

Abstract
Background
Enterochromaffin cells and enteric neurons synthesize and release serotonin (5-HT). Reuptake, mediated by a plasmalemmal transporter (SERT) terminates the action of released 5-HT. Serotonin secretion and serotonin reuptake transporter (SERT) expression have been reported to be decreased in TNBS-induced experimental colitis and in patients with ulcerative colitis. The present study was designed to utilize the transgenic deletion of SERT as a gain-of-function model to test the hypothesis that 5-HT is a pro-inflammatory mediator in experimental colitis.
Methods
Colitis was compared in animals with IL10+/+SERT+/+ (wild-type), IL10−/−SERT+/+, IL10−/−SERT+/−, and IL10−/−/SERT−/− (double knockout) genotypes. Macroscopic and histological damage scores were evaluated after a time period of up to 15 weeks.
Key Results
Serotonin reuptake transporter expression was significantly increased in the inflamed colons of IL-10−/− mice, which displayed intestinal damage and a minor decrement in general health. General health was significantly worse and intestinal inflammation was more severe in IL-10−/−SERT+/−, and IL-10−/−SERT−/− mice than in IL-10−/−SERT+/+ or wild-type animals. Regardless of the associated SERT genotype, the number of 5-HT-immunoreactive cells was decreased by ~55–65% in all mice lacking IL-10.
Conclusions & Inferences
Our observations indicate that colitis associated with IL-10 deficient mice is enhanced when the IL-10 deficiency is combined with a SERT deficiency. The data support the concept that 5-HT is a pro-inflammatory mediator in the gut.
Keywords: colitis, inflammation, interleukin 10, serotonin, serotonin reuptake transporter
Introduction
Inflammatory bowel disease (IBD) is an idiopathic disorder that is usually diagnosed on the basis of characteristic clinical, pathological, endoscopic, and radiological observations. The pathogenesis of IBD is not yet understood but there is evidence that excessive innate and/or adaptive immune responses to commensal bacteria are involved (reviewed in 1). The susceptibility to IBD, and its variable severity, may be due to abnormalities in innate and adaptive immunity, its regulation by pro- and anti-inflammatory cytokines, and in neuroimmune interactions.2,3 Neuroimmune interactions are significant because cytokines affect neuronal activity and neurotransmitters affect the activity of immuno-effector cells.
Serotonin (5-hydroxytryptamine, 5-HT) is a neurotransmitter that promotes lymphocyte activation and secretion of pro-inflammatory cytokines. 4–6 Serotonin is also highly concentrated in the bowel that stores ~95% of the body’s 5-HT (reviewed in 7). The bulk of this 5-HT is present in enterochromaffin (EC) cells of the mucosal epithelium, but other significant sources of enteric 5-HT are the serotonergic neurons of the enteric nervous system and, in rats and mice, mast cells. Enterochromaffin cells function as sensory transducers. In response to increases in intraluminal pressure or chemical stimuli, EC cells secrete 5-HT, which stimulates intrinsic primary afferent neurons to initiate peristaltic and/or secretory reflexes. Enteric bacteria are another possible stimulator of 5-HT secretion because EC cells express Toll-like receptors. 8 Because dendritic cells, 9 lymphocytes, 10–12 macrophages, endothelial cells, 4,13 and enteric epithelial cells 7 all express 5-HT receptors, the secretion of 5-HT is potentially able to influence intestinal inflammation.
Once 5-HT acts on its receptors, it has to be inactivated. This function depends on the plasmalemmal 5-HT transporter (SERT; SLC6A4) that mediates the reuptake of 5-HT and that is expressed by serotonergic neurons in the brain and in the gut, and by enterocytes. 14–16 Inhibition or deletion of SERT potentiates the effects of endogenous 5-HT in the bowel.15–17 Mucosal SERT expression is decreased during 2,4,6-trinitrobenzene sulfonic acid (TNBS)-mediated colitis in guinea pigs and mice 18–20 moreover, SERT transcription has also been reported to be decreased in the rectum of human patients with ulcerative colitis. 21 Recent studies have revealed that deletion of SERT potentiates the inflammatory reactions induced by TNBS in mice 22 and that deletion of tryptophan hydroxylase-1 (TPH1) ameliorates intestinal inflammation 23 supporting the concept that 5-HT is a potential modifier of inflammation, which could play a role in IBD, either in its pathogenesis or as a mediator of symptoms.
