

Small molecules that perturb the function of their targets on fast-time scales can be powerful probes of dynamic cellular processes, such as intracellular transport.[1] In the past decade, inhibitors for motor proteins, ATPases that drive movements of cellular cargoes, have been reported.[2] These chemical inhibitors (with μM potency) have served as valuable tools to dissect complex cellular mechanisms and have even provided an impetus for developing chemotherapeutics that target motor proteins.[3] However, chemical inhibitors are available for only ~ 6% of the motor proteins (over 100 in humans[4]) involved in a variety of biological processes, ranging from development, hearing, intracellular signaling, and muscle function.
Myosins are motor proteins that move along the actin cytoskeleton (Fig. 1). Since their initial characterization as proteins that drive muscle contraction, eighteen different classes of myosins have been characterized and it is now known that myosins are involved in almost every aspect of biological motion.[5] However, specific small molecule probes are available only for class II myosins.[2b,c] Therefore, we have set our long-term goal to develop chemical probes for members of the other myosin classes.
Figure 1.

Schematic showing myosin V walking on actin filaments and structures of ‘privileged’ scaffold (bold)-based compounds as potential inhibitors (X=NH or CH2; R1–3 are various aliphatic or aromatic groups).
Analysis of myosin structures reveals that while these enzymes bind ATP, their structures are similar to that of GTPases.[6] Good inhibitors for GTPases have been generally difficult to obtain. We reasoned that the high nucleotide affinity (low nM), and not the structure of the nucleotide binding pocket itself, is a key factor limiting GTPase inhibitor identification. This has also been noted in the context of inhibitor development for kinases that have unusually high ATP affinity.[7] Myosins have a KM for ATP that is typically in the μM range,[8] raising the possibility that inhibitors for these enzymes may be more readily accessible.
To test this hypothesis, we focused on class V myosins. These motor proteins are essential for survival in eukaryotes and mutations impairing activity give rise to pigmentation and neurological defects in mice and humans.[9] Thus far, myosin V has been the focus of intense research examining its micromechanics and mechano-chemistry.[10] However, myosin V’s precise cellular functions remain poorly characterized, particularly in vertebrates, and a small molecule inhibitor would be a valuable tool.
We have recently shown that small molecules based on ‘privileged’ chemical scaffolds, which map to the region of chemical space occupied by known bioactive compounds, can yield diverse cellular phenotypes.[11] These data, along with other studies,[12] suggest that privileged scaffold-based compounds may provide efficient starting points for developing inhibitors of different target proteins. We noted that such scaffolds include pyrimidines (1), oxindoles (2), pyrrolopyrimidines (3) and pyrazolopyrimidines (4) (Fig. 1). We also noted that such scaffolds are common to many known kinase inhibitors.[13] The specificity of kinase inhibitors is typically examined in vitro against a large panel of known kinases. However, the ability of these inhibitors to target motor proteins has not been examined systematically. To determine if kinase inhibitors can inhibit myosin V, we assembled a small collection of known kinase inhibitors, focusing on various ‘privileged’ scaffolds (Fig. 1).
Myosin V exists as a multi-protein complex of over twelve polypeptides that possesses two catalytic ATPase motor domains, called heads, which bind actin filaments and generate force (Fig. 1). To test our compounds of interest, we used a recombinant protein comprised of a single ATPase motor domain of chicken Myosin Va. This construct, from a representative member of the Myosin V class, has been extensively characterized in vitro.[14] Maximum activation of myosin ATPase requires actin polymer. Using an enzyme-coupled steady-state ATPase assay,[15] we found that two compounds (5 and 6, Fig. 2) of 10 tested, reduced actin-activated enzyme activity by ~35–40% at 100 μM (Fig. 2).[16] Compound 5 is a known inhibitor of CHK1 kinase (IC50 7 nM)[13b] while compounds similar to 6 inhibit various cell cycle kinases (IC50 low μM).[13d]
Figure 2.
