

Abstract
Studies have shown that thalidomide exerts modest activity as a single agent in the therapy of acute myeloid leukemia (AML). The present investigation was conducted to test the hypothesis that the cytotoxic effect of thalidomide is enhanced when properly combined with other chemotherapeutic agents. The human AML cell line KG-1a was used in this study. Cells were cultured for 48 h in the presence or absence of thalidomide, arsenic trioxide and a combination of the two substances. Results obtained indicate that thalidomide at concentrations of 1, 2 and 5 mg/l produced a dose-dependent cytotoxic effect and at 5 mg/ml resulted in late apoptosis in 49.39% of the total cell population (as compared to 5.35% in the control cells). When the cells were incubated with arsenic trioxide alone (4 μM), late apoptosis was detected in 16.97% of the total cell population. However, when cells were incubated with a combination of thalidomide (5 mg/l) and arsenic trioxide (4 μM), late apoptosis was noted to be 80.6% in the total cell population. This percentage of late apoptosis was statistically significant from that observed when cells were incubated with thalidomide alone. These findings clearly indicate that arsenic trioxide enhances the cytotoxic effects of thalidomide.
Keywords: thalidomide, arsenic trioxide, acute myeloid leukemia
Introduction
Acute myeloid leukemia (AML) is a heterogeneous malignant disease with diverse biological features. AML occurs in approximately 60% of patients, most of whom are older than 60 years (1). These elderly patients normally do not respond as well to the current conventional chemotherapy as their younger counterparts. This is due to the intrinsic resistant nature of their leukemic cells and/or poor tolerance to conventional chemotherapy regimens (2). Progress has been ongoing in the treatment outcome of AML, especially in elderly patients. However, outcome has been poor with only 25–30% of adult patients being cured (3–5). Although prognosis varies among AML subtypes, the majority of patients relapse following an initial complete response (CR) and ultimately succumb due to resistant disease. Patients with AML who experience a particularly short first CR and those who fail to achieve CR after two induction attempts are unlikely to respond to any currently available chemotherapeutic agents. Similarly, patients with high-risk myelodysplastic syndromes likely to progress to AML [refractory anemia with excess blasts (RAEB) or refractory anemia with excess blasts in transformation (RAEBT)] have an estimated survival of less than one year (6). Supportive therapy remains the standard care for this population since intensive chemotherapy regimens, such as those used in AML, were reported to produce high rates of treatment-related mortality with rare durable remissions (7,8).
Therefore, novel approaches and alternative therapeutic strategies need to be explored. The role of angiogenesis in hematologic malignancies has been elucidated by several investigators; thus, inhibitors of angiogenesis are currently under investigation in these disorders. Thalidomide has antiangiogenic and immunomodulatory properties. However, thalidomide has shown only modest activity as a single agent in the therapy of AML. In addition, no study in the literature identifies a dose or schedule that should be followed when using thalidomide. Therefore, more studies are required to design a strategy to enhance the antileukemic effect of thalidomide. Such an approach is beneficial if the proper combination with other chemotherapeutic agents, such as interleukin-2 (IL-2) and arsenic trioxide (As2O3), is used. The current study examined the effect of the combination of thalidomide and the suggested chemotherapeutic agents on the KG-1a (human AML with early phenotype) cell line. The present study aimed to test the hypothesis that combining thalidomide with the correct chemotherapeutic agent/agents may be more efficient in the therapy of AML.
Materials and methods
Human KG-1a acute myeloid leukemia cells
The KG-1a cells, which are an early phenotype of human AML (American Type Culture Collection, Manassas, VA, USA), were grown in complete growth medium (Iscove’s Modified Dulbeco’s Medium; American Type Culture Collection) supplemented with 20% fetal bovine serum (Sigma-Aldrich, UK) and 1% penicillin-streptomycin (Gibco Invitrogen Corporation, Carlsbad, CA, USA) at 37°C in a humidified 5% CO2 incubator.
Treatment of human KG-1a acute myeloid leukemia cells
The KG-1a cells were cultured for 48 h in 12-well tissue culture plates, each containing complete growth medium at a concentration of 2×106 cells/ml. Each well contained a total volume of 2 ml. Thalidomide (Tocris Bioscience, Ellisville, MO, USA) was added at concentrations of 5 mg/l, either alone or in combination with other chemotherapeutic agents. IL-2 (Proleukin®, Aldesleuken for injection) (Chiron Therapeutics, Emeryville, CA, USA) was added at a concentration of 200 IU/ml, either alone or in combination with thalidomide and As2O3. As2O3 (Sigma-Aldrich, Inc., St. Louis, MO, USA) was added at two concentrations of 2 and 4 μM with and without 100 μM of ascorbic acid (AA) in the first flow cytometry study, and at 4 μM in the remaining studies; either alone or combined with thalidomide and IL-2. A control culture containing neither thalidomide nor IL-2 nor As2O3 was established in conditions that were otherwise identical. Duplicate control and treated cultures were established and incubated for 48 h at 37°C in a humidified 5% CO2 incubator. The incubation time was selected to allow adequate time for apoptosis and necrosis to occur in the KG-1a human myeloid leukemia cells [modified from Lu and Hassan (9)].
