

Abstract
Objective
We have previously reported a defect in neutrophil activation in children with polyarticular juvenile idiopathic arthritis (JIA). The current study was undertaken to determine whether gene expression abnormalities persist in JIA in remission and to use systems biology analysis to elucidate pathologic pathways in polyarticular JIA.
Methods
We performed gene expression profiling on neutrophils from children with polyarticular JIA. Children were grouped according to disease status. We studied 14 children with active disease who were taking medication, 8 children with clinical remission of disease who were taking medication (CRM status), and 6 children with clinical remission of disease who were not taking medication (CR status). We also studied 13 healthy children whose age ranges overlapped those of the patients.
Results
Neutrophil abnormalities persisted in children with polyarticular JIA even after disease remission was achieved. Children with active disease and those with CRM status showed no differences in expression of specific genes, although they could be separated on cluster analysis. A comparison of children with CR status and healthy control children revealed networks of pro- and antiinflammatory genes that suggested that remission is a state of homeostasis and balance rather than a return to normal immune function. Furthermore, gene overexpression in patients with CR status supports the hypothesis that neutrophils play a role in regulating adaptive immunity in this disease.
Conclusion
Neutrophil gene profiling in polyarticular JIA suggests important roles for neutrophils in disease pathogenesis. These findings suggest the presence of complex interactions between innate and adaptive immunity, that are not easily modeled in conventional, linear, reductionist systems.
Juvenile idiopathic arthritis (JIA) is a term used to denote a family of diseases of unknown etiology characterized by chronic inflammation of synovial membranes (1). Distinct phenotypes are recognized clinically, with specific immunogenetic markers associated with each of the phenotypes (2,3).
While the JIA subtypes have commonly been assumed to have an “autoimmune” origin, our growing understanding of biologic complexity makes any such simple, linear hypothesis of disease pathogenesis unlikely (4). We have hypothesized that the pathogenesis of a common JIA subtype, polyarticular disease, involves complex interactions between innate and adaptive immunity not readily subsumed under a simple “autoimmunity” model (5,6). In support of this notion, we have demonstrated the presence of a population of hyperreactive neutrophils in children with polyarticular-onset JIA (7). Given our growing knowledge of how neutrophils regulate adaptive immunity (8), it is plausible to hypothesize that these abnormal neutrophils have a significant effect on adaptive immune mechanisms and the disease course in polyarticular JIA.
The disease process in polyarticular JIA as seen in the normal clinical setting is not static. That is, children can be categorized based on their disease activity and response to therapy (i.e., active disease, inactive disease, remission of disease while taking medication, remission of disease while not taking medication), as Wallace and colleagues have shown (9). We have recently demonstrated that these clinically derived criteria for disease state have objective biologic identities, based on gene transcription profiling in peripheral blood mononuclear cells (PBMCs) (10). Thus, we have hypothesized that a practical way of gaining insight into the potential role of neutrophils in JIA pathogenesis is to study their function in specific disease states in conjunction with PBMCs. In the current study, we used a systems biology approach (gene transcription profiling and in silico modeling) to determine whether and how the neutrophil function may be altered in polyarticular JIA at different stages of the disease.
PATIENTS AND METHODS
Patient population and definition of disease states
We studied 14 children with active, polyarticular, rheumatoid factor–negative JIA as defined by the criteria of the International League of Associations for Rheumatology (11); all of these children were taking medication. We also studied 8 children who met the criteria for clinical remission of disease and who were taking medication (CRM status, further defined below). Finally, we studied 6 children who had clinical remission of disease and who were not taking medication (CR status, further defined below). All children except those with CR status were receiving oral or subcutaneous (SC) methotrexate, and 5 of these children were also receiving SC etanercept. The age range of the subjects was 3–18 years. Blood was obtained at the time of routine clinical monitoring using standard precautions, and topical anesthesia with 2.5% lidocaine/2.5% prilocaine cream was offered to all children prior to the procedure.
Disease states were defined according to the consensus criteria developed by Wallace and colleagues (12). Children with active disease had synovitis and/or fever, rash, lymphadenopathy, splenomegaly, uveitis, an elevated erythrocyte sedimentation rate (ESR) or C-reactive protein (CRP) level, or a physician’s global assessment score indicating active disease. Children with inactive disease (taking or not taking medication) had no evidence of synovitis and no fever, rash, lymphadenopathy, splenomegaly, or active uveitis, as well as a normal ESR and CRP level and a physician’s global assessment score indicating no active disease. Children with CRM status were those with inactive disease (disease in remission) who were taking medication and who had maintained that state for 6 continuous months. Children with CR status were those with inactive disease (disease in remission) who were not taking medication and who had maintained that state for 12 continuous months.