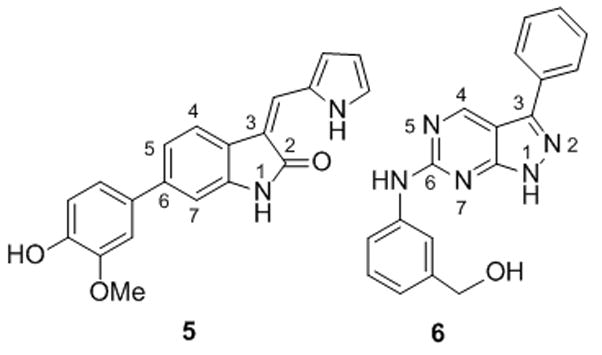
Initial hits 5 and 6 and their effects on the steady-state rate of actin-activated ATP hydrolysis by single-headed myosin V. (n=3, uncertainty bars= SD).
Encouraged by these data, we synthesized a series of compounds based on 5 and 6 to derive inhibitors that would have reduced activity towards kinases but have increased potency for myosin V.[16] To create structural diversity, we employed an interchangeable two-step sequence of Knoevenagel condensation and Suzuki coupling on commercially available oxindoles.[13b] For the synthesis of pyrazolopyrimidines analogues, a four-step sequence involving a metal-halogen exchange reaction, followed by hydrazine-mediated ring closure and nucleophilic aromatic substitution was adopted.[13d] A total of 60 compounds (39 oxindoles and 21 pyrazolopyrimidines) were obtained with >95% purity and characterized.[16]
Testing oxindole-based compounds indicated that while substitutions at position 6 had a modest effect on inhibitor potency, changes at postion 3 had significant effects, with a biaryl moiety being most favourable (see supporting information for SAR data). Further modification of the biaryl moiety led to 7, which is the most potent compound in this series (KI ~14 μM) (Fig. 3). Testing pyrazolopyrimidine-based compounds revealed that changes in substitutions at position 6 improved potency, with aminobenzylthiobenzoic acid (ABTA) moiety being favored. Our SAR study (see supporting information) indicated that a meta-meta substitution pattern of the ABTA provides highest potency. With this position fixed, changes at position 3 further improved potency, leading to the compound 8 as the most potent Myosin V inhibitors in each series (KI ~6 μM) (Fig. 3). To the best of our knowledge, such extended aromatic substitution on both oxindole and pyrazolopyrimidine has not been reported. Importantly, we could show that compound 8 also inhibits double-headed myosin V, an active construct that contains two catalytic domains and twelve associated light chains,[17] with equal efficiency as single-headed motor (KI = 5.5 ± 2.4 μM).[16]
Figure 3.
Myosin V inhibitors 7, 8 and 9. Dose-dependent reduction in the rate of steady-state actin-activated ATP hydrolysis rate (s−1head−1) by single-headed myosin V (n=3, uncertainty bars= SD).
It has been noted that protein aggregation is a common mechanism underlying promiscuous chemical inhibitors.[18] Detergent (e.g. 0.02–0.1% triton X) has been proposed to reduce aggregation and can be used in assays to exclude inhibitors that act via such mechanisms. The presence of 0.1% triton X in the assay buffer led to a ~10-fold increase in the KI value for compound 7, consistent with aggregation-based inhibition of myosin V.[16] In contrast, compound 8 (named MyoVin-1, for myosin V inhibitor-1) showed ~1.25 fold increase in KI in presence of 0.1% triton X.[16] This sensitivity to detergent is not likely to be significant for a hydrophobic compound and potentially arises from the increase in myosin V’s ATPase activity in the presence of detergent (data not shown).
In the course of our SAR studies we also identified compounds that are structurally similar to MyoVin-1, but significantly less potent inhibitors of myosin V.[16] One such compound 9 (Fig. 3), is a positional isomer of MyoVin-1, and is ~12-fold less active (KI=72±8 μM). These data indicate that the observed inhibition of myosin V by pyrazolopyrimidine-based compounds depends on specific features in the target protein, and are not merely a consequence of changes in the physical properties of the compound (e.g. solubility).