Detection of apoptosis and necrosis using flow cytometry
The detection of apoptosis and necrosis by Annexin V-FITC assay using flow cytometry was performed as described in the Annexin V-FITC apoptosis detection kit manual (BioVision, Inc., Mountain View, CA, USA). Staurosporine (Sigma-Aldrich, Inc.) was used as a positive control at a concentration of 10 μM and was incubated for only 24 h with the leukemia cells. Briefly, following incubation for 48 h, the KG-1a cells were harvested by centrifugation at 1,300 rpm for 5 min. Cells were resuspended in 500 μl of 1X binding buffer. Annexin V-FITC (5 μl) and propidium iodide (PI) (5 μl) were added to the tubes (except the negative control which contained no staining nor treatment where only Annexin V-FITC was added for the Annexin V-FITC controls and only PI was added for the PI control). Tubes were incubated at room temperature for 5 min in the dark. Samples were then checked by the FACSCalibur Flow Cytometer (BD-Biosciences, San Jose, CA, USA) and apoptotic and necrotic cells were counted according to their staining with Annexin V or PI, respectively [modified from Yang et al (10)].
Statistical analysis
Results were subjected to one-way ANOVA. Statistical significant differences between means was set at p<0.05.
Results
First flow cytometry study
This study was mainly designed to determine whether AA enhances As2O3-induced cytotoxicity in the KG-1a human AML cell line. When the KG-1a cells were incubated with the chemotherapeutic agents for 48 h, results obtained indicated that 2 μM of As2O3 alone resulted in early and late apoptosis, and necrosis (14.5%, 6.88% and 2.68%, respectively) of the total cell population (Fig. 1). Moreover, 2 μM of As2O3 in the presence of 100 μM of AA resulted in 15.69% early apoptosis, 7.12% late apoptosis and 1.16% necrosis of the total cell population (Fig. 2).
Figure 1.
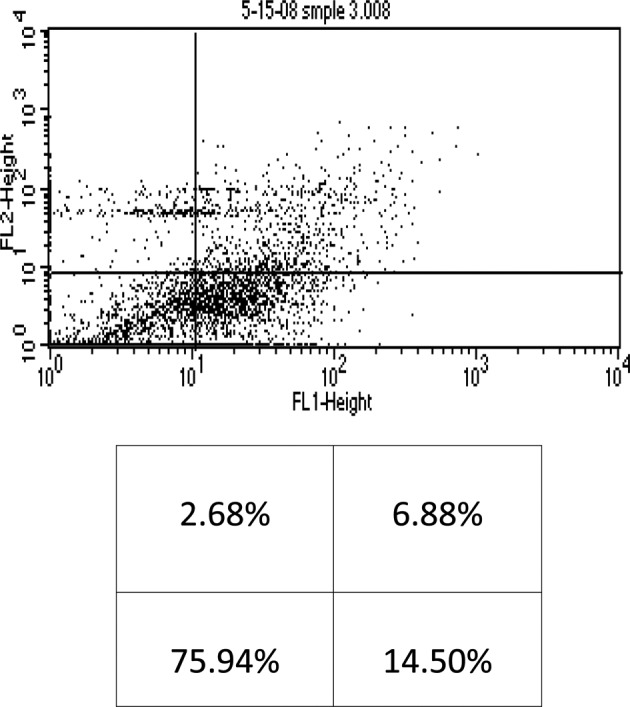
Flow cytometry analysis of KG-1a cells treated with arsenic trioxide (2 μM). The lower left quadrant shows live cells; the lower right, early apoptotic cells; the upper right, late apoptotic cells and the upper left quadrant shows necrotic cells.
Figure 2.
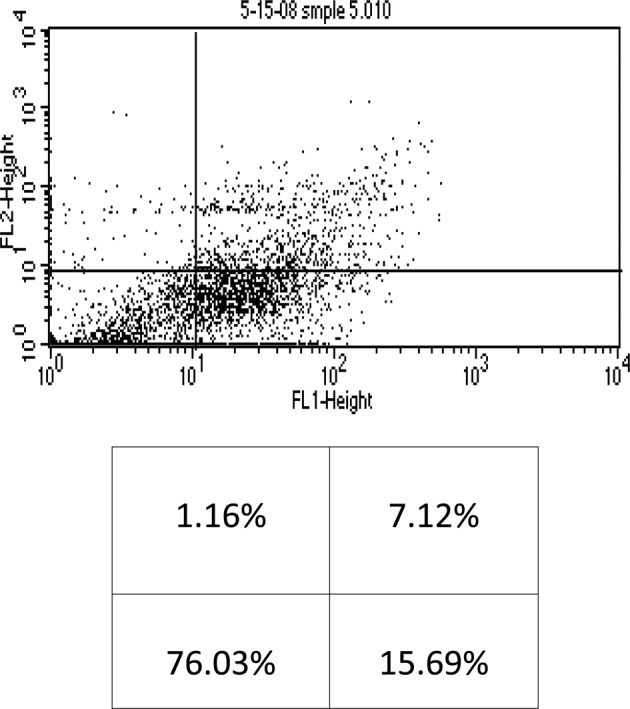
Flow cytometry analysis of KG-1a cells treated with arsenic trioxide (2 μM) + ascorbic acid (100 μM). The lower left quadrant shows live cells; the lower right, early apoptotic cells; the upper right, late apoptotic cells and the upper left quadrant shows necrotic cells.
