

Abstract
Techniques of tissue engineering and cell and molecular biology were used to create a biodegradable scaffold for transfected cells to produce complex proteins. Mullerian Inhibiting Substance (MIS) causes regression of Mullerian ducts in the mammalian embryo. MIS also causes regression in vitro of ovarian tumor cell lines and primary cells from ovarian carcinomas, which derive from Mullerian structures. In a strategy to circumvent the complicated purification protocols for MIS, Chinese hamster ovary cells transfected with the human MIS gene were seeded onto biodegradable polymers of polyglycolic acid fibers and secretion of MIS confirmed. The polymer-cell graft was implanted into the right ovarian pedicle of severe combined immunodeficient mice. Serum MIS in the mice rose to supraphysiologic levels over time. One week after implantation of the polymer-cell graft, IGROV-1 human tumors were implanted under the renal capsule of the left kidney. Growth of the IGROV-1 tumors was significantly inhibited in the animals with a polymer-cell graft of MIS-producing cells, compared with controls. This novel MIS delivery system could have broader applications for other inhibitory agents not amenable to efficient purification and provides in vivo evidence for a role of MIS in the treatment of ovarian cancer.
Keywords: tissue engineering, Mullerian Inhibiting Substance
It has been known for decades that genes coding for secreted proteins can be transfected into cells and the proteins produced in vitro. A more recent development uses techniques of tissue engineering to grow cells on a support scaffold for implantation (1). We combine these two techniques to make a device that continually produces a protein that could be used to treat a wide variety of medical problems, including cancers. This technology could circumvent the need for protein purification and would be particularly useful for complex molecules requiring parenteral delivery. Mullerian Inhibiting Substance (MIS), a molecule being considered for the treatment of ovarian and other reproductive cancers, is a model for such a delivery system. To test this possibility, we used MIS-transfected cells grown on polymer scaffolds to produce and deliver MIS, and showed that MIS inhibits growth of an ovarian cancer cell line in vivo.
The antiproliferative activity of certain naturally occurring molecules suggests their use as chemotherapeutic agents. For example, members of the transforming growth factor-β family regulate tissue growth and differentiation (2, 3). MIS is a member of this family, which includes Inhibin (4), Activin (5, 6), and bone morphogenesis proteins (7) among others. MIS is a sexually dimorphic glycoprotein homodimer of 70-kDa disulfide-linked subunits (8, 9). In males, MIS is a fetal and postnatal Sertoli cell product that causes regression of embryonic Mullerian ducts, the forerunners of the female reproductive tract (10). Its presence in males after sexual differentiation and in adult females suggests additional roles for MIS. MIS is involved in growth-regulation in the ovary because it inhibits proliferation of and steroid production by human granulosa-luteal cells in vitro (11) and of oocyte meiosis in the rat (12). Even more compelling is the ablation of the ovary in transgenic mice producing high levels of MIS (13).
Because the most common human ovarian cancers are derived from the coelomic epithelium (14) and because MIS causes regression of the precursor of this tissue, it follows that MIS may block growth of ovarian tumors (15, 16). Epithelial ovarian cancer is a common and highly lethal malignancy that is often diagnosed after metastases have occurred. Despite advances in treatment, mortality remains high (17). Most patients with advanced disease recur after standard therapy (18). There are limited treatment options for recurrences. MIS could offer a new therapeutic approach for this disease.
Bovine MIS inhibited the growth of a large number of primary ovarian, Fallopian tube, and uterine carcinomas obtained at surgery (19) as well as an ovarian cancer cell line (20). Once the human MIS gene was cloned (21), purified human MIS was produced (22), and it inhibited human ovarian cancer cell lines of Mullerian origin (23). The MIS type II receptor (24, 25) is expressed in human ovarian cancer cell lines, in ascites cells from patients with stage III or IV ovarian papillary serous cystadenocarcinoma (26), and in a cell line derived from coelomic epithelium (27). MIS inhibited each of these cell lines in vitro. These observations justify more extensive preclinical in vivo testing of MIS against human ovarian cancer.
