

Abstract
The quinoxaline core is considered a privileged scaffold as it is found in a variety of biologically relevant molecules. Here we report the synthesis of a quinoxalin-6-amine library, screening against a panel of cancer cell lines and a structure activity relationship (SAR). This resulted in the identification of a bisfuranylquinoxalineurea analog (7c) that has low micromolar potency against the panel of cancer cell lines. We also show that cells treated with quinoxalineurea 7c results in caspase 3/7 activation, PARP cleavage and Mcl-1 dependent apoptosis.
Keywords: Quinoxaline urea, Antiproliferative, Mcl-1 dependent apoptosis
The definition of privileged scaffold has evolved over the past two decades from ligands for a diverse array of receptors to multiple compounds with similar core structure having biological activity.1 Quinoxaline is an important class of nitrogen containing heterocycle that is found in drugs (Figure 1) that aid in smoking cessation (Varenicline), have antiglaucoma activity (Brimonidine) and have antibacterial properties (Quinacillin).2–4 Not surprisingly, recent high throughput screening (HTS) campaigns against a variety of biological targets have identified quinoxaline analogs as hits. Quinoxaline analogs have been identified as inhibitors of heparin-induced tau fibril formation, cyclophin A, kinases (p110δ of PI3 kinase, JSP-1), phosphatases (cdc25B, MAPK phosphatase-1) and isomerases (peptidyl-prolyl-cis-trans isomerase).5–11 Europium and Indium complexes containing substituted quinoxalines show large fluorescence enhancement through ligand to metal transfer, which has been exploited in developing imaging agents.12 Together these studies indicate that the quinoxaline core is a privileged scaffold.
Figure 1.
Drugs containing the quinoxaline core.
Our lab is focused on the identification and characterization of small molecule inhibitors of protein-protein interactions.13, 14 The early onset of breast cancer gene 1 (BRCA1) contains multiple functional domains that interact with a plethora of proteins to mediate key signal transduction events that are critical for normal cellular functions. Abnormalities of BRCA1 result in the dysfunction of the corresponding signaling networks and have been implicated in the onset and progression of cancer. A HTS using a fluorescence polarization (FP) assay to identify small molecule inhibitors of the carboxy terminus domains of BRCA1 (BRCT) resulted in the identification of a quinoxalineurea as a validated hit.10 This prompted us to generate a focused chemical library to explore this scaffold as a BRCT inhibitor. The library was screened using the BRCT FP assay.15 However, this did not result in the identification of either a compound with improved potency or a discernable structure activity relationship (SAR).
Recent studies from other labs have shown that quinoxaline analogs have antiproliferative activity with μM potency against breast and prostate cancer cell lines.16, 17 Based on these studies and the privileged scaffold status of the quinoxalines we screened our library against a panel of cancer cell lines and estimated their growth inhibitory effects. We identified a compound (7c) with anti-tumor activity against a panel of cancer cell lines. We also show that this compound (7c) was able to induce activation of caspase 3/7, poly-ADP-ribose polymerase (PARP) cleavage and Mcl-1 dependent apoptosis.
A small focused library with substitutions at the 2,3- and 6- positions on the quinoxaline core was envisioned to probe the scaffold for biological activity (Scheme 1). The various analogs and yields for the final step are summarized in Table 1. The variations at the R1 position were methyl, furan, thiophene and phenyl groups. At the 6- position acetyl, phenylurea and tolylsulfonamide were explored in the 5 compound series. The compounds (5a-l) were generated from 2,3-substituted-6-aminoquinoxaline analogs (4, Scheme 1)11 with the amino group functionalized with acetyl chloride, phenylisocyanate and tosylchloride respectively. The synthesis of acetylated and phenyl urea analogs (5a-h) was accomplished using reported methods. However reaction of the quinoxalineamine analogs (4) with tosyl chloride yielded rearranged, monosubstitued and disubstituted products depending on the substitution at the 2,3-positions (5i-l). The amines (4a-d) were also reacted with isothiocyanates to generate the corresponding quinoxalinylthiourea analogs (6a-f). In order to generate quinoxaline urea compounds (6g-m) with secondary amines, 2,3-substituted quinoxaline isocyanates were generated in situ using triphosgene and reacted with the corresponding secondary amine.