When the cells were incubated with 4 μM of As2O3 alone, early and late apoptosis, as well as necrosis (20.87%, 8.31% and 1.26%, respectively) of the total cell population were observed (Fig. 3). Conversely, when 4 μM of As2O3 was used in the presence of 100 μM of AA, 21.84% early apoptosis, 8.14% late apoptosis and 0.77% necrosis of the total cell population were noted (Fig. 4). These findings indicate that AA did not significantly enhance the cytotoxicity of As2O3, and as a result our study utilized the higher dose of As2O3 (4 μM) without the addition of AA.
Figure 3.
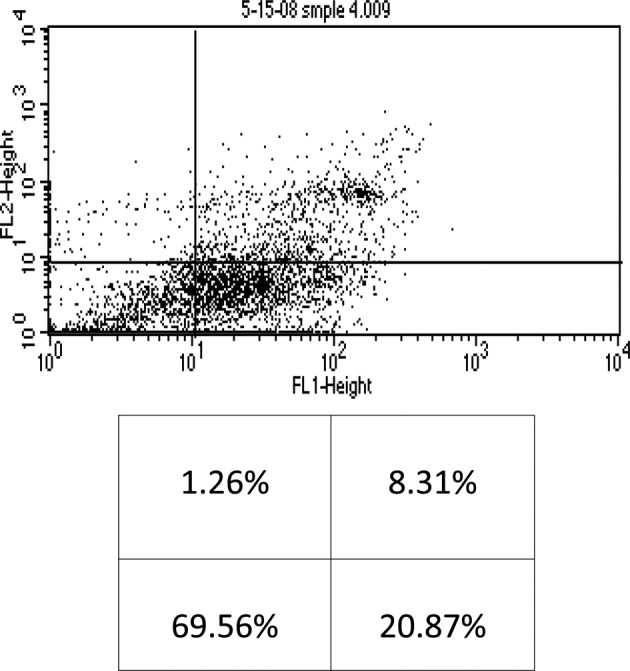
Flow cytometry of KG-1a cells treated with arsenic trioxide (4 μM). The lower left quadrant shows live cells; the lower right, early apoptotic cells; the upper right, late apoptotic cells and the upper left quadrant shows necrotic cells.
Figure 4.
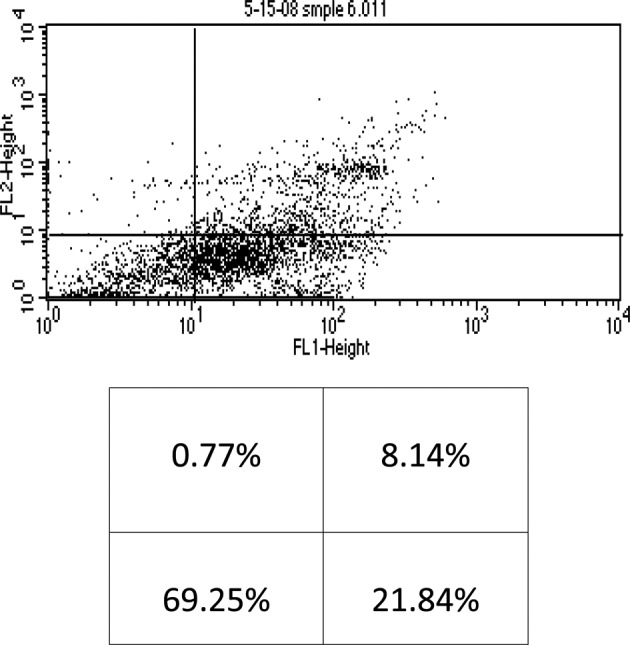
Flow cytometry of KG-1a cells treated with arsenic trioxide (4 μM) + ascorbic acid (100 μM). The lower left quadrant shows live cells; the lower right, early apoptotic cells; the upper right, late apoptotic cells and the upper left quadrant shows necrotic cells.
Cytotoxic effects of thalidomide, arsenic trioxide and interleukin-2 alone or in combination in KG-1a leukemia cells
Subsequent flow cytometry studies were conducted to test the cytotoxicity of chemotherapeutic agents both individually and in combination in KG-1a human leukemia cells in order to evaluate whether thalidomide cytotoxicity was enhanced by either IL-2 and/or As2O3.
Control KG-1a cells that were incubated for 48 h showed 0.775% necrosis, 9.89% early apoptosis and 5.345% late apoptosis in the total cell population (Figs. 5-7). When the cells were incubated with thalidomide only in a concentration of 5 mg/l, the percentages noted were: necrosis. 1.155%; early apoptosis, 7.92% and late apoptosis, 49.385% in the total cell population (Figs. 5-7). These results indicate that thalidomide exerted significant cytotoxicity in the KG-1a cells, and this cytotoxicity was mainly due to late apoptosis.
Figure 5.

Necrosis of KG-1a cells incubated with various chemotherapeutic agents. Bars represent mean ± SE. Cells were incubated for 48 h with thalidomide (Th) (5 mg/ml), arsenic trioxide (As) (4 μM) and interleukin-2 (IL-2) (200 IU/ml) alone or in combination.
Figure 7.

Late apoptosis of KG-1a cells incubated with various chemotherapeutic agents. Bars represent mean ± SE. Cells were incubated for 48 h with thalidomide (Th) (5 mg/ml), arsenic trioxide (As) (4 μM) and interleukin-2 (IL-2) (200 IU/ml) alone or in combination. *Significantly different from control (p<0.05); **significantly different from the control and thalidomide-treated cells (p<0.05).
