

Abstract
Glial tumor necrosis factor-α (TNFα) is essential for scaling up of synapses during prolonged activity blockade, but whether TNFα is an instructive or permissive signal is not known. Here we show in rat cortical neurons that the effects of TNFα and activity blockade are not additive; whereas TNFα increased AMPA quantal amplitude at control synapses, TNFα reduced quantal amplitude at prescaled synapses, demonstrating state-dependent effects of TNFα signaling on the scaling process. Whereas synaptic scaling during prolonged activity blockade [24 h tetrodotoxin (TTX)] was prevented by blocking TNFα signaling, early scaling (6 h TTX) was not, unless TNFα signaling was first blocked for 24 h. Moreover, when synapses were prescaled, prolonged (24 h) but not brief (6 h) blockade of TNFα signaling reversed scaling. Finally, prolonged block of TNFα signaling modified the synaptic localization of several scaffold proteins, suggesting that maintenance of postsynaptic density composition is TNFα dependent. Together, these data suggest that TNFα is not an instructive signal for scaling but rather is critical for maintaining synapses in a plastic state in which synaptic scaling can be expressed.
Introduction
To function properly, neuronal circuits require homeostatic plasticity mechanisms such as synaptic scaling that provide stability to circuit output. Synaptic scaling has been well documented at a variety of central synapses both in vitro and in vivo and is characterized by bidirectional changes in miniature EPSC (mEPSC) amplitude that are in the correct direction to compensate for prolonged changes in activity (Burrone and Murthy, 2003; Turrigiano and Nelson, 2004; Turrigiano, 2008). There is widespread agreement that scaling is expressed through changes in AMPA receptor (AMPAR) accumulation at postsynaptic sites (O'Brien et al., 1998; Turrigiano et al., 1998, 2005; Wierenga et al., 2005), but how changes in activity are sensed and translated into changes in AMPAR accumulation is still controversial.
Several activity-dependent signals have been suggested to mediate scaling (Turrigiano, 2008). We reported recently that scaling up is a function of postsynaptic firing and calcium-dependent changes in transcription, arguing for a cell-autonomous induction mechanism (Ibata et al., 2008). In contrast, a recent study argued for a non-cell-autonomous mechanism mediated by glial release of the proinflammatory cytokine tumor necrosis factor-α (TNFα) (Stellwagen and Malenka, 2006). This apparent contradiction prompted us to reexamine the role of TNFα in scaling and in particular to ask whether TNFα is an “activity signal” that instructs neurons to scale synapses up when activity drops, or alternatively plays a permissive role by maintaining synapses in a plastic state.
TNFα levels are elevated by prolonged (48 h) activity blockade, acute application of TNFα increases mEPSC amplitude, and scaling up in response to prolonged activity blockade is prevented by blocking TNFα signaling (Beattie et al., 2002; Stellwagen et al., 2005; Stellwagen and Malenka, 2006). These observations led to the proposal that prolonged activity blockade induces scaling by increasing glial release of TNFα, which then acts on neurons to enhance AMPAR insertion (Stellwagen and Malenka, 2006). Inconsistent with this model are the observations that scaling is a gradual and cumulative process evident after as little as 4–6 h of activity blockade (Sutton et al., 2006; Ibata et al., 2008), whereas TNFα-dependent scaling was observed only after prolonged activity block (Stellwagen and Malenka, 2006). This raises the possibility that the early phase of scaling is not mediated by TNFα, or, alternatively, that TNFα is permissive rather than instructive for scaling.
To examine these possibilities, we blocked activity while preventing TNFα signaling for brief (6 h, early scaling) or prolonged (24 h, prolonged scaling) periods of time. Prolonged scaling was prevented by blocking TNFα signaling, but early scaling was not unless TNFα signaling was first blocked for 24 h. Moreover, when synapses were prescaled, prolonged but not brief blockade of TNFα signaling could reverse scaling. Finally, prolonged block of TNFα signaling modified the synaptic localization of several scaffold proteins, suggesting that maintenance of postsynaptic density (PSD) composition is TNFα dependent. Together, these data suggest that TNFα signaling is critical for maintaining synapses in a plastic state in which synaptic scaling can be expressed.