Pharmacologic MIS delivery requires large quantities of purified recombinant protein. To provide an alternative MIS source, we adapted a technology that employs a biodegradable polyglycolic acid polymer (28, 29) as a scaffold for cells transfected with the human MIS gene. Synthetic materials such as lactic or polyglycolic acid are often used as substrates for mammalian cells in tissue engineering (1) and provide a structure on which the MIS-producing cells can proliferate. The IGROV-1 human ovarian cancer cell line that contains the MIS receptor and is growth inhibited by MIS in vitro (26) was implanted in the subrenal capsule of immunosuppressed mice containing the MIS-secreting polymer to test the in vivo bioactivity of MIS delivered in this manner.
It is conceivable that MIS, alone or in addition to existing therapy for ovarian cancer, could be delivered clinically by such an innovative, minimally invasive system. Furthermore, this technology could be expanded to a large number of complex proteins to control abnormal tissue growth.
Materials and Methods
Animals.
Female severe combined immunodeficient (SCID, ref. 30) or athymic nude mice (6 wk old, 18–20 g) were obtained from the Edwin L. Steele Laboratory (Massachusetts General Hospital, Boston, MA). All experiments conformed to Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines and were approved by the Review Board-Institutional Animal Care and Use Committee.
Cells.
The IGROV-1 human ovarian cancer cell line (American Type Cell Culture) was maintained in DMEM with glutamine, penicillin, streptomycin, and 10% MIS-free female FCS. Cells were grown at 37°C in a humidified chamber in 5% CO2 in air. At 80% confluency, cells were passed 1:4 for up to 12 passages.
The CHO-B9 cell line contains the normal human MIS gene as described (21) and a mutant MIS gene that produces a bioinactive form of MIS is in CHO-L9 cells (31). Both lines were maintained in DMEM with glutamine, penicillin, streptomycin, methotrexate, and 5% MIS-free female FCS.
Subrenal Capsule Assay.
Following established methods (32–34), the IGROV-1 tumor cell line was tested for growth in vivo in a subrenal capsule assay. Ten million cells were mixed with 300 μg of fibrinogen in PBS, pH 7.4, and 0.16 units of thrombin in double-strength DMEM and incubated at 37°C for 15 min. The cell clot was cut into 50 fragments (200,000 cells each). Selected fragments were dissolved with trypsin and cells counted to confirm uniformity of cell number.
Following ketamine/xylazine (100/10 mg/kg body weight, i.m.) anesthesia, a subcapsular space was developed in the left kidney with a 19-gauge needle trocar, and a cell clot of approximately 1 × 1 ocular micrometer units was introduced. The longest diameter (L1) of the implant and the diameter perpendicular to the longest diameter (W1) were measured with the ocular micrometer of a dissecting microscope. The graft size was estimated as L1 × W1 × W1. After 2–3 wk in vivo, tumors were remeasured at the same focal distance as the initial measurement to calculate the graft size ratio ([L2 × W2 × W2]/[L1 × W1 × W1]). Tumor growth was measured at weekly intervals and the duration of the experiment chosen so controls grew 3- to 4-fold.
Preparation of Polymer-Cell Graft and in Vitro Production of MIS.
Biodegradable polymer of 1-mm sheets of nonwoven fibers of polyglycolic acid (density 70 mg/cc, fiber diameter 14 μm, and average pore size 250 μm) was obtained from Smith and Nephew (York County, U.K.). Then, 0.5-cm2 squares were placed in 12-well tissue culture plates (Costar), sterilized with 95% ethanol, and washed with PBS. Sterile filtered 1 M NaOH was added for 60 s to make the polymer hydrophilic and the polymer washed with distilled water and coated with collagen (Vitrogen 0.0 3 mg/ml in sterile PBS) for 1 h at room temperature. The polymer squares were further washed with sterile PBS. Then, 1–2 × 106 CHO cells suspended in DMEM with 10% female FCS were seeded onto each polymer square and adsorbed on the interstices of the polymer for 1–2 h at 37°C in 5% CO2 in air. Fresh medium was added and the incubation continued for 3–7 days. MIS in the media was measured serially by using an ELISA (35), and an MIS-specific organ culture assay (36) was used to assess MIS bioactivity.
Implantation of Polymer-Cell Graft and IGROV-1 Tumor into SCID Mice (Fig. 1).
Figure 1.