Scheme 1. Synthesis of 2,3-substituted quinoxalin-6-amine analogs.

Reagents and conditions: (i) Ethanol, reflux, 36–48h; (ii) Pd/C, H2, ethanol, room temperature, 6–8h; (iii) R2NCO, DIPEA, DCM 24–72h; (iv) R2COCl, DCM, 4h; (v) TsCl, TEA, DCM 6h; (vi) R5NCS (2 eq), DCM, reflux; (vii) Triphosgene, DIPEA, DCM, 4h; Amine, DCM, 8–24h; (viii) R6C6H4NCO (1.5 eq), DIPEA, DCM, 12–24h.
Table 1.
Isolated yields of 2,3-substituted quinoxalin-6-amine analogs.
| Entry | R1 | R2 | R3 | R4 | % Isolated yield |
|---|---|---|---|---|---|
| 5a | Methyl | -COCH3 | -H | -H | 64 |
| 5b | 2-Furanyl | -COCH3 | -H | -H | 84 |
| 5c | 2-Thienyl | -COCH3 | -H | -H | 77 |
| 5d | Phenyl | -COCH3 | -H | -H | 70 |
| 5e | Methyl | -C(=O)-NHPh | -H | -H | 83 |
| 5f | 2-Furanyl | -C(=O)-NHPh | -H | -H | 77 |
| 5g | 2-Thienyl | -C(=O)-NHPh | -H | -H | 70 |
| 5h | Phenyl | -C(=O)-NHPh | -H | -H | 80 |
| 5i | Methyl | -H | -H | -SO2Tol | 65 |
| 5j | 2-Furanyl | -SO2Tol | -SO2Tol | -H | 66 |
| 5k | 2-Thienyl | -SO2Tol | -SO2Tol | -H | 69 |
| 5l | Phenyl | -SO2Tol | -H | -H | 65 |
| R1 | R5 | ||||
| 6a | 2-Furanyl | -C(=S)-NHPh | 70 | ||
| 6b | 2-Thienyl | -C(=S)-NHPh | 74 | ||
| 6c | Phenyl | -C(=S)-NHPh | 75 | ||
| 6d | 2-Furanyl | -C(=S)-NH-(4-Nitro)-Phenyl | 78 | ||
| 6e | 2-Thienyl | -C(=S)-NH-(4-Nitro)-Phenyl | 80 | ||
| 6f | Phenyl | -C(=S)-NH-(4-Nitro)-Phenyl | 80 | ||
| 6g | Methyl | -CO-Pyrrolidine | 55 | ||
| 6h | 2-Furanyl | -CO-Pyrrolidine | 57 | ||
| 6i | Phenyl | -CO-Pyrrolidine | 40 | ||
| 6j | Methyl | -C(=O)-NH-(4-Benzyl)-Piperidine | 50 | ||
| 6k | 2-Furanyl | -C(=O)-NH-(4-Benzyl)-Piperidine | 53 | ||
| 6l | Methyl | -CO-Piperdine | 54 | ||
| 6m | 2-Furanyl | -CO-Morpholine | 80 | ||
| R1 | R6 | ||||
| 7a | 2-Furanyl | -F | 80 | ||
| 7b | 2-Furanyl | -Cl | 81 | ||
| 7c | 2-Furanyl | -Br | 76 | ||
| 7d | 2-Furanyl | -Phenyl | 71 | ||
The compounds were screened in a growth inhibition assay at 20 μM over a 72 h period in a panel of cancer cell lines (lung-A549; pancreatic-Aspc1; colon-HT29; breast-MDAMB231; prostate-PC3; ovarian-SKOV3 and bone-U2OS) and the results are summarized in Table 2. In the 5 compound series only 5a, 5b and 5f inhibited growth of the various cancer cell lines. This suggests that the furan substitutions at the 2,3-positions are clearly better than the other three. It also suggests that sulfonamide substitution at the 6-position is not suitable for the growth inhibitory activity. The screening results in the 6 compound series again show that all the furan compounds (6a, 6d, 6h, 6k and 6m) were active. It also shows that substitution at the 4-position of the phenyl thioureas plays a role in the biological activity (6b and 6c vs. 6e and 6f). This size effect was also observed with the urea compounds from the secondary amine (6j vs. 6l and 6k vs. 6m).