When the cells were incubated with 4 μM of As2O3 the percentages of total cell population noted were: necrosis, 0.59%; early apoptosis, 10.095% and late apoptosis, 16.965% (Figs. 5-7). These results indicate that As2O3 alone had a modest cytotoxic effect since the percentages of cells in early and late apoptosis as well as necrosis were not statistically significantly different from values noted in the control cells. A similar conclusion was drawn with regard to cytotoxicity resulting from incubation of the KG-1a cells with 200 IU/ml of IL-2 alone. IL-2 alone resulted in 0.325% necrosis, 8.54% early apoptosis and 4.585% late apoptosis (Figs. 5-7). These values are not significantly different from those observed in the control KG-1a cells incubated under the same conditions.
However, when thalidomide and As2O3 were combined, the percentages observed were: necrosis, 0.87%; early apoptosis, 3.455% and late apoptosis, 80.6% (Figs. 5-7). These findings indicate that As2O3 enhanced the cytotoxicity induced by thalidomide since the percentage of cells in late apoptosis was 49.385% when the cells were incubated with thalidomide alone, but increased to 80.6% when thalidomide was combined with As2O3.
When IL-2 was combined with thalidomide no enhancement of cytotoxicity was noted as the percentages of total cell population showing necrosis, early apoptosis and late apoptosis were: 1.155, 7.92 and 49.385%, respectively, with thalidomide alone and 1.47, 6.58 and 35.385%, respectively, when a combination of thalidomide and IL-2 were used (Figs. 5-7).
IL-2 was also unable to enhance the cytotoxicity of the combination of thalidomide and As2O3 when the chemotherapeutic agents were used together since the percentages of total cell population showing necrosis, early and late apoptosis were 1.69, 5.655 and 70.135%, respectively. These values were not statistically significantly different from those observed with thalidomide and As2O3.
Discussion
This study was conducted to evaluate the efficacy of thalidomide in the management of AML, as well as to examine the possibility of increasing its cytotoxicity by combining it with other chemotherapeutic agents, such as IL-2 and As2O3. The variant subline KG-1a of the human acute myelogenous leukemia cell line KG-1 (11) was used as a test model.
The present study investigated the effect of AA on As2O3 cytotoxic activity. Glutathione (GSH) was shown to be an inhibitor of As2O3-induced cell death either by conjugating As2O3 or by sequestering reactive oxygen induced by As2O3. Consistent with this possibility, increasing GSH levels with N-acetylcysteine attenuated As2O3 cytotoxicity (12). Decreases in GSH levels were associated with AA metabolism. Clinically relevant doses of AA decreased GSH levels and potentiated the As2O3-mediated cell death of the four multiple myeloma cell lines. Similar results were obtained in freshly isolated human multiple myeloma cells (12). AA is widely heralded as an antioxidant (13). However, it was shown that AA acts as an oxidizing agent, particularly in the presence of compounds that increase the production of reactive oxygen species (14). The pro-oxidant effects of AA and potentiation of cell death induced by free radicals appear to involve the production of hydrogen peroxide (H2O2) (14,15). However, AA alone had no effect on cell viability suggesting that AA does not produce a sufficient level of H2O2 to initiate oxidative damage. Instead, AA treatment increases basal levels of cellular H2O2 (12). Grad et al showed that clinically-relevant doses of AA decreased GSH levels and potentiated the As2O3-mediated cell death of four types of multiple myeloma cell lines (12).
Accordingly, the likelihood that AA increases the cytotoxicity of As2O3 in the KG-1a human leukemia cell line was investigated. Two doses of As2O3, 2 and 4 μM, were tested in the presence or absence of AA in a concentration of 100 μM. The results obtained indicate that 2 μM of As2O3 alone resulted in 6.88% late apoptosis compared to 7.12% of late apoptosis in the presence of AA. In addition, late apoptosis induced by 4 μM of As2O3 alone was 8.31 compared to 8.14% in the presence of AA. These findings indicate that in our protocol, AA did not enhance the cytotoxicity of As2O3. Therefore, subsequent studies were conducted without adding AA to As2O3.
Thalidomide (Thalomid®), α-(N-phthalimido) glutarimide, is an immunomodulatory agent. Thalidomide has numerous characteristics that may contribute to its role as a potential agent in the treatment of malignant and immunological diseases. The drug inhibits angiogenesis by blocking basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) (16), modulates various cytokines (17), enhances cell-mediated immunity by directly co-stimulating T-cells (18) and alters adhesion molecule expression (19).
Thalidomide at 5 mg/l (19 μM) resulted in 49.385% late apoptosis compared to 5.345% of the control. These results were consistent with Du et al (20), who stated that thalidomide inhibited tumor growth in a concentration-dependent manner in MCF-7 and HL-60 cell lines. Moreover, 1C50S (inhibitory concentration of 50%) for the cell lines were 18.36±2.34 and 22.14±2.15 μM, respectively (20).
Efforts were made in our study to increase the cytotoxicity of thalidomide by combining it with other chemotherapeutic agents. The first agent used was IL-2. IL-2 (Proleukin, Aldesleuken for injection), currently being used to treat adults with metastatic melanoma and renal cell carcinoma, was also examined in this study to see whether it was able to enhance thalidomide cytotoxicity. IL-2 alone resulted in 4.59% late apoptosis compared to 5.35% late apoptosis in the control. The combination of thalidomide and IL-2 resulted in 35.39% late apoptosis compared to 49.39% late apoptosis for thalidomide alone. Thus, it was shown that IL-2 neither enhanced thalidomide toxicity nor was cytotoxic itself when used as a single therapeutic modality.