The current experiments utilized a gain-of-function paradigm to test the hypothesis that 5-HT contributes, not only to the manifestation of the symptoms of IBD, but to the severity of the inflammatory process itself. Therefore, double knockout mice were generated that lack both IL-10 and SERT. Intestinal inflammation develops spontaneously in mice that lack only IL-10 or its receptor. 24 The additional deletion of SERT is supposed to act as a ‘second hit’ that causes amplification of 5-HT functions and thereby enhancement of the severity of intestinal inflammation.
Material and Methods
Animals
Interleukin-10−/− mice (C57Bl6-IL-10tmlCgn, originally obtained from Jackson Laboratories, Bar Harbor, ME, USA) were crossbred with SERT−/− mice (C57Bl6 background, generous gift from Dr. Murphy, NIH, Bethesda, MA, USA). 22 Male and female littermates from the F1 generation were bred to obtain IL-10−/−/SERT−/− double homozygous mice (F2) and IL-10−/−/SERT+/− mice and controls with an IL-10−/−/SERT+/+ genotype. The effect of the deletion of SERT on the development of inflammatory lesions in the bowel of mice that lack IL-10 was evaluated in the double knockout IL-10−/−/SERT−/− animals. Comparisons were made with mice that had the genotypes IL-10−/−/SERT+/−, IL-10−/−/SERT+/+, and IL-10+/+/SERT+/+ (wild-type). Both male and female siblings up to the age of 15 weeks were investigated. Genomic DNA was purified from tail biopsies and used for the genotyping of mice at 4 weeks of age. Polymerase chain reaction (PCR) was used for genotype analysis. The PCR protocol used to determine the IL-10 genotype was obtained from Jackson Laboratory (Bar Harbor, ME, USA). The following primers were used for genotyping SERT: oIMR086 5′-GTG GGT GCA GTT ATT GTC TTC CCG-3′, oTMR087 5′-GCC TTC AGT ATA AAA GGG GGA CC-3′, and oIMR088 5′-CCT GCG TGC AAT CCA TCT TG-3′. All three primers were used in a single PCR reaction. Mice were housed behind a barrier in a pathogen-free environment in an AAALAC-accredited facility. Animal studies were performed according to the German guidelines governing the use of experimental animals and procedures have been approved by the appropriate regulatory body. Animals had ad libitum access to food and water and were maintained at 23–24 °C on a 12 : 12-h light–dark cycle. The animals were euthanized after completing an experimental protocol or after reaching the age of 15 weeks.
Assessment of colonic inflammation
A global assessment score was used to assess the severity of colitis. The score ranged from 1 to 6. Normal/healthy mice were assigned a score of 1, mice with a dull coat a score of 2, mice with a dull coat and curved back or rectal prolapse a score of 3, mice with the features of 3 plus reduced movement a score of 4, mice with severe illness without body movement necessitating immediate euthanasia, a score of 5, and mice that died spontaneously a score of 6. The global assessment score was assigned and body weight was documented at weekly intervals from birth to 15 weeks of age. Stool water content was calculated from the wet weight of stool pellets, measured immediately after collection, and the dry weight, determined after overnight dehydration at 60 °C. Stool water content was calculated by using the formula: 1-dry weight/wet weight and was expressed as a percent.