Materials and Methods
Neuronal cultures, drug treatments, and immunostaining.
Dissociated cultures were prepared from the visual cortex of postnatal d 2–4 Long–Evans rat pups as described previously (Pratt et al., 2003). All experiments were performed after 6–10 d in vitro on pyramidal neurons identified morphologically as described previously (Watt et al., 2000). Drug concentrations were tetrodotoxin (TTX), 1 μm; TNFα (410-MT, R&D Systems), 100 ng/ml; and soluble form of the TNFα receptor 1 (sTNFR) (425-RI, R&D Systems), 2 μg/ml. Antibody localization of PSD95 (MA1-046, Pierce Biotechnology), SAP102 (75-058, NeuroMab), pan-TARP (07-577, Millipore), and VGlut-1 (135 304, Synaptic Systems) was performed as described previously (Rutherford et al., 1997). Antibody labeling of synaptic GluR1 (PC246, Calbiochem) and GluR2 (sc-7611, Santa Cruz Biotechnology) was performed live under nonpermeant conditions as described previously (Wierenga et al., 2005). To ensure uniformity, data were acquired from dendrites branching off of the apical-like dendrite. All experimental conditions were run in parallel on sister cultures from the same dissociations. All images in Figure 3 were quantified with MetaMorph software (Molecular Devices).
Figure 3.
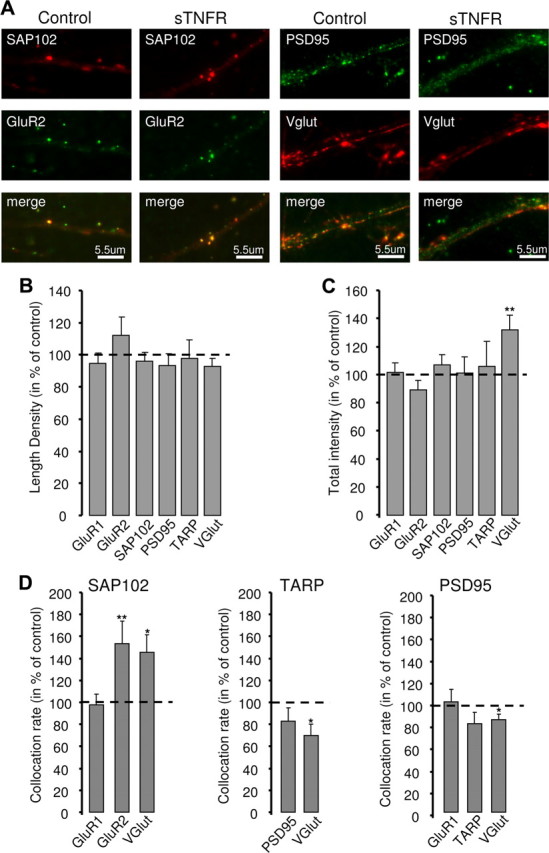
Prolonged blockade of TNFα signaling has subtle effects on the composition of the postsynaptic density. A, Example images showing colocalization between SAP102 and GluR2 (left) or PSD95 and VGlut (right) in control and after 24 h of sTNFR treatment. B, Summary of changes in length density after 24 h of sTNFR treatment. C, Summary of changes in puncta fluorescence intensity after 24 h of sTNFR treatment. D, Summary of the changes in colocalization rates for SAP102 (left), TARP (middle), and PSD95 (right). *p < 0.05, **p < 0.01.
Culture electrophysiology.
Whole-cell AMPA-mediated mEPSCs were recorded and analyzed as described previously (Watt et al., 2000) at 25°C from a holding potential of –70 mV. Recordings with resting membrane potential (Vm) > –54 mV, series resistances > 20 MΩ, input resistance (Rin) < 100 MΩ, or < 20 mEPSCs were excluded. None of the conditions reported resulted in significant changes in mEPSC frequency, rise or decay times, Rin, or Vm.