Healthy control subjects
Healthy control subjects consisted of 13 healthy children ages 3–15 years. These children were undergoing elective surgery for noninflammatory conditions (e.g., minor orthopedic procedures) or were being seen for routine health maintenance in the Oklahoma University Health Services Center Children’s Physicians’ general pediatrics clinic. Healthy children were excluded if they had experienced fever (38°C) in the 48 hours prior to phlebotomy. Topical anesthesia with 2.5% lidocaine/2.5% prilocaine cream was applied to the phlebotomy site for all children for at least 30 minutes before the procedure. Participation of all human subjects was reviewed and approved by the University of Oklahoma Health Sciences Center Institutional Review Board.
Specimens and specimen handling
Whole blood was drawn into 10-ml citrated CPT tubes (no. 362760; Becton Dickinson, Franklin Lakes, NJ). Blood was carried immediately to the laboratory, and specimen processing was started within 60 minutes of obtaining the blood. PBMCs were separated from granulocytes and red blood cells by subjecting the CPT tubes directly to density-gradient centrifugation. Granulocytes, which sediment with red blood cells in the CPT tubes, were collected and placed in TRIzol reagent (Invitrogen, Carlsbad, CA) after hypotonic lysis of the red blood cells. Cell lysates were stored at −80°C in TRIzol reagent until used for RNA isolation, always within 48 hours after preparation.
RNA isolation, labeling, hybridization, and scanning
Total RNA extractions from TRIzol reagent were carried out according to the manufacturer’s directions. For the Affymetrix arrays (Affymetrix, Santa Clara, CA), RNA was further purified by passage through RNeasy mini-columns (Qiagen, Valencia, CA) according to the manufacturer’s protocols for RNA clean-up. Final RNA preparations were suspended in RNase-free water. The RNAs were quantified spectrophotometrically. RNA integrity was assessed using capillary gel electrophoresis (Agilent 2100 Bioanalyzer; Agilent Technologies, Palo Alto, CA) to determine the ratio of 28S:18S ribosomal RNA in each sample. Complementary DNA (cDNA) synthesis, hybridization, and staining were performed as specified by Affymetrix using Affymetrix human U133 Plus 2.0 Arrays, an Affymetrix automated GeneChip 450 fluidics station, and an Affymetrix 3000 7G scanner.
Statistical analysis
For clarity of analysis, children taking medication were only compared with children taking medication (i.e., children with active disease were compared with children with CRM status), and children not taking medication (i.e., those who had achieved CR status) were compared with healthy controls. All Affymetrix array data preprocessing was performed with the R/Bioconductor Package, “Affy.” The raw Affymetrix perfect match probes were normalized by the robust multichip analysis method combined with median-polish (13). The marginal data distributions were adjusted through quantile normalization. The resulting normalized values were imported into JMP Genomics version 3.2 (SAS Institute, Cary, NC), where they were then logtransformed. Genes were filtered using the “Log Expression Variation Filter” to screen out genes that were not likely to be informative, based on the variance of each gene across the arrays. In this case, the filter was set to exclude genes that fell below the 50th percentile of gene variance.
We identified genes that were differentially expressed between the 2 classes by using a Student’s 2-sample t-test (14). We used the Student’s t-test to provide a false discovery rate (FDR) of 5% (15). The FDR is the proportion of the list of genes claimed to be differentially expressed that are false positives. Data were exported to Excel (Microsoft, Redmond, WA), where averages of the classes were used to calculate expression ratios. Genes that simultaneously were differentially expressed (<5% FDR), had a ratio of ≥2-fold, and had minimum normalized average intensity of >64 units in at least 1 group were retained for further analysis. Unsupervised hierarchical clustering was performed with Spotfire (TIBCO Software, Somerville, MA) using Ward’s minimum variance method (16). Differences between cluster groups were tested using a chi-square test. P values less than 0.05 were considered significant.
Real-time quantitative reverse transcription–polymerase chain reaction (RT-PCR) validation
Total RNA extractions from TRIzol reagent were carried out according to the manufacturer’s directions. RNA was further purified by passage through RNeasy mini-columns with DNase I (Qiagen) according to the manufacturer’s protocols. Final RNA preparations were suspended in RNase-free water. The RNAs were quantified spectrophotometrically. Primers were designed with a 60°C melting temperature and a length of 9–40 nucleotides to produce PCR products with lengths of 50–150 bp using Primer Express 2.0 software (Applied Biosystems, Foster City, CA). First-strand cDNA was generated from 1.8 μg of total RNA per sample using OmniScript Reverse Transcriptase according to the directions of the manufacturer (Qiagen). Complementary DNA was diluted 1:20 in water. PCR was run with a 4-μl cDNA template in 20-μl reactions in duplicate on an ABI SDS 7000 (Applied Biosystems) using ABI SYBR Green I Master Mix (Applied Biosystems) and gene-specific primers at a concentration of 0.2 μM each. The temperature profile consisted of an initial step of 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute, and then a final melting curve analysis with a ramp from 60°C to 95°C over 20 minutes.