We next examined the specificity of MyoVin-1. The known SAR data for kinase inhibition by pyrazolopyrimidine-based compounds suggests that MyoVin-1 should have limited activity against kinases.[13d] We directly tested this using CDK1/cyclin B, a likely target for this chemical scaffold.[13d] In vitro kinase assays revealed that MyoVin-1 inhibited CDK1/cyclin B with at least 30-fold lower potency (Fig 4a, IC50 > 200 μM, precise measurement being limited by compound solubility in assay buffer). MyoVin-1 at 100 μM concentration did not significantly inhibit (<5–10%) many representative kinases including CHK1, PLK1, Abl kinase, p42 MAP kinase, Casein kinase II and Aurora kinase (Fig. 4a–g). Known inhibitors of these kinases efficiently suppressed kinase activities in our assay (Fig. 4a–g).[19]
Figure 4.

Analysis of MyoVin-1 (8) specificity against: a) CDK1, b) CHK1, c) PLK1, d) Abl kinase, e) p42 MAP kinase, f) casein kinase II, and g) Aurora kinase (n=3, uncertainty bars=SD.; DAP-81= Diaminopyrimidine-81,[11] Stauros.= straurosporine,[19a,b] CKII In= casein kinase II inhibitor,[19c] Hesp.= hesperadin[19d]).
We next examined the specificity of MyoVin-1 towards other myosins. As representative examples in other classes, we tested skeletal muscle myosin II and a non-muscle myosin, myosin VI. In in vitro ATPase assays we did not observed any measurable inhibition by MyoVin-1 at 50 μM (Fig. 5a, b), while blebbistatin, a known small-molecule inhibitor of class II myosins,[2c] significantly inhibited muscle myosin II (Fig. 5a). This specificity of MyoVin-1 towards myosin V is quite remarkable given that ATPase domains in members of the myosin superfamily are well conserved.[5a]
Figure 5.
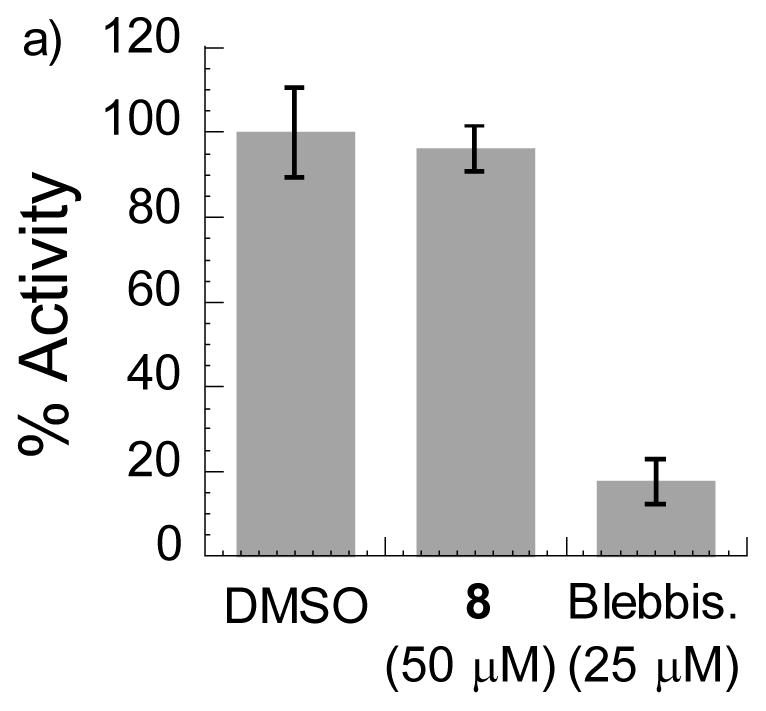
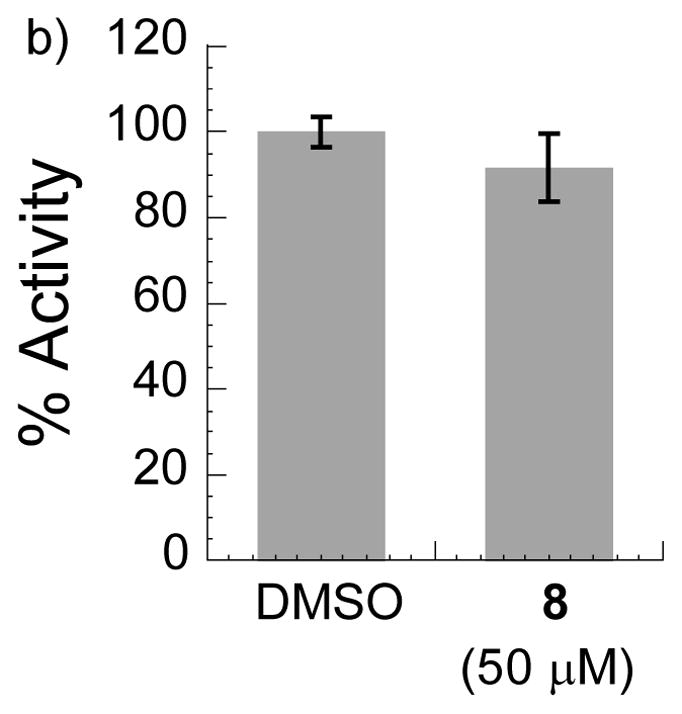
Analysis of MyoVin-1 (8) specificity against ATPase activities of: a) Myosin II, b) Myosin VI (n=3, uncertainty bars= SD).
To identify the mechanism of inhibition by MyoVin-1 and identify which ATPase cycle transition(s) are targeted, we performed a series of steady-state and transient kinetic experiments. MyoVin-1 lowers the maximum turnover rate (kcat) and apparent Michealis constant for actin (KM) (i.e. uncompetitive inhibition; Fig. 6a), which can be explained by a reduction in rate limiting ADP release from actomyosin, i.e. the complex of actin and myosin V.