Time line of the experimental protocol. On day 0, cells are seeded onto the polymer. Then, 3–7 days later, the polymer graft is implanted on the right ovarian pedicle. On day 10–14, the IGROV-1 cell clot is implanted under the left renal capsule. Then, 2–3 wk later, the tumor sizes are measured. The kidney and the ovarian pedicle are removed for immunohistochemical and/or histologic analysis. Serum is collected throughout the protocol to measure MIS.
Three to seven days after seeding with CHO-B9 or CHO-L9 cells, a 5 × 5 × 1-mm polymer square was implanted into the right ovarian pedicle of SCID mice by using existing methods (37). After ketamine-xylazine anesthesia, a 1-cm incision was made in the right flank. The polymer-cell graft was sutured onto the ovarian pedicle with 6-0 prolene. Six to thirteen days later, serum MIS was measured and, as it reached supraphysiologic levels, a fibrin/thrombin cell clot containing the IGROV-1 cells was prepared and implanted under the left renal capsule as described above. Different sized polymers (0.125, 0.25, 0.5, and 1.0 cm2) seeded with CHO-B9 cells were implanted in SCID mice and serum MIS levels measured to determine the optimal size of the polymer implant. Two to three weeks after implantation of the IGROV-1 cell clot, the tumors were measured. The graft size ratio was calculated and comparisons made between animals receiving the B9, L9, or empty polymer. Also, the polymer was measured examined by immunohistochemistry or routine histology. The animals implanted with CHO-B9 seeded polymer were the experimental group and compare with the controls implanted with CHO-L9 seeded or empty polymer.
Serum MIS and Bioassay.
MIS was measured after polymer implantation by using a human MIS-specific ELISA (35). MIS-containing serum was placed in the MIS organ culture assay (36) to correlate the bioactivity of the MIS in serum with the ELISA values.
Tissue Analysis.
The tissue formed from the cell-polymer implant and IGOV-1 cell clot were fixed in 5% picric acid and 15% formalin in PBS. Eight-micrometer sections were stained for routine histological analysis or for immunohistochemistry (38). Viability of the tumors was confirmed because they lacked central necrosis or an inflammatory infiltrate. Selected ovarian pedicles were harvested and examined histologically to determine the rate of biodegradation of the polymer.
Statistics.
Graft-size ratios are expressed as mean +/− SEM. An unpaired t test performed by STATVIEW and ANOVA performed by EXCEL were used to determine statistical significance.
Results
Production of MIS.
The polymer-cell grafts were incubated in vitro for 3–7 days, at which time media MIS levels reached 100–400 ng/ml and the grafts implanted into ovarian pedicles. Serum MIS was measured in animals implanted with different sized polymer squares (Fig. 2A). MIS levels in animals implanted with polymers measuring 1.0 (n = 4) or 0.5 (n = 4) cm2 were supraphysiologic at 2 wk post implantation and exceeded 1 μg/ml 1 wk later. In animals with squares measuring 0.25 (n = 4) and 0.125 (n = 4) cm2, the MIS levels were supraphysiologic by 3 wk after implantation. Then, 0.5 cm2 was selected as the optimal size of seeded graft for implantation. The CHO cell line did not grow in animals made immunosuppressed by inactivation of the RAG-2 gene; hence, SCID mice were used.
Figure 2.

(A) MIS in serum is influenced by polymer implant size. Polymer squares were seeded with MIS-producing cells 3 days before implantation. After 21 days, marked differences in mean MIS serum (n = 4 per polymer size) were seen in the animals implanted with polymers of varying sizes. Then, 0.5 cm2 grafts were used for further study. (B) Increasing serum MIS was detected on days 7, 14, 21, and 28 in SCID mice after the CHO-B9 seeded polymer was implanted. This composite figure shows the mean +/− the SEM for 40 animals. (C) The accumulation of MIS in the serum of polymer-bearing mice is reversible. Two weeks after implantation, serum MIS was approached 1200 ng/ml. Upon polymer removal, MIS falls to undetectable levels; thus there is no migration of cells from the implantation site. This figure is representative of six experiments.
Serum MIS was measured at 7, 14, 21, and 28 days after implantation of 0.5 cm2 a polymer-cell graft (n = 40). At 14 days, MIS was 100–500 ng/ml, and, at 28 days, the levels reached 7–10 μg/ml (Fig. 2B). Upon removal of the polymer, serum levels were undetectable within 7 days (n = 6) (Fig. 2C).