Statistics.
Data are presented as mean ± SEM for the number of neurons indicated. Each experiment was repeated on at least three separate dissociations and the n values represent number of neurons. To determine statistical significance, unpaired two-tailed Student's t tests, or for multiple comparisons single-factor ANOVAs followed by a Tukey test, were run as appropriate. P values ≤0.05 were considered significant. To analyze whether changes in distribution of mEPSCs were multiplicative, 20 events were randomly selected from each neuron in each condition, each distribution was linearly interpolated to contain the same number of events, and experimental conditions were then plotted against baseline conditions. A linear change in slope from the unity line is indicative of multiplicative scaling (Turrigiano et al., 1998; Gainey et al., 2009).
Results
Synaptic scaling modulates but does not occlude the effect of TNFα application
Scaling grows in magnitude as a function of the length of deprivations and is not saturated after 24 h of TTX treatment (Turrigiano et al., 1998; Wierenga et al., 2005). If TTX scales synapses up by gradually increasing the release of TNFα (Stellwagen and Malenka, 2006), then the effects of TTX and TNFα on mEPSCs should be additive, either linearly or sublinearly if there is some occlusion due to partial saturation of scaling. To test whether the effects of TNFα and TTX are additive, we treated cultures with TTX alone, TNFα alone, or TTX and TNFα together and measured mEPSC amplitude. First, we examined the effects of acute (90 min) application of TNFα in the presence or absence of TTX. TNFα alone significantly increased mEPSC amplitude (n = 11; p = 0.018), similar to previous reports (Stellwagen et al., 2005), whereas 90 min of TTX alone had no significant effect (Fig. 1A–C). Interestingly, TNFα was unable to increase mEPSC amplitude in the presence of acute TTX (Fig. 1B,C) (n = 14, p = 0.97), indicating that this effect requires ongoing action potential firing. This result is inconsistent with the idea that TTX increases mEPSC amplitude by enhancing TNFα release.
Figure 1.

The effects of TTX and TNFα on mEPSC amplitude are not additive. A, D, Schematic of experimental protocol for coapplication of TTX and TNFα. B, E, left, Examples of mEPSC recordings for the indicated conditions; right, average mEPSC waveforms for the same conditions. C, F, Average mEPSC amplitude ± SEM for indicated conditions. *p < 0.05, **p < 0.01. G, Analysis of change in distribution of mEPSC amplitude following TTX (left), TNFα (middle,), or TTX + TNFα (right) treatment. Black line is unity line (baseline plotted against itself), symbols are experimental condition (experimental plotted against baseline), and dashed line is best linear fit for the experimental condition.
Next, we examined the effects of TNFα after synapses had been prescaled by 24 h of TTX treatment. When applied separately, both 24 h of TTX and acute TNFα application increased mEPSC amplitude (Fig. 1D–F). Surprisingly, prolonged TTX treatment not only prevented acute TNFα from increasing mEPSC amplitude but actually reversed the effects of TNFα; acute TNFα after 24 h TTX (applied in the continued presence of TTX) reduced mEPSC amplitude relative to either TNFα or TTX alone (Fig. 1D–F; TTX: 175.6 ± 27% of control, n = 10, p = 0.015; TNFα: 171.1% ± 17.7% of control, n = 18, p = 0.01; TTX +TNFα: 122 ± 9.7% of control, n = 13, p = 0.024). Together, our data demonstrate that the effects of TNFα and TTX on AMPA quantal amplitude are not additive. Instead, the effects of TNFα are complex and depend both on ongoing activity and the state of the synapse. These data are not consistent with the simple model that activity blockade scales synapses up by gradually enhancing the release of TNFα.