Gene-specific amplification was confirmed by a single peak using the ABI Dissociation Curve software (Applied Biosystems). Average threshold cycle (Ct) values for GAPDH (run in parallel reactions to the genes of interest) were used to normalize average Ct values of the gene of interest. These values were used to calculate averages for each group (healthy control or patient subsets), and the relative ΔCt was used to calculate fold-change values between the groups.
Physiologic pathway modeling
Pathways of potential interactions between gene products were generated by placing only the genes that were statistically significantly differentially expressed between groups into Ingenuity Pathways Analysis (Ingenuity Systems, Redwood City, CA). Each Affymetrix gene identifier was mapped to its corresponding gene object in the Ingenuity knowledge base. These “focus” genes were overlaid onto a global molecular network developed from information contained in the Ingenuity knowledge base. Networks of these focus genes were then algorithmically generated based on their “connectivity” derived from known interactions between products of these genes.
RESULTS
Validation of array results
We chose 8 genes to validate each of the array comparisons. Results of the RT-PCR experiments are shown in Table 1. In all cases, the PCR findings corroborated results from the arrays.
Table 1.
PCR confirmation of differentially expressed genes*
| GenBank accession no. | Gene symbol | Primer, (5′–3′) | Fold change in array (comparison) | Fold change in PCR |
|---|---|---|---|---|
| AA682425 | DECR1 | Sense: CAAAAAGCGATGCTACCACCTA | 2.5 (active JIA versus CRM) | 1.56 |
| Antisense: GGCCAGTACCTCCCCCAGTA | ||||
| NM_001767 | CD2 | Sense: CACCTCTGCATCTTCGAACTCA | 4 (CR versus controls) | 4.6 |
| Antisense: CTGGTGTGATGGAGCTCTCTGA | ||||
| U66146 | CD6 | Sense: CCCCAGGTGTTTTCTTCAGAGA | 2 (CR versus controls) | 2.84 |
| Antisense: GCCGGCCAGCTCCAA | ||||
| AF043337 | IL8 | Sense: TGTTGAATTACGGAATAATGAGTTAGAAC | 4.4 (controls versus CR) | 4.25 |
| Antisense: CAAGTTTCAACCAGCAAGAAATTACT | ||||
| AY151286 | PTGS2 | Sense: AATGTGCCATAAGACTGACCTTTTAA | 3.1 (controls versus CR) | 2.97 |
| Antisense: CACAGTGCTTGACACAGAATATTTTC | ||||
| NM_000732 | CD3D | Sense: CTACCGTGCAAGTTCATTATCGAA | NA | 3.41 |
| Antisense: AGTGGCAATGACATCAGTGACAA | ||||
| NM_000733 | CD3E | Sense: GCAAACCAGAAGATGCGAACTT | 3.5 (CR versus controls) | 2.09 |
| Antisense: ACATCACATCCATCTCCATGCA | ||||
| NM_000073 | CD3G | Sense: GCTTCAGACAAGCAGACTCTGTTG | 5.4 (CR versus controls) | 1.76 |
| Antisense: TGTACTGGTCATCTTCTCGATCCTT |
Children with CRM status were those with inactive disease (disease in remission) who were taking medication and who had maintained that state for 6 continuous months. Children with CR status were those with inactive disease who were not taking medication and who had maintained that state for 12 continuous months. For clarity of analysis, children taking medication were only compared with children taking medication (i.e., children with active juvenile idiopathic arthritis [JIA] were compared with children with CRM status), and children with CR status were compared with healthy controls. PCR = polymerase chain reaction; NA = not available.
Hierarchical cluster analysis: neutrophils
Figure 1 shows a hierarchical cluster analysis in neutrophils from children with JIA. These samples clustered into 3 groups with some overlap between children in the different disease states, as summarized in Table 2. As we noted in our previous study (7), control subjects segregated out as a separate group. Similarly, 4 of the 6 children who had achieved CR status fell into a distinct group, with 2 clustering with controls. Consistent with the previous study, which compared children with inactive disease and those with active disease (7), children with CRM status clustered together with children with active disease (n = 5) and with healthy control children (n = 3). Across the groups, there were no statistically significant differences in gene expression between active disease and CRM groups, although the clustering suggests that there may be a trend toward the normalization of gene expression profiles in children who achieve CRM status.
Figure 1.