[15],[20] This was confirmed by measuring ADP release with a fluorescent methylanthraniloyl-nucleotide analogs (MANT-ATP/MANT-ADP).[21] ADP release from actomyosin-ADP prepared by equilibrating actomyosin and ADP is unaffected by myoVin-1 (data not shown). However, ADP release is inhibited by myoVin-1 if actomyosin V-ADP is prepared by mixing with ATP, indicating that myoVin-1 binds to an actomyosin V intermediate populated during ATPase cycling. When ADP release is measured in this way, myoVin-1 lowers the amplitude, but not the rate constant of ADP release (Fig. 6b), indicating that MyoVin-1 exchanges slowly on the timescale of ADP release (i.e. MyoVin-1 binding cannot be treated as a rapid equilibrium). A myoVin-1 affinity of 24±4 μM was obtained from the best fit of the [myoVin-1]-dependence of the amplitudes. The slightly weaker KI measured with this assay compared to steady-state kinetics is likely due to the use of a modified nucleotide analog.[21]
Figure 6.


Steady-state and transient kinetic analysis. a) Effect of 12 μM of MyoVin-1 (8) on steady-state rate of ATP hydrolysis by myosin V at various concentrations of actin (n=3, uncertainty bars= SD). b) Effect of MyoVin-1 (8) on mantADP release amplitude. Data points represent the normalized amplitudes of MANT-ADP release from actomyosin V-ADP. The solid line is the best fit to a rectangular hyperbola, and uncertainty bars represent standard errors of the best fits of dissociation time courses. In the absence of myoVin-1, a residual slow phase was observed as reported (Ampfast represents amplitude of the fast phase).[21]
These experiments indicate that myoVin-1 slows the actin-activated myosin V ATPase by specifically inhibiting ADP release from the actomyosin complex (Fig. 7). Single MANT-ATP turnover measurements[22] indicate that ATP binding and rate-limiting Pi release from myosin V in the absence of actin are unaffected by myoVin-1 (data not shown), consistent with myoVin-1 binding to the actomyosin complex. This represents a unique mechanism of inhibition of myosins by a chemical inhibitor. We anticipate that myoVin-1 will be a powerful tool to analyze motor protein mechanochemistry.