Further supporting the distinction between the effects of TNFα and TTX, TNFα failed to induce multiplicative changes in synaptic strength (Fig. 1G). When the ranked TTX amplitude distribution was plotted against the ranked control distribution, the data were well fit by a line with increased slope relative to the unity line (control versus control), indicative of multiplicative synaptic scaling (Fig. 1G, left) (Turrigiano et al., 1998). In contrast, the increase induced by TNFα deviated from scaling (Fig. 1G, middle), as did the reduction in mEPSC amplitude when TNFα was applied after 24 h of TTX (Fig. 1G, right).
Prolonged but not brief blockade of TNFα signaling prevents or reverses synaptic scaling
sTNFR that scavenges endogenous TNFα blocks the ability of prolonged TTX application (48 h) to scale up mEPSC amplitude (Stellwagen and Malenka, 2006). To determine if TNFα signaling is also required for the early phase of scaling, we coapplied TTX and sTNFR for 6 or 24 h. When TTX and sTNFR were coapplied for 24 h, scaling up was blocked (Fig. 2A) (mEPSC amplitude was 108.8 ± 7.7% of control; n = 18; p = 0.9). In marked contrast, sTNFR was not able to block scaling when coapplied with TTX for 6 h (Fig. 2B) (mEPSC amplitude was 162.8 ± 12.4% of control; n = 17; p = 0.002). Coapplication for 12 h had an intermediate effect (mEPSC amplitude was 138.8 ± 12% of control; n = 15; p = 0.01), indicating that the ability of sTNFR to block scaling develops gradually. sTNFR application had no significant effect on baseline mEPSC amplitude when applied for either 6 or 24 h (Fig. 2A,B) (sTNFR 6 h, 101.4 ± 7.3% of control, n = 11, p = 0.9; sTNFR 24 h, 106 ± 11.4% of control, n = 17, p = 0.9), indicating that maintenance of baseline mEPSC amplitude is not dependent on ongoing TNFα signaling.
Figure 2.

Prolonged but not brief blockade of TNFα signaling prevents synaptic scaling. A, B, Average mEPSC amplitude for the indicated conditions. C, Schematic of experimental conditions for data shown in D. D, Average mEPCS amplitude for the various conditions. *p < 0.05, **p < 0.01.
TNFα signaling could be essential for prolonged but not early synaptic scaling because early and prolonged scaling are mediated by different signaling pathways. Alternatively, prolonged treatment with sTNFR might prevent synapses from being able to express or maintain scaling. To differentiate between these possibilities, we first treated cultures with sTNFR for 24 h, followed by 6 h of TTX (Fig. 2Cb). If TNFα signaling is not involved in early scaling, then this condition should resemble 6 h TTX alone. Alternatively, if prolonged block of TNFα signaling compromises the ability of synapses to express synaptic scaling, then early scaling should be prevented by this pretreatment. We found that pretreatment with sTNFR for 24 h completely blocked the ability of 6 h TTX to scale up mEPSC amplitude (Fig. 2Db) (mEPSC amplitude was 90.1 ± 6.5% of control; n = 20; p = 0.9; compare with 2B, 6 h TTX alone), supporting the interpretation that prolonged block of TNFα signaling prevents synapses from expressing synaptic scaling.
As a second test of the model that TNFα signaling is instructive for prolonged scaling, we asked whether blockade of TNFα signaling after scaling has been induced is able to reverse prolonged scaling. Scaling was first induced by 24 h of TTX treatment and then sTNFR was applied for either 6 h (brief) or 24 h (prolonged) in the continued presence of TTX (Fig. 2Cc,d). A brief blockade of TNFα signaling was not able to reverse late scaling (Fig. 2Dc) (mEPSC amplitude was 163.2 ± 22.3% of control; n = 12; p = 0.001), indicating that late scaling does not depend upon continued TNFα release. In contrast, prolonged application (24 h) of sTNFR (again in the continued presence of TTX) was able to reverse late scaling (Fig. 2Dd) (mEPSC amplitude was 119.9 ± 6.5% of control; n = 19; p = 0.46). Together, our data strongly suggest that TNFα signaling is not instructive for synaptic scaling but rather is required to maintain the ability of synapses to express this form of plasticity.