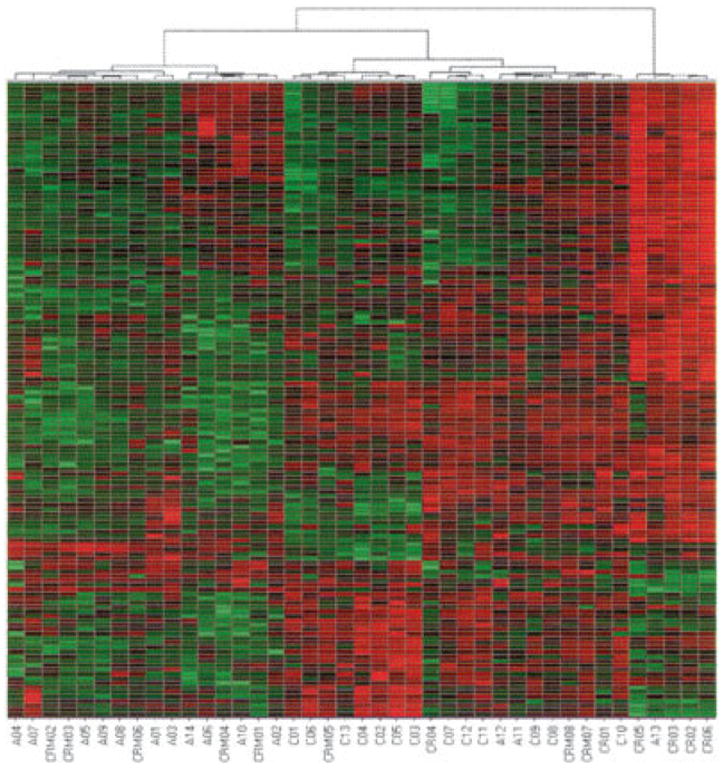
Hierarchical clustering analysis derived from gene expression profiling of neutrophils from children with distinct polyarticular juvenile idiopathic arthritis disease states and of neutrophils from healthy controls. Each block represents an individual gene, and individual patients are depicted on the x-axis. Numbers prefixed with A indicate patients with active disease. Numbers prefixed with CRM represent patients with clinical remission of disease who were taking medication. Numbers prefixed with CR represent patients with clinical remission of disease who were not taking medication. Numbers prefixed with C represent healthy control children.
Table 2.
Summary of hierarchical cluster analysis in neutrophils from children with JIA and in neutrophils from healthy controls*
| Subject status | Left cluster | Middle cluster | Right cluster |
|---|---|---|---|
| Active JIA | 11 | 2 | 1 |
| CRM | 5 | 3 | 0 |
| CR | 0 | 2 | 4 |
| Control | 0 | 13 | 0 |
Neutrophil gene expression signatures did not normalize when patients with disease in remission were compared with healthy control subjects. There were 81 different genes whose levels of expression differed between patients with disease in remission and healthy control subjects, represented by 85 different probe sets (IL8, PTGS2, SMC3, and TNFRSF25 are represented by 2 probes each) (see Supplementary Table 1, available on the Arthritis & Rheumatism Web site at http://www3.interscience.wiley.com/journal/76509746/home). Of these 81 genes, 76 were overexpressed in JIA neutrophils. These differentially expressed genes comprised 4 large overlapping networks. It is interesting to note that genes that link the largest 2 networks, phosphodiesterase 4B (PDE4B) and IL32, have both been implicated either in animal models of arthritis (17,18) or in human rheumatic disease (19-21).
Figure 2A shows the largest of these networks. It is interesting to note the presence of RNA for T cell surface antigens such as CD3 and CD2 in this network. Overexpression of each of these genes in children with JIA was confirmed using RT-PCR (see Table 1). Furthermore, this finding is peculiar to polyarticular JIA, since we did not see these genes overexpressed in a cohort of children with pauciarticular JIA (n = 10) compared with the same control cohort (Frank MB, et al: unpublished observations). The presence of T cell markers on neutrophils has been previously described, and CD3-expressing neutrophils make up ~5–8% of the peripheral blood neutrophils of healthy adults (22). Engagement of CD3 on these cells stimulates interleukin-8 (IL-8) secretion and inhibits neutrophil apoptosis.
Figure 2.
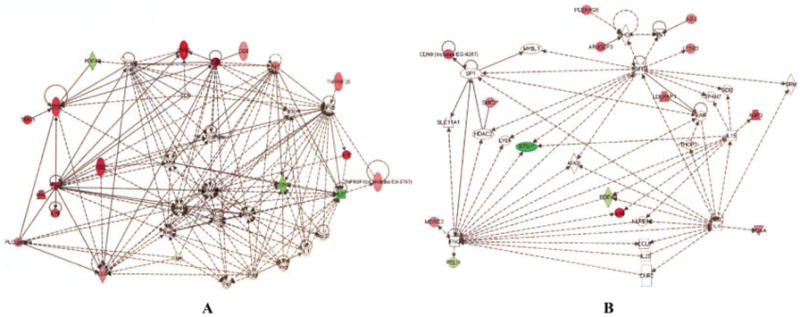
Networks derived from Ingenuity software analysis of differential gene expression, comparing neutrophils from children with juvenile idiopathic arthritis in remission who were not taking medication with those from healthy controls. Genes showing higher expression in patients are shown in red, and those expressed at higher levels in controls are shown in green. A, Note the presence of genes normally associated with the regulation of adaptive immunity (e.g., CD3, CD2) in this network. B, Note that this network demonstrates overlapping groups of interleukin-4– and interferon-γ–regulated genes that we have previously described (23).