Figure 7.
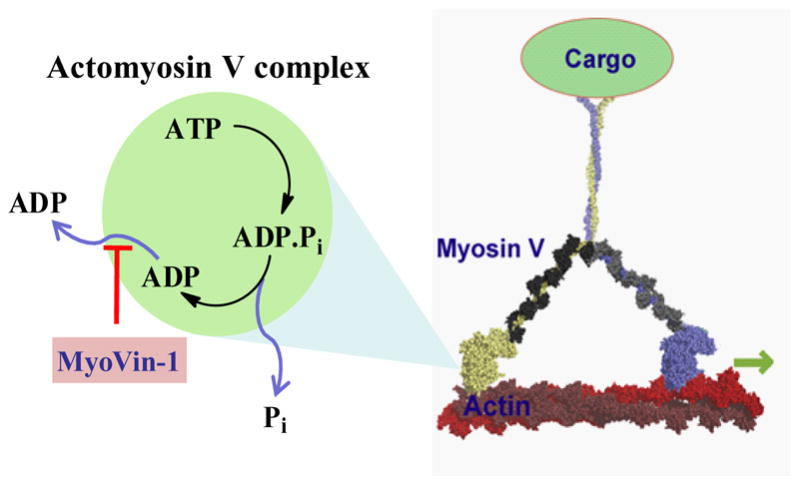
Schematic showing mechanism of inhibition of myoVin-1. MyoVin-1 inhibits ADP release from the actomyosin complex.
In summary, starting from a collection of privileged chemical scaffolds, we developed a selective myosin V inhibitor that does not directly compete with nucleotide binding. It is noteworthy that the potency of MyoVin-1 is comparable to that of other known motor protein inhibitors that are extensively used as chemical biology probes.[2a,c] There are many reported examples suggesting that small changes in an inhibitor’s chemical structure can alter its mechanism of action. In particular, very minor changes in the chemical structure of GSK923295, an inhibitor of CENP-E, a microtubule-based motor protein that like myosin has a GTPase-like fold, changes its mechanism of inhibition from ATP-uncompetitive to ATP-competitive.[23] Therefore, it is possible that the initial pyrazolopyrimidines we tested may be ATP-competitive, as they are for kinases, and the SAR-guided changes we introduced to obtain MyoVin-1 altered the binding mode and inhibitory mechanism. Further structural studies will be needed to determine if MyoVin-1 binds near the nucleotide-binding pocket, or at a remote site. Overall, we expect that the strategy we have used to identify MyoVin-1 should not only be applicable to other myosin superfamily members, but may be an attractive entry-point into the inhibitor-discovery cycle, complementing high-throughput screening of large compound libraries. This is particularly useful when protein supply is limited or when assays are complex and involve multiple components (e.g. myosins), as fewer compounds need to be tested in the primary screens. In addition, our findings also suggest that analysis of kinase inhibitor specificity should not be limited to members of this superfamily, and off-target effects resulting from the inhibition of proteins with significant structural divergence, such as motor proteins, also need to be systematically tested. While examples of inhibitor design through ‘scaffold hopping’ have been reported in the literature,[24] our work presents the first example of using kinase inhibitors to develop compounds that target a protein with a GTPase-like fold.
Supplementary Material
Footnotes
This work was supported by NIH (GM71772 and GM65933 to T.M.K.; GM071688 and GM071688-03S1 to E.M.D.L.C.). TMK is a Scholar of the Leukemia and Lymphoma Society. HFC was supported by NIH predoctoral fellowship (F31 DC009143) and, in part, by a grant from the Yale Institute for Quantum Engineering (awarded to E.M.D.L.C.). EMDLC is an American Heart Association Established Investigator (0940075N) and an NSF-CAREER Award recipient (MCB-0546353). We thank Drs. Benjamin H. Kwok and Alexander Kelly for providing us recombinant PLK1 and Aurora kinase.