The reason that IL-2 was not cytotoxic to the leukemic cell line in this setting may be due to an unfavorable microenvironment since this was an in vitro study. Other studies investigated the development of an approach aimed at preventing relapse after autologous hemopoietic cell transplantation (AHCT). Post-transplant relapse is due to minimal residual disease in the body of the patient that persists after the conditioning regimen and/or presence of leukemic cells in the auto graft (21). Following an allogeneic transplant, the rate of leukemic relapse is lower for two reasons: the graft is free of leukemic cells and, more importantly, the graft vs. leukemia (GVL) effect associated with the transfer of donor T-and natural killer-cells is able to immunologically eradicate host leukemic cells (22). Therefore, efforts have been made to lower leukemic relapse after AHCT by either purging the leukemic cells in the auto graft (23) or inducing an autologous GVL effect by immunotherapy (24). Since the results of the two approaches were not always satisfactory, other approaches which actively eradicated the residual disease from the patient’s body, as opposed to those that only eliminated the contaminating leukemic cells in the graft, were adopted to ensure long-term disease-free survival (25). Reported immunotherapeutic approaches are diverse but the majority include the use of in vivo IL-2 with or without adoptively transferred lymphokine-activated killer (LAK) cells. However, this approach is limited by the fact that the doses of IL-2 required to maintain LAK activity in vivo cause undesirable side effects (26). Alternatively, several other cytokines including IL-1 (27), IL-4 (28), IL-7 (29), IL-12 (29) and granulocyte-macrophage colony-stimulating factor (GM-CSF) (30) have shown to be able to induce LAK activity themselves or in combination with IL-2. Rojas et al (31) demonstrated that immunotherapy with IL-2 + GM-CSF after total body irradiation results in a net improvement in survival in BALB/C mice injected with LSTRA leukemic cells (31). Other investigators reported the in vitro and in vivo generation of LAK activity by the synergistic effects of IL-2 and GM-CSF (30,32). However, the ability of this approach to cure leukemia or reduce the post-transplant relapse rate has yet to be conclusively demonstrated. Thus, IL-2 would be more effective if combined with a cytokine such as GM-CSF.
As2O3 is a chemotherapeutic agent used in the current study to enhance the cytotoxicity of thalidomide. As2O3 (Trisenox®) is currently used in the treatment of the first relapse of acute promyelocytic leukemia. The detailed mechanisms of As2O3 cytotoxicity are not completely known, but various preclinical studies have provided insight into the processes involved. The mechanisms include cellular differentiation, induction of apoptosis, and degradation of specific acute promyelocytic leukemia (APL) transcripts, antiproliferation, and inhibition of angiogenesis (33). Numerous studies examining the activity of arsenic utilized the prototype APL cell line NB4, which carries the t(15;17) translocation involving the RAR-α and PML genes (34). This activity generates a PML/RAR-α fusion protein between RAR-α, a nuclear receptor for retinoic acid and PML, a growth suppressor localized on nuclear-matrix-associated bodies. In APL studies, As2O3 induced a differential effect that was shown to be dose-dependent in that it induced partial differentiation at low concentrations (0.1–0.5 mmol/l) and induced apoptosis at relatively high concentrations (0.5–2.0 mmol/l) (35). Apoptosis in APL patients was shown to be, in part, secondary to the down-regulation of bcl-2 gene expression at the protein and mRNA levels as well as through modulation of PML-RAR-α and PML (36,37). The down-regulation of bcl-2 protein is independent of PML and PML/RAR-α expression (38). Davison et al showed that the apoptotic effect of As2O3 in APL is partially dependent on JNK activation (39). The antileukemic effects of all-trans retinoic acid and As2O3 target RAR-α and PML, respectively, while both induce the degradation of PML/RAR-α fusion proteins in NB4 cells (35,40). As2O3 induces the degradation of PML/RAR-α (as well as the wild-type PML) over a wide range of concentrations (0.5–2.0 mmol/l) (35). Moreover, As2O3 was shown to induce the degradation of the PML/RAR-α fusion protein in retinoic acid-resistant cells (35,41).
In related hematologic malignancies, such as multiple myeloma and lymphoma, preclinical studies of As2O3 have demonstrated similar apoptotic effects (15,36,42,43). Park et al demonstrated that As2O3-induced G1 and/or G2M phase arrest in myeloma cells (36). Simultaneous induction of cyclin-dependent kinase inhibitor, p21 was also noted. Liu et al showed that As2O3 induced apoptosis in resistant cell lines and fresh myeloma cells through p53-dependent cell cycle arrest and activation of extrinsic and intrinsic caspase pathways (43). Evidence exists of an immune mechanism with As2O3 in myeloma cells with elevated LAK and other immune cells (44). A similar immune mechanism has yet to be demonstrated in As2O3-treated APL.
The anti-leukemic effect of As2O3 may partially be related to the inhibition of angiogenesis due to interruption of the reciprocal stimulant loop between endothelial cells releasing cytokines which stimulate leukemic cells to release growth factors such as VEGF (through apoptosis of the two cell types) (45). Another mechanism of As2O3-induced apoptosis is through the activation of caspases. As2O3 activates these proteases, which play an important role in the degradation phase of apoptosis, in NB4 cell lines (43,46,47). As2O3 is able to lead to potential changes in the membrane and increased membrane permeability with the resultant degradation phase of apoptosis. Furthermore, the ability of As2O3 to induce apoptosis is dependent on the generation of reactive oxygen species (46,48). This suggests that the effect of As2O3 is potentiated through modulation of the glutathione redox system.