To obtain ‘Swiss rolls’ of colon for histological assessment, the colon was rinsed with PBS and opened along the mesenteric attachment. The preparation was then rolled with the luminal side facing out; the proximal end was located at the center of the roll. The rolled tissue was embedded as a block in embedding medium (OCT) and frozen with liquid N 2. Sections were cut at 10 μm in a cryostat-microtome, air dried, and fixed in ice-cold acetone. Sections were stained with hematoxylin and eosin. The severity of mucosal destruction was assessed by two independent pathologists who were blinded to the genotypes of the mice. The following parameters were assigned numerical scores: (i) severity of mucosal destruction (0, normal; 1, mild; 2, moderate; 3, extensive), (ii) extent of cellular infiltration (0, absent; 1, mild; 2, pronounced), (iii) thickening of the muscle layer (0, absent; 1, mild; 2, moderate; 3, extensive), (iv) the presence or absence of crypt abscesses (0, absent; 1, present), and (v) the presence or absence of goblet cell mucus (0, absent, 1, present). The individual scores (i–v) were integrated to provide an overall histological inflammation score for each animal (0, normal; 1, mild; 2, moderate; 3, extensive). 22
Immunocytochemistry
Frozen fixed tissue was sectioned at 10 μm, air-dried and fixed in ice-cold acetone. Sections were rehydrated in TBST (0.1 M Tris pH 7.5, 0.15 M NaCl, 0.1% Tween20) and blocked with rat serum, avidin, and biotin blocking solutions (avidin/biotin activity must be blocked to eliminate background) (Zymed Laboratories, San Francisco, CA, USA). Sections were then incubated for 60 min with combinations of primary antibodies. These combinations included biotinylated anti-CD45.2 (Chemicon/Millipore GmbH, Schwalbach/Ts., Germany) and anti-B220 (Southern Biotech, Birmingham, AL, USA); biotinylated anti-CD3 (BioLegend, San Diego, CA, USA) and anti-B220 (Southern Biotech) or serotonin (Sigma, Schnelldorf, Germany). Following incubation with primary antibodies, sections were washed with TBST, incubated with streptavidin Cy3 (Dianova, Germany) for 45 min and washed again. Alternatively sections were incubated peroxidase-labeled secondary antibodies and sites of immunoreactivity were visualized with 3, 3′-diaminobenzidine (Peroxidase Envision Kit; DAKO, Hamburg, Germany). Slides were coverslipped with VectaShield mounting media containing 4′,6-Diamidino-2-phenylindol (DAPI) (Vector Laboratories, Burlingame, CA, USA). A Zeiss microscope (Axioplan; Zeiss, Göttingen, Germany) was used to examine slides. To quantify the total number of 5-HT-immunoreactive cells and the proportion that appeared to be dying, slides were viewed at a magnification of 400× and cells were counted in 10 defined squares (0.42 × 0.32 mm).
RNA isolation and real-time RT-PCR
Total RNA was extracted from samples of colon by RNeasy Kit (Quiagen, Hilden, Germany). RNA concentrations were determined spectrophotometrically, and 1 μg total RNA was reverse transcribed using a SuperScript® reverse transcriptase (Invitrogen, Karlsruhe, Germany) and oligo dT primers. PCR primers for SERT, IL-6, TNFα and 18S were designed using Primer 3 software (Whitehead Institute for Biomedical Research, Cambridge, MA, USA). Primers were designed to cross introns to ensure that only cDNA, and not DNA, was amplified. Specific primers were used for detection of SERT (forward 5′–3′ CAAAACCAAGAACCAAGAG; reverse 3′–5′ CATAGCCAATGACAGACAG), 18S (forward 5′–3′ GTAACCCGTTGAACCCCATT; reverse 3′–5′ CCATCCAATCGGTAGTAGCG), IL-6 (forward 5′–3′ TGCTGGTGACAA CAACGGCC; reverse 3′–5′GTACTCCAGAAGACCAGAGG) and TNFα (forward 5′–3′ CCAGGCGGTGCCTATGTCTC; reverse 3′–5′ CAGCCACTCCAGCTGCTCCT) by real-time RT-PCR. Primers were added to SyberGreen Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA) to provide a final primer concentration of 300 nmol L−1. Amplification reactions were carried out in an iQ Cycler sequence detection system (BioRad, München, Germany) with an initial hold step (95 °C for 4 min) and 42 cycles of a three-step PCR (SERT/18S, 94 °C for 60 s, 55 °C for 50 s, 72 °C for 40 s or IL-6/18S; and TNFα/18S, 95 °C for 15 s, 60 °C for 30 s, 72 °C for 30 s). The fluorescence intensity of each sample was measured at each change of temperature to monitor amplification of the target gene. The comparative CT method was used to determine fold differences between samples. The comparative CT method determines the amount of target, normalized to an endogenous reference (18S) and relative to a calibrator (2−ΔΔCt). The purity of PCR products was verified by gel electrophoresis and the melting curve.