Contributor Information
Dr. Kabirul Islam, Laboratory of Chemistry and Cell Biology, Rockefeller University, 1230 York Avenue, New York, New York 10065 (USA)
Harvey F. Chin, Department of Molecular Biophysics and Biochemistry, Yale University, 260 Whitney Avenue, New Haven, Connecticut 06520 (USA)
Adrian O. Olivares, Department of Molecular Biophysics and Biochemistry, Yale University, 260 Whitney Avenue, New Haven, Connecticut 06520 (USA).
Lauren P. Saunders, Department of Molecular Biophysics and Biochemistry, Yale University, 260 Whitney Avenue, New Haven, Connecticut 06520 (USA)
Prof. Dr. Enrique M. De La Cruz, Department of Molecular Biophysics and Biochemistry, Yale University, 260 Whitney Avenue, New Haven, Connecticut 06520 (USA).
Prof. Dr. Tarun M. Kapoor, Laboratory of Chemistry and Cell Biology, Rockefeller University, 1230 York Avenue, New York, New York 10065 (USA).
References
- 1.a) Peterson JR, Mitchison TJ. Chem Biol. 2002;9:1275–1285. doi: 10.1016/s1074-5521(02)00284-3. [DOI] [PubMed] [Google Scholar]; b) Schreiber SL. Chem Eng News. 2003;81:51–61. [Google Scholar]
- 2.a) Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]; b) Cheung A, Dantzig JA, Hollingworth S, Baylor SM, Goldman YE, Mitchison TJ, Straight AF. Nat Cell Biol. 2001;4:83–88. doi: 10.1038/ncb734. [DOI] [PubMed] [Google Scholar]; c) Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]; d) Bergnes G, Brejc K, Belmont L. Curr Top Med Chem. 2005;5:17–145. doi: 10.2174/1568026053507697. [DOI] [PubMed] [Google Scholar]
- 3.Lampson MA, Kapoor TM. Nat Chem Biol. 2006;2:19–27. doi: 10.1038/nchembio757. [DOI] [PubMed] [Google Scholar]
- 4.Vale RD. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 5.a) Sellers JA. Myosins. 2. Oxford university press; Oxford: 1999. [Google Scholar]; b) Krendel M, Mooseker MS. Physiology. 2005;20:239–251. doi: 10.1152/physiol.00014.2005. [DOI] [PubMed] [Google Scholar]
- 6.a) Smith CA, Rayment I. Biophysical J. 1996;70:1590–1602. doi: 10.1016/S0006-3495(96)79745-X. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vale RD. J Cell Biol. 1996;135:291–302. doi: 10.1083/jcb.135.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knight ZA, Shokat KM. Chem Biol. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 8.a) Hackney DD, Clark PK. J Biol Chem. 1985;260:5505–5510. [PubMed] [Google Scholar]; b) Watanabe S, Mabuchi K, Ikebe R, Ikebe M. Biochemistry. 2006;45:2729–2738. doi: 10.1021/bi051682b. [DOI] [PubMed] [Google Scholar]
- 9.a) Reck-Peterson SL, Provance DW, Jr, Mooseker MS, Mercer JA. Biochim Biophys Acta. 2000;1496:36–51. doi: 10.1016/s0167-4889(00)00007-0. [DOI] [PubMed] [Google Scholar]; b) Trybus KM. Cell Mol Life Sci. 2008;65:1378–1389. doi: 10.1007/s00018-008-7507-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Vale RD. J Cell Biol. 2003;163:445–450. doi: 10.1083/jcb.200308093. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sellers JR, Veigel C. Curr Opin Cell Biol. 2006;18:68–73. doi: 10.1016/j.ceb.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Peters U, Cherian J, Kim JH, Kwok BH, Kapoor TM. Nat Chem Biol. 2006;2:618–626. doi: 10.1038/nchembio826. [DOI] [PubMed] [Google Scholar]