As2O3 was used in the current study to test its efficacy in the enhancement of the cytotoxicity of thalidomide in an acute myelogenous human leukemia cell line. It was noted that As2O3 alone resulted in 16.97% late apoptosis compared to 5.35% late apoptosis in the control. When As2O3 was used concurrently with thalidomide it resulted in 80.6% late apoptosis compared to 49.39% late apoptosis of thalidomide alone. The combination of thalidomide, As2O3 and IL-2 resulted in 70.14% of late apoptosis. Thus, in our study As2O3 enhancesd the cytotoxic effect of thalidomide in contrast to IL-2 which did not manifest any noticeable cytotoxicity. Further studies are warranted to prove or disprove the potential role of IL-2 to enhance the cytotoxicity of thalidomide.
In conclusion, this study showed that the cytotoxic effect of thalidomide is significantly enhanced when combined with As2O3 in the KG-1a human AML cell line. These findings point to the potential use of As2O3 in combination with thalidomide for a more efficient management of AML by increasing the rate of complete remission for AML patients and the least chance of relapse. Further studies are needed to confirm the current findings. However, the results obtained are promising since thalidomide effectiveness in the therapy of AML may be significantly enhanced by the concurrent use of As2O3. This may aid patients who suffer from this disease.
Figure 6.

Early apoptosis of KG-1a cells incubated with various chemotherapeutic agents. Bars represent mean ± SE. Cells were incubated for 48 h with thalidomide (Th) (5 mg/ml), arsenic trioxide (As) (4 μM) and interleukin-2 (IL-2) (200 IU/ml) alone or in combination.
Acknowledgements
This study was supported in part by NIH grant RR03020.
References
- 1.Godwin JE, Smith SE. Acute myeloid leukemia in the older patient. Crit Rev Oncol Hematol. 2003;48:S17–S26. doi: 10.1016/j.critrevonc.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Harousseuau JL. Acute myeloid leukemia in the elderly. Blood Rev. 1988;12:145–153. doi: 10.1016/s0268-960x(98)90012-1. [DOI] [PubMed] [Google Scholar]
- 3.Cortes JE, Kantarjian HM. Acute lymphocytic leukemia: A comprehensive review with emphasis on biology and therapy. Cancer. 1995;76:2393–2417. doi: 10.1002/1097-0142(19951215)76:12<2393::aid-cncr2820761203>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 4.Estey EH, Kantarjian H, Keating MJ. Therapy for acute myeloid leukemia. In: Hoffman R, Benz E Jr, Shattil S, Cohen H, Silberstein L, editors. Hematology: Basic Principles and Practice. 2nd edition. Churchill Livingstone; New York: 1994. pp. 1014–1028. [Google Scholar]
- 5.Thomas DA, Kantarjian H, Smith TL. Primary refractory and relapsed adult acute lymphoblastic leukemia: characteristics, treatment results, and prognosis with salvage therapy. Cancer. 1999;86:1216–1230. doi: 10.1002/(sici)1097-0142(19991001)86:7<1216::aid-cncr17>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 6.Beran M, Estey E, O’Brien S. Topotecan and cytarabine is an active combination regimen in myelodysplastic syndromes and chronic myelomonocytic leukemia. J Clin Oncol. 1999;17:2819–2830. doi: 10.1200/JCO.1999.17.9.2819. [DOI] [PubMed] [Google Scholar]
- 7.Estey EH, Kantarjian HM, O’Brien S. High remission rate, short remission duration in patients with refractory anemia with excess blasts (RAEB) in transformation (RAEB-t) given acute myelogenous leukemia (AML)-type chemotherapy in combination with granulocyte-CSF (G-CSF) Cytokines Mol Ther. 1995;1:21–28. [PubMed] [Google Scholar]
- 8.Thomas D. Pilot studies of thalidomide in acute myelogenous leukemia, myelodysplastic syndromes, and myeloproliferative disorders. Semin Oncol. 2000;37(Suppl 3):26–34. [Google Scholar]
- 9.Lu C, Hassan HT. Human stem cell factor-antibody (anti-SCF) enhances chemotherapy cytotoxicity in human CD34+ resistant myeloid leukemia cells. Leuk Res. 2006;30:296–302. doi: 10.1016/j.leukres.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 10.Yang H, Hoshino K, Sanchez-Gonzalez B, Kantarjian H, Garcia-Manero G. Antileukemia activity of the combination of 5-aza-2′-deoxycytidine with valproic acid. Leuk Res. 2005;29:739–748. doi: 10.1016/j.leukres.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 11.Koeffler HP, Billing R, Lusis AJ, Sparkes R, Golde DW. An undifferentiated variant derived from the human acute myelogenous leukemia cell line (KG-1) Blood. 1980;56:265–273. [PubMed] [Google Scholar]