Statistical analysis
Results are reported as mean ± SEM (n = 6–10). For comparison of means of continuous data derived from >2 groups univariate ANOVA was used; individual pairs of groups were further analyzed by Bonferroni’s post hoc test, provided that ANOVA yielded a P < 0.05. Non-continuous data were analyzed by chi-square tests. Statistical analyses were carried out with computer assistance (SPSS software, release 15.0, SPSS Inc., Chicago, IL, USA). Fisher’s exact test was used to compare the frequency of macroscopic signs of inflammation among groups. P values of <0.05 were accepted as the level of significance.
Results
SERT expression is enhanced in IL-10−/−SERT+/+ mice
Transcripts encoding SERT were quantified in the colons from each group of animals. The abundance of transcripts encoding SERT was increased over that in wild-type colon by eightfold in IL-10−/−SERT+/+ mice, and by fivefold in IL-10−/−SERT+/− mice (Fig. 1). As expected, transcripts encoding SERT were not detectable in IL-10−/−SERT−/− mice. The effect on SERT expression of the spontaneous inflammation that arises in the colons of mice lacking IL-10 thus seems to differ from that induced by TNBS. Inflammation induced by TNBS has been reported to decrease, rather than increase, SERT expression.19,22
Figure 1.
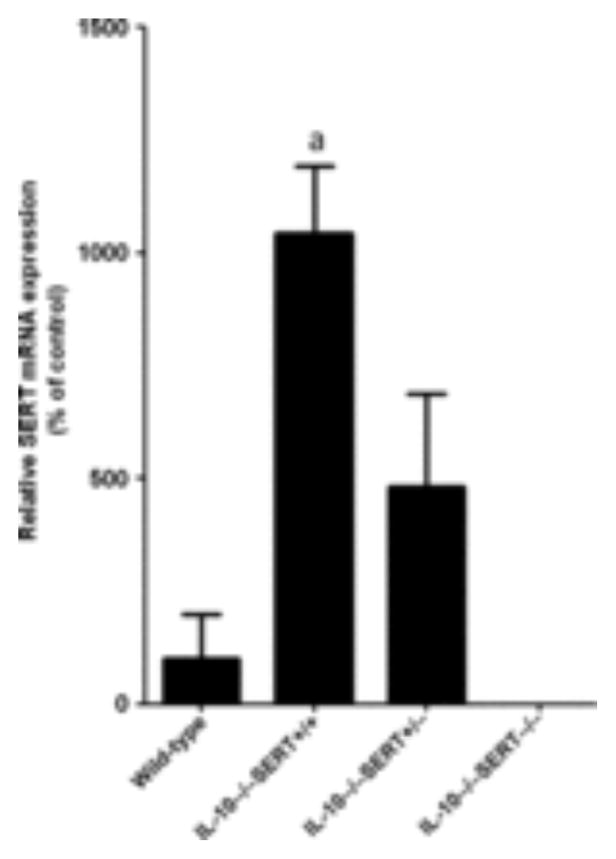
Expression of the serotonin reuptake transporter (SERT) in the colon of IL-10−/− mice and controls. Real-time RT-PCR was used to quantify transcripts encoding SERT. Data are mean ± SEM (n = 3–4). aP < 0.05 compared with wild-type mice.
Gross signs of inflammation are evident in IL-10−/−SERT+/+, IL-10−/−SERT+/−, and IL-10−/−SERT−/− mice
To investigate the role of SERT in the development and progression of IL-10-dependent colitis, SERT−/− and IL-10−/− mice were crossbred. Global health assessment scores indicated significant signs of illness were present in IL-10−/−SERT+/− and IL-10−/−SERT−/− mice, some signs of illness in IL-10−/−SERT+/+, but no illness in wild-type animals (Fig. 2). Body weights of IL-10−/−SERT+/− and IL-10−/−SERT−/− mice, but not of IL-10−/−SERT+/+ mice, were also significantly lower than those of wild-type animals; the SERT-deficient mice lost almost half of their weight. The mean water content of stool, which was 57% in wild-type mice, was also significantly higher in IL-10−/−SERT+/+, IL-10−/−SERT+/− and IL-10−/−SERT−/− animals, suggesting that diarrhea was present in all of the mutant animals; moreover, a significant proportion of mice with IL-10−/−SERT+/+, IL-10−/−SERT+/− and IL-10−/−SERT−/− genotypes displayed colonic prolapse and macroscopic signs of inflammation (Table 1 and Fig. 2), neither of which occurred in wild-type animals. Gross evidence of inflammation thus was present in all animals that lacked IL-10, including those in which expression of SERT was also deficient.