Sera from mice with high MIS levels were analyzed in the MIS bioassay to determine the bioactivity of the MIS produced by the implant. Complete regression of the Mullerian duct was achieved, indicating that the MIS in the serum of the animals retained bioactivity (Fig. 3).
Figure 3.
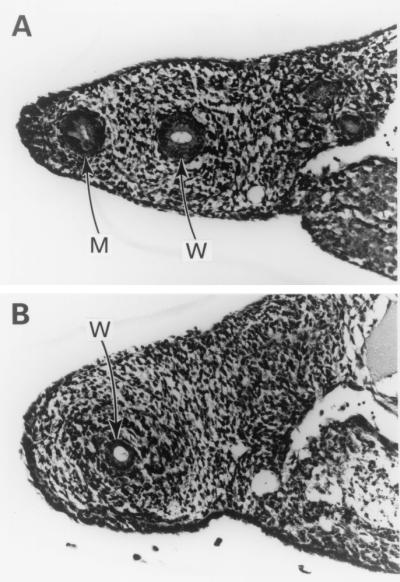
MIS produced in vivo is bioactive. MIS in serum from the animals with a CHO-B9 polymer-cell graft causes complete regression of the rat Mullerian duct, leaving only the Wolffian duct (W) (A). Control cultures show Mullerian (M) and Wolffian (W) ducts (B). (Magnification, ×250.)
After 2 wk in vivo, the polymer-cell graft formed a firm, living mass of tissue throughout the polymer fibers, which began resorbing (Fig. 4A). After 4 wk, the biodegradable polymer had disappeared and the cell graft grew into a rounded, well-contained mass with an approximate diameter of 1.0 cm (Fig. 4B). There was no evidence of spread of CHO-B9 cells beyond the pedicle during the experiment. The tissue mass consisted of epithelial cells with evidence of ingrowth of blood vessels from the ovarian pedicle. Immunohistochemical analysis confirmed the cells growing on the polymer continued to synthesize MIS (Fig. 5).
Figure 4.

(A) Two weeks after implantation of polymer-cell graft in the ovarian pedicle (OP), the CHO-B9 cells form a solid mass on the resorbing polymer fibers (P). (B) Four weeks following implantation, the graft is a mass of CHO-B9 cells incorporated in the ovarian pedicle (OP) with no remaining polymer. (Magnification, ×250.)
Figure 5.

Immunohistochemistry of the polymer-cell graft after 4 wk in vivo shows production of MIS by the cells. The polymer-cell graft stained with a human MIS antibody (A) is in marked contrast to the staining pattern seen using a control antibody (B). (Magnification, ×250.)
Inhibition of Tumor Growth by MIS Produced by Polymer-Cell Implant.
The IGROV-I cells formed measurable tumors in the subrenal capsule of SCID mice, reaching a graft-size ratio of 3–4 three weeks after implantation. Histologic analysis showed well-formed growths with neovascularization from the underlying kidney. There was minimal necrosis and inflammatory infiltrate (Fig. 6). The IGROV-1 tumor cell line failed to grow in athymic nude mice; hence, SCID mice were selected for subsequent experiments.
Figure 6.

(A) IGROV-1 tumor (T) implanted under the renal capsule of a SCID mouse after 3 wk in vivo shows neovascularization (V) and minimal necrosis or inflammation (K = kidney). (Magnification, ×250). (B) IGROV-1 tumor (T) after 3 wk of MIS exposure is about one-third the size of the control (A) and has minimal necrosis or inflammation. (Magnification, ×250).
Animals implanted with the bioactive MIS-producing CHO-B9 polymer (n = 8) showed very little net growth of the IGROV-1 implant, achieving a mean graft-size-ratio 1.62 +/− 0.218 SEM. Tumors implanted into the bioinactive MIS-producing CHO-L9 polymer-bearing mice (n = 10) achieved a mean graft-size-ratio of 3.25 +/− 0.506. This difference was statistically significant (P = 0.016, Fig. 7A).
Figure 7.