Prolonged blockade of TNFα signaling has subtle effects on the localization of postsynaptic density proteins
The expression of synaptic scaling likely depends upon the proper synaptic localization of numerous signaling and scaffold proteins that regulate synaptic function and plasticity (Turrigiano, 2008). To test whether prolonged block of TNFα signaling affects PSD composition, we treated cultures for 24 h with sTNFR and examined the punctate accumulation and synaptic localization of a number of synaptic proteins (including GluR1, GluR2, SAP102, PSD95, the TARPs, and VGlut) by doing pairwise immunohistochemistry against these various markers (Fig. 3A). For all of these markers, the puncta densities and fluorescent intensities were similar between control and sTNFR-treated cultures (Fig. 3B,C); the only significant change was in VGlut puncta intensity, which increased significantly (Fig. 3C) (132.4% ± 10.6% of control; n = 88; p = 0.003). Thus, block of TNFα signaling does not affect overall synapse density or expression levels of postsynaptic proteins.
Next, to look for effects on the synaptic localization of scaffolding proteins, we examined the colocalization rates of postsynaptic proteins with the excitatory presynaptic marker VGlut. STNFR application increased the colocalization of SAP102 with both GluR2 and VGlut, but not GluR1 (Fig. 3D) (SAP102-GluR2: n = 22, p = 0.010; SAP102-VGlut: n = 23, p = 0.017; SAP102-GluR1: n = 26, p = 0.43), suggesting that SAP102 selectively increases at GluR2 containing putative synaptic sites. In contrast, sTNFR decreased the colocalization rates of TARPs and PSD95 with VGlut; (Fig. 3D) (PSD95-VGlut: n = 22, p = 0.032; TARP-VGlut, n = 15, p = 0.04). Thus, blockade of TNFα signaling enhances the synaptic localization of SAP102 but reduces the fraction of excitatory synapses expressing PSD95 and TARPs. Overall, these data show that blocking TNFα signaling modifies the distribution of synaptic proteins at excitatory synapses.
Discussion
Glial TNFα is essential for the expression of synaptic scaling up during activity blockade (Stellwagen and Malenka, 2006; Kaneko et al., 2008), but whether TNFα acts as an instructive “activity signal” that induces scaling up, or is instead necessary for maintaining synapses in a plastic state, is unclear. Here, we show that 1) the effects of activity blockade and TNFα on mEPSC amplitude are not simply additive; 2) blocking TNFα signaling does not affect the induction or maintenance of synaptic scaling unless this blockade is maintained for >12 h; and 3) prolonged block of TNFα signaling modifies the composition of the postsynaptic density. Together, our data suggest that, rather than being an instructive signal for synaptic scaling, TNFα is critical for maintaining synapses in a plastic state that allows the expression of synaptic scaling.
It was recently proposed that prolonged activity blockade increases glial release of TNFα, which then acts on neurons to enhance AMPAR insertion (Stellwagen and Malenka, 2006). However, this model is inconsistent with several known properties of synaptic scaling. First, synaptic scaling is a gradual process that becomes evident after as little as 4–6 h of activity blockade and continues to grow in magnitude as the period of activity blockade increases (Turrigiano et al., 1998; Wierenga et al., 2005; Ibata et al., 2008). In contrast, TTX treatment does not increase the levels of TNFα in the culture medium (nor is conditioned medium from TTX-treated cultures able to increase AMPA mEPSC amplitude) until after >24 h (Stellwagen and Malenka, 2006). Second, TTX treatment increases the synaptic accumulation of GluR1 and GluR2 proportionally (Wierenga et al., 2005; Ibata et al., 2008) and GluR2 is necessary for the expression of TTX-induced synaptic scaling (Gainey et al., 2009), whereas TNFα induces a rapid increase in GluR2-lacking AMPARs (Leonoudakis et al., 2008). Finally, there is strong evidence that at least the early phase of synaptic scaling is induced in a cell-autonomous manner (Ibata et al., 2008), arguing against the requirement for global release of a soluble factor in its induction.