Figure 2.
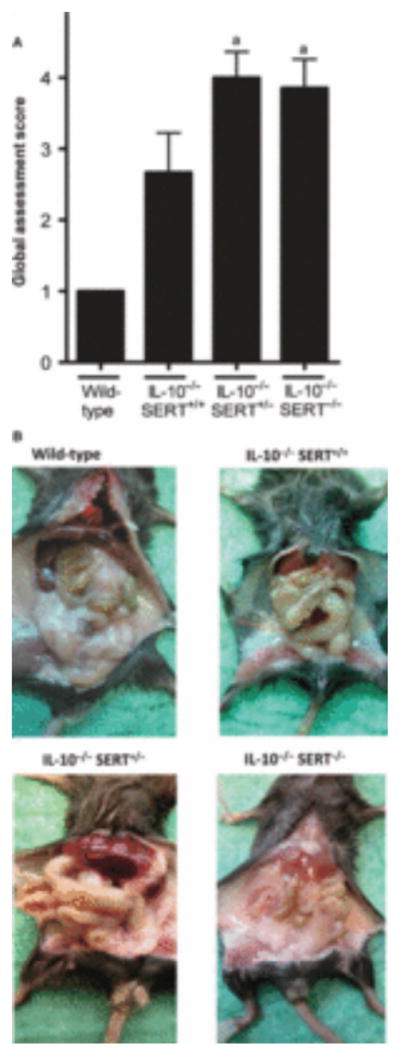
Global assessment of health and macroscopic inflammation of colon in wild-type, IL-10−/−SERT+/+, IL-10−/−SERT+/−, and IL-10−/−SERT−/− mice. (A) The global assessment score is increased in IL-10−/−SERT+/− and IL-10−/−SERT−/− mice when compared to wild-type mice (aP < 0.05), but not when compared to IL-10−/−SERT+/+ mice. (B) Representative photomicrographs of abdomen of wild-type mice, IL-10−/−SERT+/+, IL-10−/−SERT+/−, and IL-10−/−SERT−/− mice are shown. Global assessment scores were obtained as described in Material and Methods. Data are mean ± SEM (n = 4).
Table 1.
Body weight, stool water, and macroscopic signs of inflammation in wild-type, IL-10−/−SERT+/+, IL-10−/−SERT+/−, and IL-10−/−SERT−/− mice
| Wild-type | IL-10−/− SERT+/+ | IL-10−/− SERT+/− | IL-10−/− SERT−/− | |
|---|---|---|---|---|
| N | 11 | 10 | 13 | 6 |
| Body weight‡ (g) | 21.6 ± 0.9 | 15.5 ± 2.0 | 11.2 ± 0.8† | 12.7 ± 2.1† |
| Stool water (%) | 56.8 ± 1.6 | 78.7 ± 2.7† | 77.4 ± 1.3† | 80.7 ± 4.2† |
| Prolapse‡ (%) | 0/11 (0) | 4/10 (40)† | 6/13 (46)† | 3/6 (50)† |
| Visible signs of colitis‡ (%) | 0/11 (0) | 6/10 (60)† | 9/13 (69)† | 2/6 (33)† |
SERT, serotonin reuptake transporter. Data are mean ± SEM.
P < 0.05 compared to wild-type mice.
At time of death.
Colonic inflammation is increased in IL-10−/−SERT−/− double knockout mice
Classical signs of inflammation (e.g., cellular infiltration, muscular thickening) were present in the intestines of IL-10−/−SERT+/+, IL-10−/−SERT+/− and IL-10−/−SERT−/− mice, but not in wild-type littermates (Fig. 3A,B). Histopathology scores were investigated for distal, middle, and proximal colon, and combined in a ‘total score’. In distal, middle, and proximal colon the histopathology score was significantly higher in IL-10−/−SERT+/− and IL-10−/−SERT−/− mice than in either wild-type or IL-10−/−SERT+/+ animals (Fig. 3C). The total histopathology score indicates that significant inflammation was present in the colons of IL-10−/−SERT+/+ mice, but inflammation was much more prominent in the colons of IL-10−/−SERT+/− and IL-10−/−SERT−/− mice, than in IL-10−/−SERT+/+ mice or their wild-type littermates (Fig. 3C).