Table 2.
Screen results of the 2,3-substituted quinoxalin-6-amine analogs.
| Entry | % Growth inhibition at 20μM | ||||||
|---|---|---|---|---|---|---|---|
| A549 | AsPC1 | HT29 | MDA-MB-231 | PC3 | SKOV3 | U2OS | |
| 5a | 23.4 ± 3.3 | 26.2 ± 2.8 | 31.7 ± 2.8 | 25.7 ± 5.1 | 21.8 ± 15.1 | 4.0 ± 3.6 | 31.4 ± 3.6 |
| 5b | 57.8 ± 7.0 | Inactive | 7.6 ± 5.1 | 20.3 ± 5.9 | 57.7 ± 9.4 | 9.0 ± 3.4 | Inactive |
| 5c | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 5d | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 5e | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 5f | 54.6 ± 2.6 | 9.6 ± 6.6 | 52.1 ± 11.6 | 31.3 ± 10.7 | 70.6 ± 1.3 | 24.7 ± 1.1 | 59.6 ± 1.6 |
| 5g | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 5h | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 5i | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 5j | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 5k | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 5l | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 6a | 18.0 ± 0.1 | Inactive | 26.1 ± 15.0 | 19.1 ± 12.5 | 56.8 ± 1.4 | Inactive | 21.4 ± 8.4 |
| 6b | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 6c | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 6d | 19.2 ± 8.9 | Inactive | 42.0 ± 1.9 | 9.0 ± 4.7 | 43.4 ± 9.3 | Inactive | 32.2 ± 0.1 |
| 6e | Inactive | Inactive | 23.9 ± 27.8 | 53.3 ± 7.2 | 19.5 ± 37.4 | Inactive | 50.6 ± 3.9 |
| 6f | 16.6 ± 11.2 | 55.5 ± 25.4 | 81.2 ± 0.7 | 72.6 ± 0.7 | 81.0 ± 8.5 | 55.8 ± 30.7 | 76.1 ± 0.4 |
| 6g | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 6h | 23.3 ± 2.9 | 11.6 ± 4.2 | 16.7 ± 3.7 | 18.2 ± 7.0 | 53.1 ± 9.9 | 16.9 ± 4.9 | 46.5 ± 4.4 |
| 6i | 11.0 ± 1.3 | 29.4 ± 9.8 | Inactive | 16.7 ± 3.9 | 56.9 ± 2.7 | 18.5 ± 4.3 | 54.2 ± 2.2 |
| 6j | 40.3 ± 1.9 | Inactive | 16.3 ± 10.1 | 12.4 ± 5.0 | 30.6 ± 11.6 | 8.9 ± 5.1 | 33.5 ± 0.4 |
| 6k | 93.1 ± 7.8 | >100 | 55.2 ± 4.1 | 90.8 ± 4.5 | >100 | 47.6 ± 2.5 | 80.3 ± 2.4 |
| 6l | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 6m | 39.7 ± 12.8 | Inactive | 19.4 ± 3.6 | 10.9 ± 5.0 | 48.9 ± 7.5 | Inactive | Inactive |
| 7a | >100 | >100 | 74.2 ± 5.2 | 38.4 ± 2.7 | 90.7 ± 4.1 | Inactive | 55.6 ± 6.6 |
| 7b | 99.2 ± 5.2 | 81.1 ± 2.0 | 86.3 ± 7.7 | 92.5 ± 6.6 | 86.4 ± 1.1 | 55.8 ± 24.1 | 87.4 ± 1.1 |
| 7c | >100 | >100 | 88.6 ± 4.6 | >100 | >100 | 94.9 ± 7.9 | >100 |
| 7d | 13.8 ± 9.8 | Inactive | 72.4 ± 7.6 | 50.2 ± 8.9 | 88.5 ± 1.9 | Inactive | 46.9 ± 7.4 |