The second network that was derived from comparing children with disease in remission (and not taking medication) with healthy control subjects (Figure 2B) demonstrated clusters of IL-4– and interferon-γ (IFNγ)–regulated genes that we have previously described in polyarticular JIA (23). In addition, this network consists of a cluster of transforming growth factor β–regulated genes. This finding corroborates our recent work demonstrating that CR status does not represent a return to normalcy in polyarticular JIA, but instead represents a state of homeostasis in which pro- and antiinflammatory pathways are held in balance (10). This hypothesis is supported by the structure of the network shown in Figure 3A, showing persistence of JUN- and MYC-regulated genes in JIA neutrophils. At the same time, a counterinflammatory network (Figure 3B) emerges. In this network, retinoic acid, a known modulator of inflammation (24,25), appears as a system “hub” (26).
Figure 3.
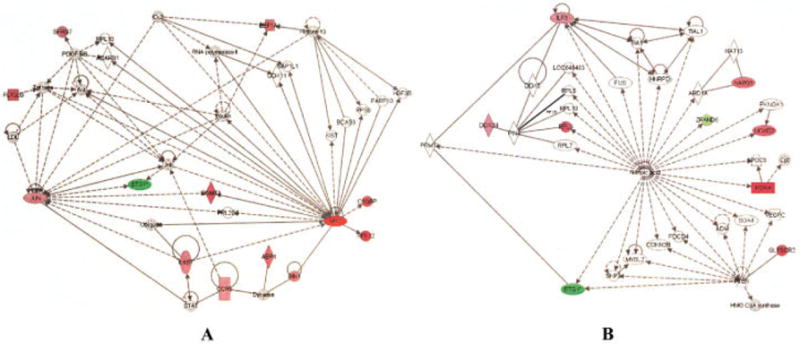
Networks derived from Ingenuity software analysis of genes differentially expressed in neutrophils from children with polyarticular juvenile idiopathic arthritis (JIA) who have achieved remission and are not taking medication and in neutrophils from healthy control children. Genes overexpressed in children with JIA are shown in red, and those expressed at higher levels in controls are shown in green. A, Shown is a network comprising of groups of proinflammatory genes (e.g., CCR5, interleukin-6 signal transducer [IL6ST]) regulated through the JUN and MYC transcription regulators. B, Shown is a network in which retinoic acid, a known modulator and attenuator of inflammation, is a prominent system “hub.”
DISCUSSION
Neutrophils have sometimes been overlooked by basic immunologists, and especially by investigators interested in the rheumatic diseases of childhood. However, advances in 2 broad areas of biology and immunology have made it imperative that we reconsider simple, linear theories of disease pathogenesis of JIA (e.g., the autoimmunity model) and seriously consider the complexity of biologic and pathologic systems that likely underlie disease pathogenesis and clinical phenotype. The first advance is our understanding that biologic and pathologic systems are enormously more complex than was previously understood. This is true even for such apparently “simple” disorders as those arising from single-gene mutations (27,28). Indeed, we are now learning that simple, linear models for understanding inflammation (e.g., receptor engagement→receptor phosphorylation→kinase engagement→transcription factor activation→gene transcription) are inadequate to explain the complex, oscillatory, mutually resonating systems that are activated in an inflammatory/immune response (4).
The second advance is our growing understanding that innate and adaptive immunity are part of such a mutually interacting system (29,30), and that neutrophils are a critical component of this system (8). Neutrophils regulate adaptive immunity in multiple ways and at multiple levels, and their effects extend beyond their influence of the early phases of antigen-presenting cell activation and immune response induction (31-35). For example, neutrophils release the tumor necrosis factor–related ligand B lymphocyte stimulator, thus regulating the expansion and maturation of B cells (36). In a similar vein, neutrophil-derived IFNγ regulates the activation and expansion of T cells (37).
We have separately examined the gene expression profiles in PBMCs from JIA patients (10). One might predict that many genes would be similarly differentially expressed in JIA patients relative to controls both in PBMCs and in neutrophils, perhaps reflecting common states of inflammation in leukocytes or common responses to medications. We found very little evidence for this. For example, the JUN oncogene in patients was found to be up-regulated only slightly more than 2-fold in both cell types, as were 2 other transcripts, the functions of whose products are currently unknown (Affymetrix probe sets 240347_at and 243509_at). These results further suggest complex interactions as opposed to triggering of common biochemical pathways, in these cell types.