- 12.Grad JM, Bahlis NJ, Reis I, Oshiro MM, Dalton WS, Boise LH. Ascorbic acid enhances arsenic trioxide-induced cytotoxicity in multiple myeloma cells. Blood. 2001;98:805–813. doi: 10.1182/blood.v98.3.805. [DOI] [PubMed] [Google Scholar]
- 13.Block G, Levine M. Vitamin C: a new look. Ann Intern Med. 1991;114:909–910. doi: 10.7326/0003-4819-114-10-909. [DOI] [PubMed] [Google Scholar]
- 14.Sakagami H, Satoh K. Modulating factors of radical intensity and cytotoxic activity of ascorbate [review] Anticancer Res. 1997;17:3513–3520. [PubMed] [Google Scholar]
- 15.Dai J, Weinberg RS, Waxman S, Jing Y. Malignant cells can be sensitized to undergo growth inhibition and apoptosis by arsenic trioxide through modulation of the glutathione redox system. Blood. 1999;93:268–277. [PubMed] [Google Scholar]
- 16.Kruse FF, Joussen AM, Rohrschneider K, Becker MD, Volcker HE. Thalidomide inhibits corneal angiogenesis induced by vascular endothelial growth factor. Graefes Arch Clin Exp Ophthalmol. 1998;236:461–466. doi: 10.1007/s004170050106. [DOI] [PubMed] [Google Scholar]
- 17.Corral LG, Haslett PAJ, Muller GW, et al. Differential cytokine modulation and T-cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J Immunol. 1999;163:380–386. [PubMed] [Google Scholar]
- 18.Haslett PAJ, Corral LG, Albert M, Kaplan G. Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J Exp Med. 1998;187:1885–1892. doi: 10.1084/jem.187.11.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geitz H, Handt S, Zwingenberger K. Thalidomide selectively modulates the density of cell surface molecules involved in the adhesion cascade. Immunopharmacology. 1996;31:213–221. doi: 10.1016/0162-3109(95)00050-x. [DOI] [PubMed] [Google Scholar]
- 20.Du GJ, Lin HH, Xu QT, Wang MW. Thalidomide inhibits growth of tumors through COX-2 degradation independent of antiangiogenesis. Vascul Pharmacol. 2005;43:112–119. doi: 10.1016/j.vph.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Heslop HE, Rooney CM, Brenner MK. Gene-marking and hemopoietic stem cell transplantation. Blood Rev. 1995;4:220–224. doi: 10.1016/s0268-960x(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 22.Barret AJ. Mechanisms of the graft-versus-leukemia reactivity. Bone Marrow Transplant. 1997;1:61–68. [Google Scholar]
- 23.Ball ED, Mills LE, Cornwell GG, Jr, Davis BH, Coughlin CT, Howell AL. Autologous bone marrow transplantation for acute myeloid leukemia using monoclonal antibody-purged bone marrow. Blood. 1990;75:1199–1206. [PubMed] [Google Scholar]
- 24.Sznol M, Parkinson DR. Interleukin-2 in therapy of hematologic malignancies. Blood. 1994;83:2020–2222. [PubMed] [Google Scholar]
- 25.Uckun FM, Kersey JH, Vallera DA, Ledbetter JA, Weisdorf D, Myers DE. Autologous bone marrow transplantation in high risk remission T-lineage acute lymphoblastic leukemia using immunotoxins plus 4-hydroperoxycyclophosphamide for marrow purging. Blood. 1990;76:1723–1733. [PubMed] [Google Scholar]
- 26.Kalland T, Belfrage H, Bhiladvala P, Hedlund G. Analysis of the murine lymphokine-activated killer (LAK) phenomenon: dissection of effectors and progenitors into NK-and T-like cells. J Immunol. 1987;38:3640–3645. [PubMed] [Google Scholar]
- 27.Ochoa AC, Gromo G, Alter BJ, Sondel PM, Bach FH. Long-term growth of lymphokine-activated killer (LAK)-cells: role of anti CD-3, beta-IL 1, interferon-gamma and -beta. J Immunol. 1987;138:2728–2733. [PubMed] [Google Scholar]
- 28.Peace DJ, Kern DE, Schultz KR, Greenberg PD, Cheever MA. IL-4-induced lymphokine-activated killer cells. Lytic activity is mediated by phenotypically distinct natural killer-like and T-cell-like large granular lymphocytes. J Immunol. 1988;140:3679–3685. [PubMed] [Google Scholar]
- 29.Naume B, Espevik T. Effects of IL-7 and IL-12 on highly enriched CD56+ natural killer cells: a comparative study. J Immunol. 1991;147:2208–2214. [PubMed] [Google Scholar]
- 30.Stewart-Akers AM, Cairns JS, Tweardy DJ, McCarthy SA. Effect of granulocyte macrophage-colony stimulating factor on lymphokine-activated killer cell induction. Blood. 1993;81:2672–2678. [PubMed] [Google Scholar]
- 31.Rojas R, Roman J, Herrera C, Alvarez MA, Ramirez R, Torres A. BALB/C mice injected with LSTRA leukemic cell line are cured by in vivo treatment with IL-2 + GM-CSF. Leuk Res. 2003;27:351–357. doi: 10.1016/s0145-2126(02)00180-7. [DOI] [PubMed] [Google Scholar]
