 10.1080/15216540252774748. [DOI] [PubMed] [Google Scholar]
- 9.Seo MS, Kang SW, Kim K, Baines IC, Lee TH, Rhee SG. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J Biol Chem. 2000;275:20346–20354. doi: 10.1074/jbc.M001943200. [DOI] [PubMed] [Google Scholar]
- 10.Fisher AB, Dodia C, Manevich Y, Chen JW, Feinstein SI. Phospholipid hydroperoxides are substrates for non-selenium glutathione peroxidase. J Biol Chem. 1999;274:21326–21334. doi: 10.1074/jbc.274.30.21326. [DOI] [PubMed] [Google Scholar]
- 11.Chen JW, Dodia C, Feinstein SI, Jain MK, Fisher AB. 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J Biol Chem. 2000;275:28421–28427. doi: 10.1074/jbc.M005073200. [DOI] [PubMed] [Google Scholar]
- 12.Manevich Y, Reddy KS, Shuvaeva T, Feinstein SI, Fisher AB. Structure and phospholipase function of peroxiredoxin 6: identification of the catalytic triad and its role in phospholipid substrate binding. J Lipid Res. 2007;48:2306–2318. doi: 10.1194/jlr.M700299-JLR200. [DOI] [PubMed] [Google Scholar]
- 13.Lehtonen ST, Ohlmeier S, Kaarteenaho-Wiik R, Harju T, Paakko P, Soini Y, Kinnula VL. Does the oxidative stress in chronic obstructive pulmonary disease cause thioredoxin/peroxiredoxin oxidation? Antioxid Redox Signal. 2008;10:813–819. doi: 10.1089/ars.2007.1952. [DOI] [PubMed] [Google Scholar]
- 14.Mo Y, Feinstein SI, Manevich Y, Zhang Q, Lu L, Ho YS, Fisher AB. 1-Cys peroxiredoxin knock-out mice express mRNA but not protein for a highly related intronless gene. FEBS Lett. 2003;555:192–198. doi: 10.1016/s0014-5793(03)01199-2. [DOI] [PubMed] [Google Scholar]
- 15.Kim TS, Dodia C, Chen X, Hennigan BB, Jain M, Feinstein SI, Fisher AB. Cloning and expression of rat lung acidic Ca(2+)-independent PLA2 and its organ distribution. Am J Physiol. 1998;274:L750–L761. doi: 10.1152/ajplung.1998.274.5.L750. [DOI] [PubMed] [Google Scholar]
- 16.Lee SB, Ho JN, Yoon SH, Kang GY, Hwang SG, Um HD. Peroxiredoxin 6 promotes lung cancer cell invasion by inducing urokinase-type plasminogen activator via p38 kinase, phosphoinositide 3-kinase, and Akt. Mol Cells. 2009;28:583–588. doi: 10.1007/s10059-009-0152-6. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Feinstein SI, Fisher AB. Peroxiredoxin 6 as an antioxidant enzyme: protection of lung alveolar epithelial type II cells from H2O2-induced oxidative stress. J Cell Biochem. 2008;104:1274–1285. doi: 10.1002/jcb.21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Phelan SA, Forsman-Semb K, Taylor EF, Petros C, Brown A, Lerner CP, Paigen B. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J Biol Chem. 2003;278:25179–25190. doi: 10.1074/jbc.M302706200. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Feinstein SI, Manevich Y, Ho YS, Fisher AB. Lung injury and mortality with hyperoxia are increased in peroxiredoxin 6 gene-targeted mice. Free Radic Biol Med. 2004;37:1736–1743. doi: 10.1016/j.freeradbiomed.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Feinstein SI, Manevich Y, Ho YS, Fisher AB. Peroxiredoxin 6 gene-targeted mice show increased lung injury with paraquat-induced oxidative stress. Antioxid Redox Signal. 2006;8:229–237. doi: 10.1089/ars.2006.8.229. [DOI] [PubMed] [Google Scholar]