We have previously reported that the neutrophils in polyarticular JIA manifest an intrinsic defect in the way in which fundamental metabolic oscillatory events are regulated (7), and that this defect persists when children’s disease becomes inactive. The present study demonstrates that neutrophil abnormalities persist even when the disease has been inactive for 6 months (i.e., when CRM status has been achieved). In the earlier study, genes of children with active and inactive disease scattered indiscriminately across a hierarchical cluster grid and showed no differences in expression. The present study corroborates that finding, although cluster analysis suggests a trend toward normalization of the neutrophil expression profile as children reach CRM status (Figure 1 and Table 2).
These data also cast light on a previously vexing and important clinical question: why do children with JIA, even those with disease in remission for extended periods of time, experience disease flares? Part of the answer appears to be the finding that CR status does not represent a return to “normal.” Rather, it reflects a homeostatic state in which proinflammatory networks (e.g., see Figure 3A) are modulated by networks of genes that balance or counter inflammation. Especially intriguing, perhaps, is the finding that both TGFβ (Figure 2B) and retinoic acid (Figure 3B) are prominent hubs in these networks, since both of these mediators have been shown to inhibit the development of autoimmunity-enhancing Th17 cells (38). This finding, in turn, suggests that neutrophils play a far more important role than has previously been recognized in regulating the known immune abnormalities that are believed to be a part of JIA pathogenesis. It is also reasonable to hypothesize that these networks reflect modulation of other parts of the innate immune system. IL-4, for example (Figure 2B), is known to modulate the maturation of macrophages into the so-called M2 phenotype; these M2 macrophages balance and attenuate the potent proinflammatory effects of classic M1 macrophages (39,40).
On the basis of these data, we cannot determine whether the neutrophil gene expression profile might be used to guide therapy. For example, it would be extremely useful to know which children with CRM status can safely discontinue therapy without risk of immediate flare. The cluster analysis suggests that there is a point at which the CRM profile begins to look more like the CR profile, but our numbers are small and this question will only be answered with large, prospective, dynamic studies.
It would be reckless to propose that adaptive immunity plays only a secondary role in the pathogenesis of JIA, but the data we report here and our previously published work (7) strongly support the concept that neutrophils are also important elements in the disease process and are not simply at the end of the pathogenic pathway. Indeed, “either-or” thinking about innate and adaptive immunity in JIA is artificial, and we have eschewed this “blind men and the elephant” approach to understanding complex traits like JIA (41). We believe that our understanding of disease pathogenesis will need to be strongly informed by a better appreciation of biologic complexity that is emerging from newer elements of network theory (42,43) and by a better understanding of the importance of nonlinear oscillators in regulating leukocyte function (44,45). Finally, there is reason to be optimistic that the rapidly expanding tools for understanding biologic processes at the systems level (46) will both revolutionize our understanding of JIA (and other complex diseases) and rapidly translate to more effective therapies for this family of diseases.
Acknowledgments
Supported by the NIH (grants RR-03145, RR-020143, RR-16478, RR-15577, AI-062629, AR-061-015, AR-081-006, and HR-07-139) and by the Oklahoma Center for the Advancement of Science and Technology. Dr. Jarvis’ work was supported by an Innovative Research grant from the Arthritis Foundation. Dr. Aggarwal’s work was supported by an Overseas Associateship grant from the Indian government, Department of Biotechnology. Mr. McKee and Dr. Chaser’s work was supported by Summer Medical Student Preceptorships from the American College of Rheumatology; Dr. Chaser also received a summer research stipend from the University of Oklahoma Health Sciences Center, Native American Center of Excellence.
Footnotes
AUTHOR CONTRIBUTIONS Dr. Jarvis had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study design. Jarvis, Knowlton.
Acquisition of data. Jarvis, Jiang, Frank, Aggarwal, Wallace, McKee, Chaser, Tung, Smith, McGhee, Chen, Osban, O’Neil.
Analysis and interpretation of data. Jarvis, Jiang, Frank, Knowlton, Aggarwal, O’Neil, Centola.
Manuscript preparation. Jiang, Frank, Knowlton, Aggarwal, Wallace, O’Neil.
Statistical analysis. Jarvis, Jiang, Frank, Knowlton.
Dr. Centola has received consulting fees from Crescendo Biosciences (more than $10,000).