Figure 3.
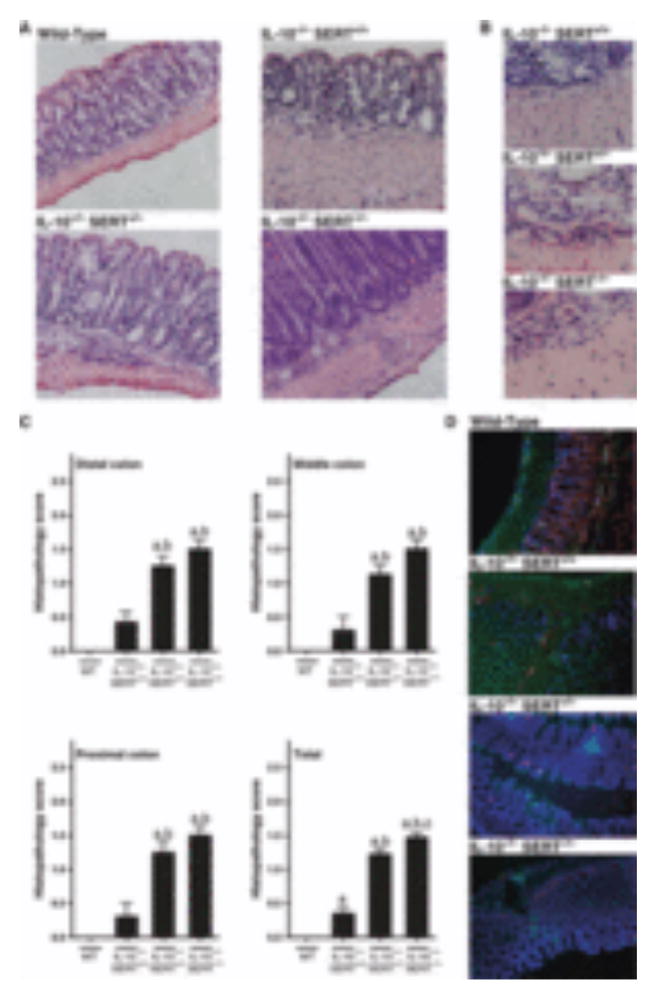
Colonic inflammation in wild-type, IL-10−/−SERT+/+, IL-10−/−SERT+/−, and IL-10−/−SERT−/− mice. (A) Representative photomicrographs showing the histological appearance of the colon (H&E staining, 100× original magnification) in wild-type mice, IL-10−/−SERT+/+, IL-10−/−SERT+/−, and IL-10−/−SERT−/− mice. (B) Representative photomicrographs at higher magnification (400×) show infiltrating neutrophils and mononuclear cells in the submucosae of IL-10−/−SERT+/+, IL-10−/−SERT+/−, and IL-10−/−SERT−/− mice. (C) Inflammation scores obtained from the distal, middle, proximal colon. Data represent mean ± SEM (n = 4–6). aP < 0.05 compared to wild-type mice, bP < 0.05 compared to IL-10−/−SERT+/+ mice, cP < 0.05 compared to IL-10−/−SERT+/− mice. (D) Representative photomicrographs showing the simultaneous immunohistological identification of T and B lymphocytes infiltrating the mucosa. T lymphocytes were labeled with primary antibodies against CD3 (red) and B lymphocytes were labeled with antibodies against B220 (green). DNA was stained with DAPI to locate nuclei. Although lymphocytic infiltration of the mucosa was greater than normal (wild-type) in each of the strains of mice that lacked IL-10, lymphocytic infiltration of the mucosa was further potentiated when the IL-10 deficiency was combined with a partial (IL-10−/−SERT+/−) or complete knockout of serotonin reuptake transporter (SERT) (IL-10−/−SERT−/−).