These results prompted us to synthesize four additional compounds to probe the size effect at the 4-position on a phenyl urea (7a-d). Evaluation of these analogs in the growth inhibition assay clearly showed a size effect and compound 7c with bromo substituion at the 4-position was identified as the best compound. A dose-response study with compound 7c shows low-μM GI50 values against a panel of cancer cell lines (Table 3). In summary this iterative synthesis and screening effort show that the furan substitution at the 2,3-position, a urea at the 6-position and the substitutent at the para-position of a phenyl urea are important for the biological activity. These studies also resulted in the identification of 7c with low-μM GI50 values against a panel of cancer cell lines.
Table 3.
Dose-response growth inhibition study with 7c.
| Cell line | GI50 (μM) |
|---|---|
| A549 | 6.4 ± 3.0 |
| AsPC1 | 17.3 ± 0.9 |
| HT29 | 12.1 ± 7.4 |
| MDA-MB-231 | 8.4 ± 0.9 |
| PC3 | 5.9 ± 2.7 |
| SKOV3 | 16.8 ± 5.2 |
| U2OS | 10.8 ± 0.2 |
Caspases are a class of cysteine proteinases that are activated during apoptosis and measuring caspase activity is often used to detect activation of apoptotic signaling. To determine if the growth inhibitory effects observed with 7c in various cancer cell lines were a result of programmed cell death, we explored the ability of 7c to induce caspase-3/7. Our results show that 7c induces caspase 3/7 much more rapidly compared to the positive control (Etoposide) in MDA-MB-231 and PC3 cells (Figure 2A and B) and the induction is sustained for 72h in these cell lines.
Figure 2.

Induction of caspase 3/7 activity by 7c and Etoposide (a known chemotherapeutic agent) in MDA-MB-231 breast cancer cells (A) and PC3 prostate cancer cells (B).
Bcl-xL, Bcl-2 and Mcl-1 are antiapoptotic proteins that are implicated in the survival of cancer cells.18, 19 Bad3SA is the endogenous inhibitor of Bcl-xL and Bcl-2 but not Mcl-1. Using HeLa cells that over express Bad3SA, we explored the mechanistic basis for the induction of apoptosis by 7c. In these cell lines expression of Bad3SA is under the control of Doxycycline (Dox). The apoptosis studies carried out in this cell line are summarized in Figure 3. As with the other cancer cell lines we observe induction of caspase 3/7 and PARP cleavage by 7c (Figure 3A and 3B respectively). We next carried out a dose response study with 7c in untreated cells [(−) Dox] and cells treated with Dox (1 μg/mL for 3 h) to induce Bad3SA [(+) Dox]. The cells were incubated with 7c and a positive control (DNA damaging agent Camptothecin, CPT) for 12h. Cell death was measured by counting the number of condensed nuclei. We observed a dose dependent increase in the induction of apoptosis in the (+) Dox cells, indicating that 7c induces apoptosis in a Mcl-1 dependent manner (Figure 3C).
Figure 3.