References
- 1.Jarvis JN. Juvenile rheumatoid arthritis: a guide for pediatricians. Pediatr Ann. 2002;31:437–46. doi: 10.3928/0090-4481-20020701-08. [DOI] [PubMed] [Google Scholar]
- 2.Forre O, Dobloug JH, Hoyeraal HM, Thorsby E. HLA antigens in juvenile arthritis: genetic basis for the different subtypes. Arthritis Rheum. 1983;26:35–8. doi: 10.1002/art.1780260106. [DOI] [PubMed] [Google Scholar]
- 3.Prahalad S. Genetics of juvenile idiopathic arthritis: an update. Curr Opin Rheumatol. 2004;16:588–94. doi: 10.1097/01.bor.0000134407.48586.b0. [DOI] [PubMed] [Google Scholar]
- 4.Buchman TG. Nonlinear dynamics, complex systems, and the pathobiology of critical illness. Curr Opin Crit Care. 2004;10:378–82. doi: 10.1097/01.ccx.0000139369.65817.b6. [DOI] [PubMed] [Google Scholar]
- 5.Jarvis JN. Mechanisms of pathogenesis and inflammation in the pediatric rheumatic diseases. Curr Opin Rheumatol. 1998;10:459–67. doi: 10.1097/00002281-199809000-00011. [DOI] [PubMed] [Google Scholar]
- 6.Jarvis JN, Jiang K, Petty HR, Centola M. Neutrophils: the forgotten cell in JIA disease pathogenesis. Pediatr Rheumatol Online J. 2007;5:13. doi: 10.1186/1546-0096-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jarvis JN, Petty HR, Tang Y, Frank MB, Tessier PA, Dozmorov I, et al. Evidence for chronic, peripheral activation of neutrophils in polyarticular juvenile rheumatoid arthritis. Arthritis Res Ther. 2006;8:R154. doi: 10.1186/ar2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–82. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 9.Wallace CA, Huang B, Bandeira M, Ravelli A, Giannini EH. Patterns of clinical remission in select categories of juvenile idiopathic arthritis. Arthritis Rheum. 2005;52:3554–62. doi: 10.1002/art.21389. [DOI] [PubMed] [Google Scholar]
- 10.Knowlton N, Jiang K, Frank MB, Aggarwal A, Wallace C, McKee R, et al. The meaning of clinical remission in polyarticular juvenile idiopathic arthritis: gene expression profiling in peripheral blood mononuclear cells identifies distinct disease states. Arthritis Rheum. 2009;60:892–900. doi: 10.1002/art.24298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390–2. [PubMed] [Google Scholar]
- 12.Wallace CA, Ruperto N, Giannini E Childhood Arthritis and Rheumatology Research Alliance (CARRA), the Pediatric Rheumatology International Trials Organization (PRINTO), and the Pediatric Rheumatology Collaborative Study Group (PRCSG) Preliminary criteria for clinical remission for select categories of juvenile idiopathic arthritis. J Rheumatol. 2004;31:2290–4. [PubMed] [Google Scholar]
- 13.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 14.Simon R, Korn E, McShane L, Radmacher M, Wright G, Zhao Y. Design and analysis of DNA microarray investigations. New York: Springer-Verlag; 2003. [Google Scholar]
- 15.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 16.Ward JH. Hierarchical grouping to optimize an objective function. J Am Stat Assoc. 1963;58:236–44. [Google Scholar]
- 17.Nyman U, Mussener A, Larsson E, Lorentzen J, Klareskog L. Amelioration of collagen II-induced arthritis in rats by the type IV phosphodiesterase inhibitor Rolipram. Clin Exp Immunol. 1997;108:415–9. doi: 10.1046/j.1365-2249.1997.3931291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh HN, Blancuzzi V, Greenwood S, Skiles JW, O’Byrne EM. Synovial fluid levels of tumor necrosis factor-α in the inflamed rat knee: modulation by dexamethasone and inhibitors of matrix metalloproteinase and phosphodiesterase. Inflamm Res. 1997;46(Suppl 2):S153–4. doi: 10.1007/s000110050151. [DOI] [PubMed] [Google Scholar]
- 19.Dinarello CA, Kim SH. IL-32, a novel cytokine with a possible role in disease. Ann Rheum Dis. 2006;65(Suppl 3):iii61–4. doi: 10.1136/ard.2006.058511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shoda H, Fujio K, Yamaguchi Y, Okamoto A, Sawada T, Kochi Y, et al. Interactions between IL-32 and tumor necrosis factor α contribute to the exacerbation of immune-inflammatory diseases. Arthritis Res Ther. 2006;8:R166. doi: 10.1186/ar2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asquith DL, McInnes IB. Emerging cytokine targets in rheumatoid arthritis. Curr Opin Rheumatol. 2007;19:246–51. doi: 10.1097/BOR.0b013e3280eec78c. review. [DOI] [PubMed] [Google Scholar]
- 22.Puellmann K, Kaminski WE, Vogel M, Nebe CT, Schroeder J, Wolf H, et al. A variable immunoreceptor in a subpopulation of human neutrophils. Proc Natl Acad Sci U S A. 2006;103:14441–6. doi: 10.1073/pnas.0603406103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jarvis JN, Dozmorov I, Jiang K, Frank MB, Szodoray P, Alex P, et al. Novel approaches to gene expression analysis of active polyarticular juvenile rheumatoid arthritis. Arthritis Res Ther. 2004;6:R15–32. doi: 10.1186/ar1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 25.Nozaki Y, Tamaki C, Yamagata T, Sugiyama M, Ikoma S, Kinoshita K, et al. All-trans-retinoic acid suppresses interferon-γ and tumor necrosis factor-α; a possible therapeutic agent for rheumatoid arthritis. Rheumatol Int. 2006;26:810–7. doi: 10.1007/s00296-005-0076-1. [DOI] [PubMed] [Google Scholar]