- 21.Fisher AB, Dodia C, Yu K, Manevich Y, Feinstein SI. Lung phospholipid metabolism in transgenic mice overexpressing peroxiredoxin 6. Biochim Biophys Acta. 2006;1761:785–792. doi: 10.1016/j.bbalip.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 22.Phelan SA, Wang X, Wallbrandt P, Forsman-Semb K, Paigen B. Overexpression of Prdx6 reduces H2O2 but does not prevent diet-induced atherosclerosis in the aortic root. Free Radic Biol Med. 2003;35:1110–1120. doi: 10.1016/s0891-5849(03)00462-3. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Phelan SA, Manevich Y, Feinstein SI, Fisher AB. Transgenic mice overexpressing peroxiredoxin 6 show increased resistance to lung injury in hyperoxia. Am J Respir Cell Mol Biol. 2006;34:481–486. doi: 10.1165/rcmb.2005-0333OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao H, Edirisinghe I, Rajendrasozhan S, Yang SR, Caito S, Adenuga D, Rahman I. Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1174–L1186. doi: 10.1152/ajplung.00439.2007. [DOI] [PubMed] [Google Scholar]
- 25.Teague SV, Pinkerton KE, Goldsmith M, Gebremichael A, Chang S, Jenkins RA, Moneyhun JH. Sidestream cigarette smoke generation and exposure system for environmental tobacco smoke studies. Inhal Toxicol. 1994;6:79–93. [Google Scholar]
- 26.Witschi H, Oreffo VI, Pinkerton KE. Six-month exposure of strain A/J mice to cigarette sidestream smoke: cell kinetics and lung tumor data. Fundam Appl Toxicol. 1995;26:32–40. doi: 10.1006/faat.1995.1072. [DOI] [PubMed] [Google Scholar]
- 27.Foronjy RF, Mercer BA, Maxfield MW, Powell CA, D’Armiento J, Okada Y. Structural emphysema does not correlate with lung compliance: lessons from the mouse smoking model. Exp Lung Res. 2005;31:547–562. doi: 10.1080/019021490951522. [DOI] [PubMed] [Google Scholar]
- 28.Imai K, Mercer BA, Schulman LL, Sonett JR, D’Armiento JM. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J. 2005;25:250–258. doi: 10.1183/09031936.05.00023704. [DOI] [PubMed] [Google Scholar]
- 29.Kawakami M, Paul JL, Thurlbeck WM. The effect of age on lung structure in male BALB/cNNia inbred mice. Am J Anat. 1984;70:1–21. doi: 10.1002/aja.1001700102. [DOI] [PubMed] [Google Scholar]
- 30.Parameswaran H, Majumdar A, Ito S, Alencar AM, Suki B. Quantitative characterization of airspace enlargement in emphysema. J Appl Physiol. 2006;100:186–193. doi: 10.1152/japplphysiol.00424.2005. [DOI] [PubMed] [Google Scholar]
- 31.Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2006;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 32.Kode A, Rajendrasozhan S, Caito S, Yang SR, Megson IL, Rahman I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;294:L478–L488. doi: 10.1152/ajplung.00361.2007. [DOI] [PubMed] [Google Scholar]
- 33.Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 34.Roede JR, Carbone DL, Doorn JA, Kirichenko OV, Reigan P, Petersen DR. In Vitro and in Silico Characterization of Peroxiredoxin 6 Modifled by 4-Hydroxynonenal and 4-Oxononenal. Chem Res Toxicol. 2008;21:2289–2299. doi: 10.1021/tx800244u. [DOI] [PubMed] [Google Scholar]