These considerations suggested either that early and prolonged scaling operate via distinct signaling pathways, or, alternatively, that TNFα signaling is permissive rather than instructive for synaptic scaling. The data presented here strongly support the latter interpretation. Most critically, robust early scaling was still induced when activity and TNFα signaling were blocked for 6 h but was prevented if TNFα signaling was first blocked for 24 h. Further, after scaling was induced, prolonged but not brief block of TNFα signaling could reverse it. Together, these two observations strongly suggest that neither brief nor prolonged scaling relies on enhanced TNFα signaling for scaling induction but rather that prolonged loss of TNFα signaling causes changes in neurons or synapses that no longer allow them to express or maintain synaptic scaling.
TNFα has pleotropic effects on neuronal function and can play both neurodegenerative and neuroprotective roles during brain injury and inflammation (Sriram and O'Callaghan, 2007). TNFα signaling via TNFR1 and TNFR2 can activate several signal transduction pathways, including the nuclear factor-κB, Jun N-terminal kinase, ceramide, and caspase pathways (Pickering et al., 2005). Differences in the levels and activation state of these receptors and their downstream effectors have been suggested to confer either neurotoxic or neuroprotective effects on TNFα (Botchkina et al., 1999). Here, we find similarly complex effects of TNFα on AMPA quantal amplitude. First, the ability of TNFα to enhance mEPSC amplitude is activity dependent and can be blocked by acute TTX. Even more surprisingly, when synapses were prescaled by prolonged TTX treatment, TNFα did not induce further scaling but instead decreased mEPSC amplitude back to control values. These observations demonstrate that the graded increase in mEPSC amplitude as a function of time during TTX treatment cannot be accounted for by a gradual enhancement of TNFα release. These state-dependent effects of TNFα on synaptic strength might contribute to the pleotropic effects of TNFα in the brain.
What role does constitutive TNFα signaling play in synaptic maintenance? Prolonged blockade of TNFα signaling (using sTNFR as we did here) has produced inconsistent effects on baseline mEPSC properties and surface AMPAR accumulation in different studies. A decrease in GluR1 surface expression following 24 h of sTNFR application has been reported (Beattie et al., 2002; Stellwagen et al., 2005), whereas effects on quantal transmission are inconsistent; Beattie et al. (2002) reported a modest decrease in mEPSC amplitude, whereas Stellwagen and Malenka (2006) saw no significant effect. In our hands, neither mEPSC amplitude nor synaptic accumulation of GluR1 and GluR2 were altered, and blocking TNFα signaling had no effect on overall synapse density or the expression levels of synaptic proteins. These somewhat subtle differences between studies may reflect differences in age or activity state of the culture preparations used, but in general it appears that baseline mEPSC amplitude is only modestly affected by prolonged block of TNFα signaling.
Chronic TTX treatment alters the synaptic levels of a number of scaffold proteins, including SAP102, PSD-95, and TARPs (Ehlers, 2003; Noritake et al., 2009). Blocking TNFα signaling does not reduce the dendritic accumulation of these proteins, suggesting that the block of scaling is not due to depletion of key scaffolds involved in synaptic scaling, although we cannot rule out changes in proteins we did not assay. We did observe a decrease in the fraction of excitatory presynaptic structures apposed to PSD95/TARP puncta and an increase in the fraction apposed to SAP102 puncta, suggesting an inverse regulation of the synaptic localization of PSD95 and SAP102. The synaptic localization of PSD95 and SAP102 is developmentally regulated (Elias and Nicoll, 2007), with SAP102 more strongly expressed at immature synapses and PSD95 more strongly expressed at mature synapses (Sans et al., 2000), raising the possibility that block of TNFα signaling may revert synapses to a less mature state. Although we do not know if these particular changes in PSD composition are related to the loss of scaling, these data do suggest that TNFα signaling plays a role in maintaining the composition of the postsynaptic density.