- 26.Barbasi AL, Albert A. Emergence of scaling in networks. Science. 1999;286:509–12. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 27.Dipple KM, McCabe ER. Phenotypes of patients with “simple” Mendelian disorders are complex traits: thresholds, modifiers, and systems dynamics. Am J Hum Genet. 2000;66:1729–35. doi: 10.1086/302938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCabe LL, McCabe ER. Complexity in genetic diseases: how patients inform the science by ignoring the dogma. Am J Med Genet A. 2006;140:160–1. doi: 10.1002/ajmg.a.31032. [DOI] [PubMed] [Google Scholar]
- 29.Fearon DT, Locksley RM. The instructive role of innate immunity in the acquired immune response. Science. 1996;72:50–3. doi: 10.1126/science.272.5258.50. [DOI] [PubMed] [Google Scholar]
- 30.Nathan C. Inflammation: points of control. Nature. 2002;420:846–52. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 31.Lefkowitz DL, Gelderman MP, Fuhrmann SR, Graham S, Starnes JD, III, Lefkowitz SS, et al. Neutrophilic myeloperoxidase–macrophage interactions perpetuate chronic inflammation associated with experimental arthritis. Clin Immunol. 1999;91:145–55. doi: 10.1006/clim.1999.4696. [DOI] [PubMed] [Google Scholar]
- 32.Pereira HA, Shafer WM, Pohl J, Martin LE, Spitznagel JK. CAP37, a human neutrophil-derived chemotactic factor with monocyte specific activity. J Clin Invest. 1990;85:1468–76. doi: 10.1172/JCI114593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennouna S, Bliss SK, Curiel TJ, Denkers EY. Cross-talk in the innate immune system: neutrophils instruct recruitment and activation of dendritic cells during microbial infection. J Immunol. 2003;171:6052–8. doi: 10.4049/jimmunol.171.11.6052. [DOI] [PubMed] [Google Scholar]
- 34.Bennouna S, Denkers EY. Microbial antigen triggers rapid mobilization of TNF-α to the surface of mouse neutrophils transforming them into inducers of high-level dendritic cell TNF-α production. J Immunol. 2005;174:4845–51. doi: 10.4049/jimmunol.174.8.4845. [DOI] [PubMed] [Google Scholar]
- 35.Brown B, Frank MB, Knowlton N, Jiang K, Chen Y, Centola M, et al. Neutrophil regulation of adaptive immunity: an in vitro model identifies hubs and nodes with similarities to those seen in polyarticular JIA. Arthritis Rheum. 2008;58(Suppl 9):S708. abstract. [Google Scholar]
- 36.Scapini P, Carletto A, Nardelli B, Calzetti F, Roschke V, Merigo F, et al. Proinflammatory mediators elicit secretion of the intracellular B-lymphocyte stimulator pool (BLyS) that is stored in activated neutrophils: implications for inflammatory diseases. Blood. 2005;105:830–7. doi: 10.1182/blood-2004-02-0564. [DOI] [PubMed] [Google Scholar]
- 37.Ethuin F, Gerard B, Benna JE, Boutten A, Gougereot-Pocidalo MA, Jacob L, et al. Human neutrophils produce interferon γ upon stimulation by interleukin-12. Lab Invest. 2004;84:1363–71. doi: 10.1038/labinvest.3700148. [DOI] [PubMed] [Google Scholar]
- 38.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–60. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 39.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 40.Naito M. Macrophage differentiation and function in health and disease. Pathol Int. 2008;58:143–55. doi: 10.1111/j.1440-1827.2007.02203.x. [DOI] [PubMed] [Google Scholar]
- 41.Jarvis JN. Gene expression arrays in juvenile rheumatoid arthritis: will the blind men finally see the elephant? Curr Probl Pediatr Adolesc Health Care. 2006;36:91–6. doi: 10.1016/j.cppeds.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 42.Jeong H, Tombor B, Oltval ZN, Barabasi AL. The large-scale organization of metabolic networks. Nature. 2000;407:651–4. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 43.Barbasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–13. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 44.Petty HR. Neutrophil oscillations: temporal and spatiotemporal aspects of cell behavior. Immunol Res. 2001;23:85–94. doi: 10.1385/IR:23:1:85. [DOI] [PubMed] [Google Scholar]
- 45.Kindzelskii AL, Petty HR. Apparent role of traveling metabolic waves in oxidant release by living neutrophils. Proc Natl Acad Sci U S A. 2002;99:9207–12. doi: 10.1073/pnas.132630999. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Kirschner MW. The meaning of systems biology. Cell. 2005;121:503–4. doi: 10.1016/j.cell.2005.05.005. [DOI] [PubMed] [Google Scholar]