MIS inhibits the growth of the human ovarian tumor cell line IGROV-1 in vivo. (A) The mean graft-size-ratio (GSR) +/− the SEM of the IGROV-1 tumors in the animals producing bioactive MIS (n = 8) was significantly smaller than the tumors exposed to bioinactive MIS (n = 10) (P = 0.016). A GSR of 1 indicates no net growth. (B) The mean GSR of the IGROV-1 tumors implanted in animals with MIS-producing CHO-B9 polymer (n = 30) was significantly smaller than the IGROV-1 tumors in the animals with empty polymer (n = 30) (P < 0.001).
Three experiments were performed in which animals were implanted with the bioactive MIS-producing CHO-B9 polymer or empty polymer. In the first experiment, the graft-size-ratios of the IGROV-1 tumors were 1.323 +/− 0.168 (n = 10) in the animals with CHO-B9 polymer and 3.427 +/− 0.682 (n = 10) in those with polymer alone. In the second, the graft-size-ratio of tumors in CHO-B9 animals and polymer alone were 2.025 +/− 0.198 (n = 10) and 3.024 +/− 0.454 (n = 10), respectively. In the third experiment, the ratios were 1.427 +/− 0.147 (n = 10) in the MIS-treated group and 2.447 +/− 0.375 in the controls (n = 10). The average graft-size ratios for the three studies combined were 1.592 +/− 0.112 in the CHO-B9 polymer animals and 2.966 +/− 0.299 in the animals with empty polymer (Fig. 7B). The difference was statistically significant (P < 0.001).
Discussion
We designed a novel system to deliver in vivo a naturally occurring molecule known to inhibit human ovarian cancers in vitro, and tested the efficacy of the protein in vivo. Ovarian cancer is a devastating and often lethal disease. Current therapeutic options include surgery and chemotherapy. Unfortunately, the disease is often in an advanced stage at the time of diagnosis and surgery is rarely curative. Chemotherapy results in an initial tumor response, but the overall 5-yr mortality rate for stage III and IV ovarian cancer remains high at 70% (17, 39).
The majority of ovarian cancers derive from the coelomic epithelium, which forms Mullerian ducts in the embryo. Because MIS ablates the Mullerian duct, we reasoned that it might control the growth of ovarian tumors. Our laboratory reported inhibition of human ovarian carcinoma cell lines (15, 23, 26, 40) and of primary human ovarian tumors (19, 26) by MIS. The normal ovary is an MIS target (12) and expresses MIS type II receptor (24, 41). Human ovarian cancer cell lines such as IGROV-1 and ascites cells from patients with stage III or IV ovarian cancers also express MIS type II receptor and are growth inhibited by MIS (26, 27). In the present study, we describe in vivo inhibition of IGROV-1 cell growth.
The MIS delivery system employs tissue engineering, gene transfer, and cell transplantation. Tissue engineering is an interdisciplinary field where principles of chemical engineering and cell biology are combined to create new organs or tissue. Most approaches use cells combined with synthetic, biodegradable polymers (42, 43). The polymer is a support scaffold for three-dimensional growth of cells, and it allows for neovascularization and the continued diffusion of nutrients and waste products. As cells grow, the polymer degrades, leaving behind no toxic or immunogenic material. In addition to forming new organs, tissue engineering could also generate tissues of genetically manipulated cells that secrete bioactive, therapeutic molecules.
Biodegradable polymer fibers support the growth of genetically modified cells in vivo (28). Cells in the present study were transfected with the human MIS gene, seeded onto biodegradable polyglycolic acid fibers, and produced biologically active MIS in vitro and in vivo (Fig. 2 A and B, and Fig. 3). There were no adverse effects because of the presence of the polymer, cells, or high levels of MIS. Removal of the polymer-cell graft resulted undetectable levels of MIS (Fig. 2C), suggesting no spread of cells from the ovarian pedicle. MIS responsive cells implanted in mice containing the MIS secreting graft were growth inhibited compared with tumors implanted in animals with a polymer secreting biologically inactive MIS or no MIS (Fig. 7). Thus, only bioactive MIS and not a CHO cell product or biopolymer component is responsible for the growth inhibition. Histologic analysis confirmed three-dimensional tumor growth and the lack of necrosis or inflammation that could alter graft size (Fig. 6).