Infiltration of leukocytes is altered in colon of IL-10−/− SERT−/− mice
Immunohistology was used to identify the cells that infiltrated the intestinal mucosa in wild-type mice and the inflamed mucosae of animals with the IL-10−/−SERT+/+, IL-10−/−SERT+/−, and IL-10−/−SERT−/− genotypes. Primary antibodies to CD3 were used to identify T lymphocytes, antibodies to B220 to identify B lymphocytes, and antibodies to CD45.2 to mark cells displaying this essential regulator of leukocyte activation and development. Most of the cells that infiltrated inflamed segments of the colon were not immunostained by antibodies to CD3, B220, or CD45.2 and thus were, as suggested by the morphology of infiltrating cells in H&E-stained sections, leukocytes in myeloid lineages (Fig. 3D). Immunostaining with a combination of antibodies against CD3 and B220 revealed that there were considerably more CD3-positive T cells in IL-10−/−SERT+/+ mice than in wild-type controls. Still more CD3-positive T cells were observed in animals lacking one or both SERT genes (Fig. 3D). In contrast, infiltration of the colonic tissue with B220-and CD45.2-immunoreactive B cells was minimal in wild-type animals and only slightly increased in IL-10−/− animals (data not shown).
Expression of transcripts encoding IL-6 and TNFα is significantly increased in the colons of IL-10−/−SERT−/− mice
The abundance of transcripts encoding IL-6 was ninefold greater in IL-10−/−SERT−/− mice than in wild-type mice (Fig. 4A; P < 0.01). Expression of transcripts encoding IL-6 was also apparently greater than wild-type in IL-10−/−SERT+/+ and in IL-10−/−SERT+/− mice. Similarly, the abundance of transcripts encoding TNFα was 50-fold greater (P < 0.01) in IL-10−/−SERT−/− than in wild-type mice (Fig. 4B). Apparent but not statistically significant increases over wild-type occurred in transcripts encoding TNFα in IL-10−/−SERT+/+ (threefold) and in IL-10−/−SERT+/− mice (eightfold).
Figure 4.
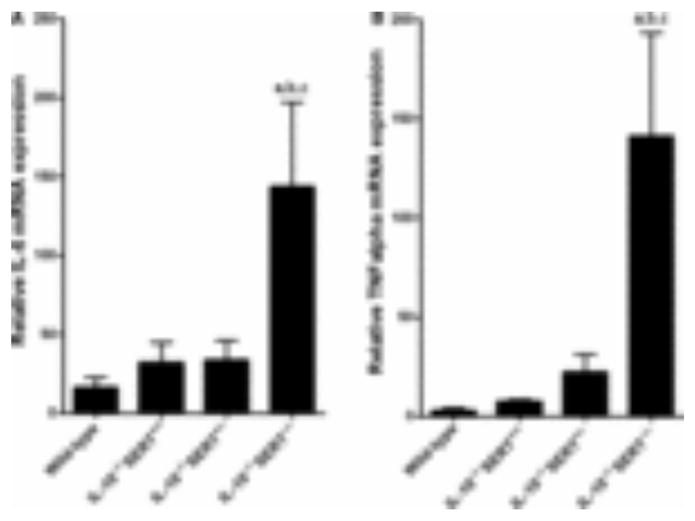
Expression of transcripts encoding IL-6 and TNFα in the colons of wild-type, IL-10−/−SERT+/+, IL-10−/−SERT+/−, and IL-10−/−SERT−/− mice. (A) Abundance of transcripts encoding IL-6 is significantly greater in IL-10−/−SERT−/− than in wild-type mice (aP < 0.01, bP < 0.01 compared to IL-10−/−SERT+/+ mice, cP < 0.01 compared to IL-10−/−SERT+/− mice). Data represent mean ± SEM (n = 4–6). (B) Abundance of transcripts encoding TNFα is significantly greater in IL-10−/−SERT−/− than in wild-type animals (aP < 0.01). Data represent mean ± SEM (n = 4–6).