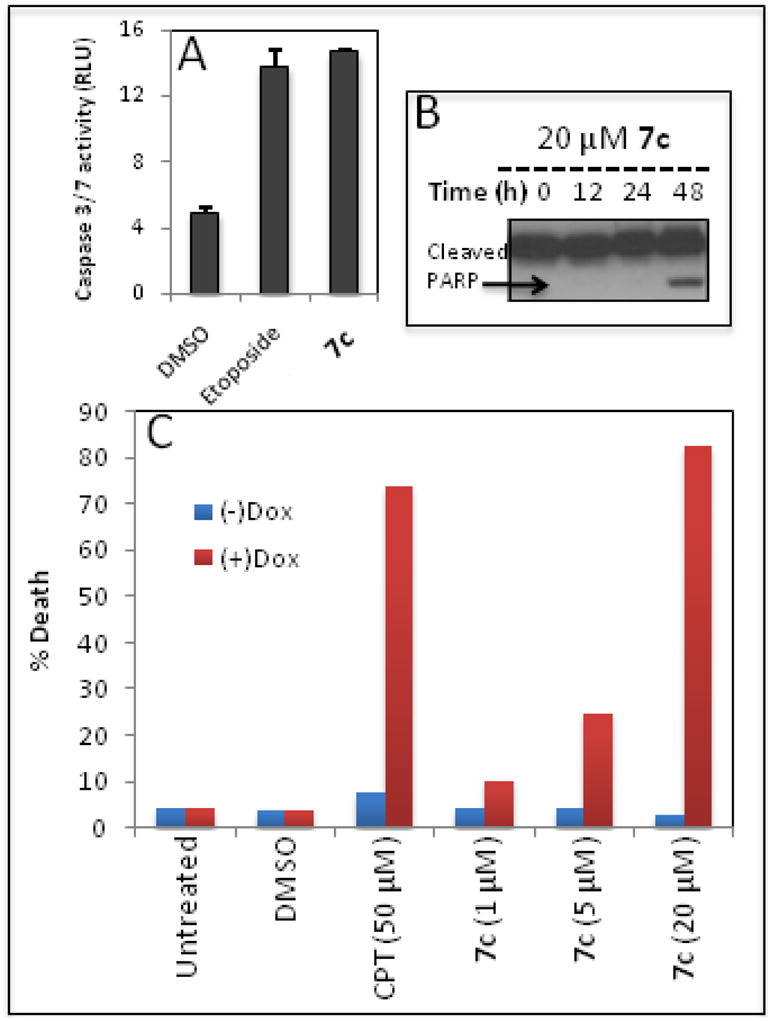
Apoptosis studies in HeLa cells: (A) Induction of caspase 3/7 by 7c and Etoposide. (B) PARP cleavage induced by 7c assessed by Western blot. (C) Mcl-1 dependent induction of apoptosis by 7c.
In summary, a focused library of 2,3-substituted quinoxalin-6-amine analogs was synthesized and evaluated in a panel of cancer cell lines for growth inhibition. The preliminary structure activity relationship (SAR) showed bis-furan substitution at the 2,3- positions was favored. A comparison of a series of linkers between the 2,3- disubstituted quinoxaline and a substituted phenyl ring showed that a urea linker was optimal for the antiproliferative activity. In addition, the size of the substituent at the 4-position of the phenyl ring was important for the activity. This led to the identification of a bisfuranylquinoxalineurea analog (7c) with low micromolar potency against the panel of cancer cell lines. The analog 7c induces caspase 3/7 activation, PARP cleavage and Mcl-1 dependent apoptosis. The molecular target of compound 7c is currently under investigation and will be reported in due course.
Supplementary Material
Acknowledgments
We thank the Eppley NMR facility. Funding in part from the National Institutes of Health (NIH R01CA127239 to AN) is gratefully acknowledged.