To be used clinically, MIS must be administered in a safe and cost-effective manner in sufficient quantities to achieve tumor inhibition. MIS is a complex molecule consisting of 70-kDa subunits that require cleavage for biological activity; therefore, production and delivery of purified MIS is complicated and costly. An alternative to production of the purified protein is the use of biodegradable polymer seeded with MIS-producing cells that can deliver biologically active MIS in quantities sufficient to achieve tumor inhibition.
The present work tests proof of principle for the device by using a partially transformed Chinese hamster ovary cell line that is tumorigenic in immunosuppressed mice. Ongoing work uses transfected fibroblasts, which grow on the polymer (M. Streit and A.E.S., unpublished observations). To avoid immunosuppression, we envision a system where the patients' own fibroblasts are transfected with the human MIS gene. The cells would be grown on the polymer in vitro and implanted in the patient, providing continual production of MIS. The efficacy of MIS in suppressing the growth of a human cancer cell line in vivo encourages the development of this technology for other complex molecules requiring parenteral delivery. In addition, the results provide further proof that MIS has potential clinical utility in vivo. This system need not be limited to the treatment of cancers. Disorders resulting from a lack of secreted proteins could also be amenable to such a delivery system. It may even be possible to regulate the transcription of such genes exogenously. These strategies could make complex molecules available for patients for replacement or therapeutic needs.
Acknowledgments
We thank members of the Edwin L. Steele Laboratory at the Massachusetts General Hospital, in particular Dr. Herman Suit, Dr. Rakesh Jain, and Sylvie Roberge for access to the animal colony and their technical expertise. We thank Thomas Manganaro for the tissue processing. We thank Drs. Robert S. Langer and Ross S. Berkowitz for critically reviewing the manuscript. A.E.S. is supported by National Institutes of Health (CA71345-04) and the Marshall K. Bartlett Fellowship from the Department of Surgery, Massachusetts General Hospital. D.L.S. is supported by the National Institutes of Health National Cancer Institute Training Grant in Cancer Biology (F32 CA77945-01A1) and Resident Research Award from the American College of Surgeons. This work was also funded by National Institutes of Health, R01 CA17393 (to P.K.D. and D.T.M.) and the Department of Defense 1200-202487 (to J.P.V.) and DAMD17-99-2-9001 (to D.T.M.). P.T.M. was supported by National Cancer Institute Grant T32 CA71345, Resident Research Award from the American College of Surgeons, and the Department of Surgery, Boston University School of Medicine. We would also like to acknowledge the Center for Innovative Minimally Invasive Therapy (Boston, MA) for their support.
Abbreviations
- MIS
Mullerian Inhibiting Substance
- SCID
severe combined immunodeficient
References
- 1.Vacanti J P, Langer R. Lancet. 1999;354:32–34. doi: 10.1016/s0140-6736(99)90247-7. [DOI] [PubMed] [Google Scholar]