- 35.Foronjy RF, Mirochnitchenko O, Propokenko O, Lemaitre V, Jia Y, Inouye M, Okada Y, D’Armiento JM. Superoxide dismutase expression attenuates cigarette smoke- or elastase-generated emphysema in mice. Am J Respir Crit Care Med. 2006;173:623–631. doi: 10.1164/rccm.200506-850OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pak JH, Manevich Y, Kim HS, Feinstein SI, Fisher AB. An antisense oligonucleotide to 1-cys peroxiredoxin causes lipid peroxidation and apoptosis in lung epithelial cells. J Biol Chem. 2002;277:49927–49934. doi: 10.1074/jbc.M204222200. [DOI] [PubMed] [Google Scholar]
- 37.Eismann T, Huber N, Shin T, Kuboki S, Galloway E, Wyder M, Edwards MJ, Greis KD, Shertzer HG, Fisher AB, Lentsch AB. Peroxiredoxin-6 protects against mitochondrial dysfunction and liver injury during ischemia-reperfusion in mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G266–G274. doi: 10.1152/ajpgi.90583.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Avila PC, Kropotov AV, Krutilina R, Krasnodembskay A, Tomilin NV, Serikov VB. Peroxiredoxin V contributes to antioxidant defense of lung epithelial cells. Lung. 2008;186:103–114. doi: 10.1007/s00408-007-9066-2. [DOI] [PubMed] [Google Scholar]
- 39.Serikov VB, Leutenegger C, Krutilina R, Kropotov A, Pleskach N, Suh JH, Tomilin NV. Cigarette smoke extract inhibits expression of peroxiredoxin V and increases airway epithelial permeability. Inhal Toxicol. 2006;18:79–92. doi: 10.1080/08958370500282506. [DOI] [PubMed] [Google Scholar]
- 40.Roede JR, Orlicky DJ, Fisher AB, Petersen DR. Overexpression of peroxiredoxin 6 does not prevent ethanol-mediated oxidative stress and may play a role in hepatic lipid accumulation. J Pharmacol Exp Ther. 2009;330:79–88. doi: 10.1124/jpet.109.152983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 42.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 44.Chang TS, Cho CS, Park S, Yu S, Kang SW, Rhee SG. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem. 2004;279:41975–41984. doi: 10.1074/jbc.M407707200. [DOI] [PubMed] [Google Scholar]
- 45.Findlay VJ, Tapiero H, Townsend DM. Sulfiredoxin: a potential therapeutic agent? Biomed Pharmacother. 2005;59:374–379. doi: 10.1016/j.biopha.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rhee SG, Jeong W, Chang TS, Woo HA. Sulfiredoxin, the cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: its discovery, mechanism of action, and biological significance. Kidney Int Suppl. 2007;106:S3–S8. doi: 10.1038/sj.ki.5002380. [DOI] [PubMed] [Google Scholar]
- 47.Bae SH, Woo HA, Sung SH, Lee HE, Lee SK, Kil IS, Rhee SG. Induction of sulfiredoxin via an Nrf2-dependent pathway and hyperoxidation of peroxiredoxin III in the lungs of mice exposed to hyperoxia. Antioxid Redox Signal. 2009;11:937–948. doi: 10.1089/ars.2008.2325. [DOI] [PubMed] [Google Scholar]
- 48.Singh A, Ling G, Suhasini AN, Zhang P, Yamamoto M, Navas-Acien A, Cosgrove G, Tuder RM, Kensler TW, Watson WH, Biswal S. Nrf2-dependent sulfiredoxin-1 expression protects against cigarette smoke-induced oxidative stress in lungs. Free Radic Biol Med. 2009;46:376–386. doi: 10.1016/j.freeradbiomed.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.March TH, Wilder JA, Esparza DC, Cossey PY, Blair LF, Herrera LK, McDonald JD, Campen MJ, Mauderly JL, Seagrave J. Modulators of cigarette smoke-induced pulmonary emphysema in A/J mice. Toxicol Sci. 2006;92:545–559. doi: 10.1093/toxsci/kfl016. [DOI] [PubMed] [Google Scholar]