- 12.a) Horton DA, Bourne GT, Smythe ML. Chem Rev. 2003;103:891–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; b) Muller G. Drug Discovery Today. 2003;8:681–691. doi: 10.1016/s1359-6446(03)02781-8. [DOI] [PubMed] [Google Scholar]; c) Bon RS, Waldmann H. Acc Chem Res. 2010 doi: 10.1021/ar100014h. [Epub. ahead of print] [DOI] [PubMed] [Google Scholar]
- 13.a) Breault GA, Ellston RPA, Green S, James SR, Jewsbury PJ, Midgley CJ, Pauptit RA, Minshull CA, Tucker JA, Pease JE. Bioorg Med Chem Lett. 2003;13:2961–2966. doi: 10.1016/s0960-894x(03)00203-8. [DOI] [PubMed] [Google Scholar]; b) Nan-Horng L, Ping X, Peter K, Chang P, Zehan C, Haiying Z, Rosenberg SH, Sham HL. Bioorg Med Chem Lett. 2006;16:421–426. doi: 10.1016/j.bmcl.2005.09.064. [DOI] [PubMed] [Google Scholar]; c) Choi H-S, Wang Z, Richmond W, He X, Yang K, Jiang T, Sim T, Karanewsky D, Gu X-J, Zhou V, Liu Y, Ohmori O, Caldwell J, Gray N, He Y. Bioorg Med Chem Lett. 2006;16:2173–2176. doi: 10.1016/j.bmcl.2006.01.053. [DOI] [PubMed] [Google Scholar]; d) Ding Q, Jiang N, Roberts JL. 277655 US Pat Appl Publ. 2005
- 14.De La Cruz EM, Wells AL, Rosenfeld SS, Ostap EM, Sweeney HL. Proc Natl Acad Sci U S A. 1999;96:13726–13731. doi: 10.1073/pnas.96.24.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De La Cruz EM, Sweeney HL, Ostap HM. Biophys J. 2000;79:1524–1529. doi: 10.1016/S0006-3495(00)76403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For details, see supporting information.
- 17.Olivares AO, Chang W, Mooseker MS, Hackney DD, De La Cruz EM. J Biol Chem. 2006;281:31326–31336. doi: 10.1074/jbc.M603898200. [DOI] [PubMed] [Google Scholar]
- 18.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 19.a) Gadbois DM, Hamaguchi JR, Swank RA, Bradbury EM. Biochem Biophys Res Commun. 1992;184:80–85. doi: 10.1016/0006-291x(92)91160-r. [DOI] [PubMed] [Google Scholar]; b) Karaman MW, et al. Nat Biotechnology. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]; c) Sarno S, Reddy H, Meggio M, Ruzzene M, Davies SP, Donella-Deana A, Shugar D, Pinna LA. FEBS Lett. 2001;496:44–48. doi: 10.1016/s0014-5793(01)02404-8. [DOI] [PubMed] [Google Scholar]; d) Hauf S, Cole RW, La Terra S, Zimmer C, Zchnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De La Cruz EM, Wells AL, Sweeney HL, Ostap EM. Biochemistry. 2000;39:14196–14202. doi: 10.1021/bi001701b. [DOI] [PubMed] [Google Scholar]
- 21.Hannemann DE, Cao W, Olivares AO, Robblee JP, De La Cruz EM. Biochemistry. 2005;44:8826–8840. doi: 10.1021/bi0473509. [DOI] [PubMed] [Google Scholar]
- 22.De La Cruz EM, Ostap HM. Methods enzymol. 2009;455:157–192. doi: 10.1016/S0076-6879(08)04206-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wood KW, et al. Proc Natl Acad Sci U S A. 2010;107:5839–5844. doi: 10.1073/pnas.0915068107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winssinger N, Barluenga S, Karplus M. 2008/021213. WO. 2008
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.