- 2.Massague J, Chen Y G. Genes Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- 3.Derynck R, Jarrett J A, Chen E Y, Eaton D H, Bell J R, Assoian R K, Roberts A B, Sporn M B, Goeddel D V. Nature (London) 1985;316:701–705. doi: 10.1038/316701a0. [DOI] [PubMed] [Google Scholar]
- 4.Mason A J, Hayflick J S, Ling N, Esch F, Ueno N, Ying S X, Guillemin R, Niall H, Seeburg P H. Nature (London) 1985;318:659–663. doi: 10.1038/318659a0. [DOI] [PubMed] [Google Scholar]
- 5.Ling N, Ying S Y, Ueno N, Shimasaki S, Esch F, Hotta M, Guillemin R. Nature (London) 1986;321:779–782. doi: 10.1038/321779a0. [DOI] [PubMed] [Google Scholar]
- 6.Vale W, Rivier J, Vauhgan J, McClintock R, Corrigan A, Woo W, Karr D, Spiess J. Nature (London) 1986;321:776–779. doi: 10.1038/321776a0. [DOI] [PubMed] [Google Scholar]
- 7.Wozney J M, Rosen V, Celeste A J, Mitsock L M, Whitters M J, Kriz R W, Hewick R M, Wang E A. Science. 1988;242:1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 8.Picard J Y, Josso N. Mol Cell Endocrinol. 1984;34:23. doi: 10.1016/0303-7207(84)90155-2. [DOI] [PubMed] [Google Scholar]
- 9.Budzik G P, Powell S M, Kamagata S, Donahoe P K. Cell. 1983;34:307–314. doi: 10.1016/0092-8674(83)90161-7. [DOI] [PubMed] [Google Scholar]
- 10.Blanchard M, Josso N. Pediatr Res. 1974;8:968–971. doi: 10.1203/00006450-197412000-00011. [DOI] [PubMed] [Google Scholar]
- 11.Kim J H, Seibel M M, MacLaughlin D T, Donahoe P K, Ransil B J, Hametz P A, Richards C J. J Clin Endocrinol Metab. 1992;75:911–917. doi: 10.1210/jcem.75.3.1517385. [DOI] [PubMed] [Google Scholar]
- 12.Ueno S, Manganaro T F, Donahoe P K. Endocrinology. 1988;123:1652–1659. doi: 10.1210/endo-123-3-1652. [DOI] [PubMed] [Google Scholar]
- 13.Behringer R R, Cate R L, Froelick G J, Palmiter R D, Brinster R L. Nature (London) 1990;345:167–70. doi: 10.1038/345167a0. [DOI] [PubMed] [Google Scholar]
- 14.Scully R. Hum Pathol. 1970;1:73–98. doi: 10.1016/s0046-8177(70)80005-3. [DOI] [PubMed] [Google Scholar]
- 15.Donahoe P K, Swann D A, Hayashi A, Sullivan M D. Science. 1979;205:913–915. doi: 10.1126/science.472712. [DOI] [PubMed] [Google Scholar]
- 16.Scully R. J Cell Biochem Suppl. 1995;23:208–218. doi: 10.1002/jcb.240590928. [DOI] [PubMed] [Google Scholar]
- 17.Cannistra S A. N Engl J Med. 1993;329:1550–1559. doi: 10.1056/NEJM199311183292108. [DOI] [PubMed] [Google Scholar]
- 18.Bookman M. Semin Oncol. 1998;25:381. [PubMed] [Google Scholar]
- 19.Fuller A F, Krane I M, Budzik G P, Donahoe P K. Gynecol Oncol. 1985;22:135–148. doi: 10.1016/0090-8258(85)90019-8. [DOI] [PubMed] [Google Scholar]
- 20.Fuller A F, Guy S R, Budzik G P, Donahoe P K. J Clin Endocrinol Metab. 1982;54:1051–1055. doi: 10.1210/jcem-54-5-1051. [DOI] [PubMed] [Google Scholar]
- 21.Cate R L, Mattaliano R J, Hession C, Tizard R, Farber N M, Cheung A, Ninfa E G, Frey A Z, Gash D J, Chow E P, et al. Cell. 1986;45:685–698. doi: 10.1016/0092-8674(86)90783-x. [DOI] [PubMed] [Google Scholar]
- 22.Ragin R C, Donahoe P K, Kenneally M K, Ahmad M F, MacLaughlin D T. Protein Expression Purif. 1992;3:236–245. doi: 10.1016/1046-5928(92)90020-w. [DOI] [PubMed] [Google Scholar]
- 23.Chin T, Parry R L, Donahoe P K. Cancer Res. 1991;51:2101–2106. [PubMed] [Google Scholar]
- 24.Baarends W M, van Helmond M J, Post M, van der Schoot P J, Hoogerbrugge J W, de Winter J P, Uilenbroek J T, Karels B, Wilming L G, Meijers J H, et al. Development (Cambridge, UK) 1994;120:189–197. doi: 10.1242/dev.120.1.189. [DOI] [PubMed] [Google Scholar]