TNFα is expressed and released by astrocytes (Bezzi et al., 1998; Leonoudakis et al., 2004) and the TNFα release that is required for the expression of synaptic scaling arises from glia (Stellwagen and Malenka, 2006). Glia are clearly critical players at synapses, with a number of glial-derived factors reported to be involved in synapse formation, maturation, and stabilization (Mauch et al., 2001; Christopherson et al., 2005), as well as synaptic transmission and plasticity (Parpura et al., 1994; Zhang et al., 2003; Panatier et al., 2006). In addition to these other roles in synapse function, our data suggest that glial-derived TNFα release is critical for maintaining synapses in a plastic state in which homeostatic synaptic scaling can be expressed.
Footnotes
This research was supported by Fondation pour la Recherche Médicale (C.C.S.) and National Institutes of Health Grant RO1 NS36853 (G.G.T.). We thank B. Baxter and L. Wang for technical assistance.
References
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Botchkina GI, Geimonen E, Bilof ML, Villarreal O, Tracey KJ. Loss of NF-kappaB activity during cerebral ischemia and TNF cytotoxicity. Mol Med. 1999;5:372–381. [PMC free article] [PubMed] [Google Scholar]
- Burrone J, Murthy VN. Synaptic gain control and homeostasis. Curr Opin Neurobiol. 2003;13:560–567. doi: 10.1016/j.conb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Elias GM, Nicoll RA. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol. 2007;17:343–352. doi: 10.1016/j.tcb.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Gainey MA, Hurvitz-Wolff JR, Lambo ME, Turrigiano GG. Synaptic scaling requires the GluR2 subunit of the AMPA receptor. J Neurosci. 2009;29:6479–6489. doi: 10.1523/JNEUROSCI.3753-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Stellwagen D, Malenka RC, Stryker MP. Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron. 2008;58:673–680. doi: 10.1016/j.neuron.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis D, Braithwaite SP, Beattie MS, Beattie EC. TNFalpha-induced AMPA-receptor trafficking in CNS neurons; relevance to excitotoxicity? Neuron Glia Biol. 2004;1:263–273. doi: 10.1017/S1740925X05000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis D, Zhao P, Beattie EC. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci. 2008;28:2119–2130. doi: 10.1523/JNEUROSCI.5159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Noritake J, Fukata Y, Iwanaga T, Hosomi N, Tsutsumi R, Matsuda N, Tani H, Iwanari H, Mochizuki Y, Kodama T, Matsuura Y, Bredt DS, Hamakubo T, Fukata M. Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J Cell Biol. 2009;186:147–160. doi: 10.1083/jcb.200903101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Pickering M, Cumiskey D, O'Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–670. doi: 10.1113/expphysiol.2005.030734. [DOI] [PubMed] [Google Scholar]
- Pratt KG, Watt AJ, Griffith LC, Nelson SB, Turrigiano GG. Activity-dependent remodeling of presynaptic inputs by postsynaptic expression of activated CaMKII. Neuron. 2003;39:269–281. doi: 10.1016/s0896-6273(03)00422-7. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG. Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci. 1997;17:4527–4535. doi: 10.1523/JNEUROSCI.17-12-04527.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, 2nd, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, O'Callaghan JP. Divergent roles for tumor necrosis factor-alpha in the brain. J Neuroimmune Pharmacol. 2007;2:140–153. doi: 10.1007/s11481-007-9070-6. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Watt AJ, van Rossum MC, MacLeod KM, Nelson SB, Turrigiano GG. Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron. 2000;26:659–670. doi: 10.1016/s0896-6273(00)81202-7. [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Ibata K, Turrigiano GG. Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci. 2005;25:2895–2905. doi: 10.1523/JNEUROSCI.5217-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]