- 32.Herrera C, Garcia-Perez MJ, Ramirez R, Martin C, Alvarez MA, Martinez F. Lymphokine-activated killer (LAK)-cell generation from peripheral blood stem cells by in vitro incubation with low dose interleukin-2 plus granulocyte macrophage-colony stimulating factor. Bone Marrow Transplant. 1997;19:545–551. doi: 10.1038/sj.bmt.1700698. [DOI] [PubMed] [Google Scholar]
- 33.Evens AM, Tallman MS, Gartenhaus RB. The potential of arsenic trioxide in the treatment of malignant disease: past, present, and future. Leuk Res. 2004;28:891–900. doi: 10.1016/j.leukres.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 34.Lanotte M, Martin-Thouvenin V, Najman S, Balerini P, Valensi F, Berger R. NB4, a maturation inducible cell line with t(15;17) marker isolated from a human acute promyelocytic leukemia (M3) Blood. 1991;77:1080–1086. [PubMed] [Google Scholar]
- 35.Chen GQ, Shi XG, Tang W, et al. Use of arsenic trioxide in the treatment of acute promyelocytic leukemia (APL): I. Arsenic trioxide exerts dose-dependent dual effects on APL cells. Blood. 1997;89:3345–3353. [PubMed] [Google Scholar]
- 36.Park WH, Seol JG, Kim ES, Hyun JM, Jung CW, Lee CC. Arsenic trioxide-mediated growth inhibition in MC/CAR myeloma cells via cell cycle arrest in association with induction of cyclin-dependent kinase inhibitor, p21, and apoptosis. Cancer Res. 2000;60:3065–3071. [PubMed] [Google Scholar]
- 37.Perkins C, Kim CN, Fang G, Bhalla KN. Arsenic induces apoptosis of multidrug-resistant human myeloid leukemia cells that express Bcr-Abl or overexpress MDR, MRP, Bcl-2, or Bcl-x(L) Blood. 2000;95:1014–1022. [PubMed] [Google Scholar]
- 38.Schor NF, Rudin CM, Hartman AR, Thompson CB, Tyurina YY, Kagan VE. Cell line dependence of Bcl-2-induced alteration of glutathione handling. Oncogene. 2000;19:472–476. doi: 10.1038/sj.onc.1203324. [DOI] [PubMed] [Google Scholar]
- 39.Davison K, Mann KK, Waxman S, Miller WH. JNK activation is a mediator of arsenic trioxide-induced apoptosis in acute promyelocytic leukemia cells. Blood. 2004;103:3496–3502. doi: 10.1182/blood-2003-05-1412. [DOI] [PubMed] [Google Scholar]
- 40.Shao W, Fanelli M, Ferrara FF, Riccioni R, Rosenauer A, Davison K. Arsenic trioxide as an inducer of apoptosis and loss of PML/RAR alpha protein in acute promyelocytic leukemia cells. J Natl Cancer Inst. 1998;90:124–133. doi: 10.1093/jnci/90.2.124. [DOI] [PubMed] [Google Scholar]
- 41.Kinjo K, Kizaki M, Muto A, Fukuchi Y, Umezawa A, Yamato K. Arsenic trioxide-induced apoptosis and differentiation in retinoic acid resistant acute promyelocytic leukemia model in hGM-CSF-producing transgenic SCID mice. Leukemia. 2000;14:431–438. doi: 10.1038/sj.leu.2401646. [DOI] [PubMed] [Google Scholar]
- 42.Rousselot P, Labaume S, Marolleau JP, Larghero J, Noguera MH, Brouet JC. Arsenic trioxide and Melarsoprol induce apoptosis in plasma cell lines and in plasma cells from myeloma patients. Cancer Res. 1999;59:1041–1048. [PubMed] [Google Scholar]
- 43.Liu Q, Hilsenbeck S, Gazitt Y. Arsenic trioxide-induced apoptosis in myeloma cells: p53-dependent G1 or G2/M cell cycle arrest, activation of caspase-8 or caspase-9, and synergy with APO2/TRAIL. Blood. 2003;101:4078–4087. doi: 10.1182/blood-2002-10-3231. [DOI] [PubMed] [Google Scholar]
- 44.Deaglio S, Canella D, Baj G, Arnulfo A, Waxman S, Malavasi F. Evidence of an immunologic mechanism behind the therapeutical effects of arsenic trioxide on myeloma cells. Leuk Res. 2001;25:227–235. doi: 10.1016/s0145-2126(00)00105-3. [DOI] [PubMed] [Google Scholar]
- 45.Roboz GJ, Dias S, Lam G, Lane WJ, Soignet SL, Warrell RP., Jr Arsenic trioxide induces dose-and time-dependent apoptosis of endothelium and may exert an antileukemic effect via inhibition of angiogenesis. Blood. 2000;96:1525–1530. [PubMed] [Google Scholar]
- 46.Gartenhaus RB, Prachand SN, Paniaqua M, Li Y, Gordon LI. Arsenic trioxide cytotoxicity in steroid and chemotherapy-resistant myeloma cell lines: enhancement of apoptosis by manipulation of cellular redox state. Clin Cancer Res. 2002;8:666–672. [PubMed] [Google Scholar]
- 47.Kitamura K, Minami Y, Yamamoto K, et al. Involvement of CD95-independent caspase 8 activation in arsenic trioxide-induced apoptosis. Leukemia. 2000;14:1743–1750. doi: 10.1038/sj.leu.2401900. [DOI] [PubMed] [Google Scholar]
- 48.Gupta S, Yel L, Kim D, Kim C, Chiplunkar S, Gollapudi S. Arsenic trioxide induces apoptosis in peripheral blood T lymphocytes subsets by inducing oxidative stress: a role of Bcl-2. Mol Cancer Ther. 2003;2:711–719. [PubMed] [Google Scholar]