Footnotes
Methods and characterization data for the compounds tested are included.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Welsch ME, Snyder SA, Stockwell BR. Curr Opin Chem Biol. 2010;14:347. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Escobar-Chavez JJ, Merino V, Lopez-Cervantes M, Rodriguez-Cruz IM, Quintanar-Guerrero D, Ganem-Quintanar A. Curr Drug Discov Technol. 2009;6:171. doi: 10.2174/157016309789054924. [DOI] [PubMed] [Google Scholar]
- 3.McLaughlin MA, Chiou GC. J Ocul Pharmacol. 1985;1:101. doi: 10.1089/jop.1985.1.101. [DOI] [PubMed] [Google Scholar]
- 4.Hugo WB, Stretton RG. Nature. 1964;202:1217. doi: 10.1038/2021217a0. [DOI] [PubMed] [Google Scholar]
- 5.Crowe A, Ballatore C, Hyde E, Trojanowski JQ, Lee VM. Biochem Biophys Res Commun. 2007;358:1. doi: 10.1016/j.bbrc.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Chen J, Zhang L, Wang F, Gui C, Zhang L, Qin Y, Xu Q, Liu H, Nan F, Shen J, Bai D, Chen K, Shen X, Jiang H. Bioorg Med Chem. 2006;14:5527. doi: 10.1016/j.bmc.2006.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berndt A, Miller S, Williams O, Le DD, Houseman BT, Pacold JI, Gorrec F, Hon WC, Liu Y, Rommel C, Gaillard P, Ruckle T, Schwarz MK, Shokat KM, Shaw JP, Williams RL. Nat Chem Biol. 2010;6:244. doi: 10.1038/nchembio0310-244b. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Qiu B, Xiong B, Li X, Li J, Wang X, Li J, Shen J. Bioorg Med Chem Lett. 2007;17:2118. doi: 10.1016/j.bmcl.2007.01.094. [DOI] [PubMed] [Google Scholar]
- 9.Johnston PA, Foster CA, Tierno MB, Shun TY, Shinde SN, Paquette WD, Brummond KM, Wipf P, Lazo JS. Assay Drug Dev Technol. 2009;7:250. doi: 10.1089/adt.2008.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simeonov A, Yasgar A, Jadhav A, Lokesh GL, Klumpp C, Michael S, Austin CP, Natarajan A, Inglese J. Anal Biochem. 2008;375:60. doi: 10.1016/j.ab.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang F, Chen J, Liu X, Shen X, He X, Jiang H, Bai D. Chem Pharm Bull (Tokyo) 2006;54:372. doi: 10.1248/cpb.54.372. [DOI] [PubMed] [Google Scholar]
- 12.Chinen LK, Galen KP, Kuan KT, Dyszlewski ME, Ozaki H, Sawai H, Pandurangi RS, Jacobs FG, Dorshow RB, Rajagopalan R. J Med Chem. 2008;51:957. doi: 10.1021/jm070842+. [DOI] [PubMed] [Google Scholar]
- 13.Kumar EA, Charvet CD, Lokesh GL, Natarajan A. Anal Biochem. 2010 doi: 10.1016/j.ab.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lokesh GL, Rachamallu A, Kumar GD, Natarajan A. Anal Biochem. 2006;352:135. doi: 10.1016/j.ab.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 15.Joseph PR, Yuan Z, Kumar EA, Lokesh GL, Kizhake S, Rajarathnam K, Natarajan A. Biochem Biophys Res Commun. 2010;393:207. doi: 10.1016/j.bbrc.2010.01.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grande F, Aiello F, Grazia OD, Brizzi A, Garofalo A, Neamati N. Bioorg Med Chem. 2007;15:288. doi: 10.1016/j.bmc.2006.09.073. [DOI] [PubMed] [Google Scholar]
- 17.Gavara L, Saugues E, Alves G, Debiton E, Anizon F, Moreau P. Eur J Med Chem. 2010;45:5520. doi: 10.1016/j.ejmech.2010.08.067. [DOI] [PubMed] [Google Scholar]
- 18.Lopez H, Zhang L, George NM, Liu X, Pang X, Evans JJ, Targy NM, Luo X. J Biol Chem. 2010;285:15016. doi: 10.1074/jbc.M109.086231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L, Lopez H, George NM, Liu X, Pang X, Luo X. Cell Death Differ. 2010 November 26; doi: 10.1038/cdd.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.