- 25.Di Clemente N, Wilson C, Faura E, Boussin L, Carmillo P, Tizard R, Picard J Y, Vigier B, Josso N, Cate R. Mol Endocrinol. 1994;8:1006–1020. doi: 10.1210/mend.8.8.7997230. [DOI] [PubMed] [Google Scholar]
- 26.Masiakos P T, MacLaughlin D T, Maheswaran S, Teixeira J, Fuller A F, Jr, Shah P C, Kehas D J, Kenneally M K, Dombkowski D M, Ha T U, et al. Clin Cancer Res. 1999;5:3488–3499. [PubMed] [Google Scholar]
- 27.Ha T U, Segev D L, Barbie D, Masiakos P T, Tran T T, Dombkowski D, Glander M, Clarke T R, Lorenzo H K, Donahoe P K, Maheswaran S. J Biol Chem. 2000;275:37101–37109. doi: 10.1074/jbc.M005701200. [DOI] [PubMed] [Google Scholar]
- 28.Gilbert J C, Takada T, Stein J E, Langer R, Vacanti J P. Transplantation. 1993;56:423–427. doi: 10.1097/00007890-199308000-00033. [DOI] [PubMed] [Google Scholar]
- 29.Fontaine M, Hansen L K, Thompson S, Uyama S, Ingber D E, Langer R, Vacanti J P. Transplant Proc. 1993;25:1002–1004. [PubMed] [Google Scholar]
- 30.Custer R P, Bosma G C, Bosma M J. Am J Pathol. 1985;120:464–477. [PMC free article] [PubMed] [Google Scholar]
- 31.Kurian M S, de la Cuesta R S, Waneck G L, MacLaughlin D T, Manganaro T F, Donahoe P K. Clin Cancer Res. 1995;1:343–349. [PubMed] [Google Scholar]
- 32.Bogden A E, Haskell P M, LePage D J, Kelton D E, Cobb W R, Esber H J. Exp Cell Biol. 1979;47:281–293. doi: 10.1159/000162947. [DOI] [PubMed] [Google Scholar]
- 33.Fingert H J, Chen Z, Mizrahi N, Gajewski W H, Bamberg M P, Kradin R L. Cancer Res. 1987;47:3824–3829. [PubMed] [Google Scholar]
- 34.Donahoe P K, Krane I, Bogden A E, Kamagata S, Budzik G P. J Pediatr Surg. 1984;19:863–869. doi: 10.1016/s0022-3468(84)80386-3. [DOI] [PubMed] [Google Scholar]
- 35.Hudson P L, Dougas I, Donahoe P K, Cate R L, Epstein J, Pepinsky R B, MacLaughlin D T. J Clin Endocrinol Metab. 1990;70:16–22. doi: 10.1210/jcem-70-1-16. [DOI] [PubMed] [Google Scholar]
- 36.Donahoe P K, Ito Y, Marfatia S, Hendren W H. J Surg Res. 1977;23:141–148. doi: 10.1016/0022-4804(77)90202-5. [DOI] [PubMed] [Google Scholar]
- 37.Kristjansen P E, Roberge S, Lee I, Jain R K. Microvasc Res. 1994;48:389–402. doi: 10.1006/mvre.1994.1063. [DOI] [PubMed] [Google Scholar]
- 38.Gustafson M L, Lee M M, Scully R E, Moncure A C, Hirakawa T, Goodman A, Muntz H G, Donahoe P K, MacLaughlin D T, Fuller A F., Jr N Engl J Med. 1992;326:466–471. doi: 10.1056/NEJM199202133260707. [DOI] [PubMed] [Google Scholar]
- 39.Landis S H, Murray T, Bolden S, Wingo P A. Cancer Statistics. 1999;49:8–31. doi: 10.3322/canjclin.49.1.8. [DOI] [PubMed] [Google Scholar]
- 40.Donahoe P K, Fuller A F, Scully R E, Guy S R, Budzik G P. Ann Surg. 1981;194:472–480. doi: 10.1097/00000658-198110000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teixeira J, He W W, Shah P C, Morikawa N, Lee M M, Catlin E A, Hudson P L, Wing J, MacLaughlin D T, Donahoe P K. Endocrinology. 1996;137:160–165. doi: 10.1210/endo.137.1.8536608. [DOI] [PubMed] [Google Scholar]
- 42.Vacanti J P. Arch Surg. 1988;123:545. doi: 10.1001/archsurg.1988.01400290027003. [DOI] [PubMed] [Google Scholar]
- 43.Vacanti J P, Morse M A, Saltzman W M, Domb A J, Perez-Atayde A, Langer R. J Pediatr Surg. 1988;23:3. doi: 10.1016/s0022-3468(88)80529-3. [DOI] [PubMed] [Google Scholar]