The numbers of 5-HT-immunoreactive cells are decreased in IL-10−/− mice with colitis
2,4,6-Trinitrobenzene sulfonic acid-induced colitis in mice has been reported to be associated with an increased number of 5-HT-immunoreactive EC cells in the colon. 18,22,25 We therefore determined whether a similar increment in EC cell numbers occurs in the inflamed colons of mice that lack IL-10. Serotonin-immunoreactive EC cells were counted in wild-type mice and in animals with the IL-10−/−SERT+/+, IL-10−/−SERT+/−, IL-10−/−SERT−/− genotypes. The number of 5-HT-immunoreactive cells was significantly lower in the colons of IL-10−/−SERT+/+, IL-10−/− SERT+/−, and IL-10−/−SERT−/− mice than in those of wild-type animals (Fig. 5). Regardless of the associated SERT genotype, the number of 5-HT-immunoreactive cells was decreased in each of the three strains of mouse lacking IL-10 by ~55–65%. The increase in EC cell number associated with TNBS-induced colitis thus does not occur when colitis occurs in the setting of IL-10 deficiency. The type of change that occurs in EC cell numbers as a result of colitis, therefore, evidently depends on the nature of the perturbation that provokes the colitis.
Figure 5.
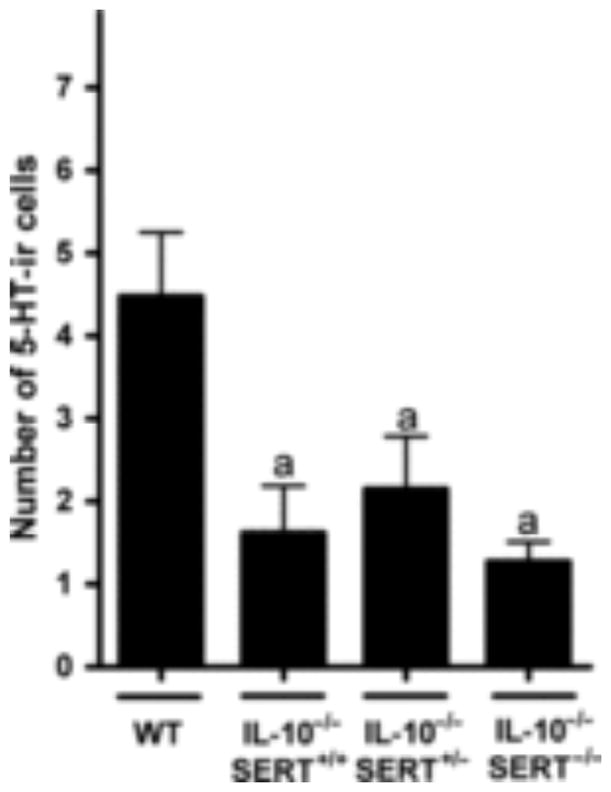
Serotonin immunoreactive cells in colon of wild-type, IL-10−/−SERT+/+, IL-10−/−SERT+/−, and IL-10−/−SERT−/− mice. Quantitative analysis of 5-HT immunoreactive cells in the proximal colon (400×). Data are shown as absolute numbers per field (mean ± SEM, n = 4–6). aP < 0.05 compared to wild-type mice.
Discussion
Serotonin has been proposed, as a result of studies in humans and animal models, to play a role in the pathophysiology of IBS and colitis (23, reviewed in 7,26). Pharmacological inhibition or genetic loss of SERT, the major regulator of extracellular 5-HT levels in tissue, leads to abnormal gastrointestinal motility in mice manifested, in the case of the SERT knockout animals, by an alternating pattern of diarrhea and constipation. 15,16 The present observations confirm that the aberrant intestinal 5-HT signaling that occurs in the SERT-deficient bowel aggravates the spontaneous inflammation of the colon that accompanies IL-10 deficiency. These data also suggest that different animal models of colitis exert different effects on SERT expression and EC cell generation.
We have previously used TNBS-induced colitis 27 in mice that lack SERT as a gain-of-function model to investigate the role of serotonergic signaling in intestinal inflammation. The enhanced serotonergic signaling associated with SERT deficiency was found to significantly increase the severity of the colitis induced by TNBS, suggesting that endogenous 5-HT exerts a proinflammatory effect in the colon. 22,23 In the current study, we employed another model, in which the deletion of IL-10 leads to the spontaneous occurrence of colitis, typically 8–10 weeks after birth. An advantage of studying colitis in mice that lack IL-10 is that it is not necessary to use a potentially confounding exogenous toxin, such as TNBS, to initiate inflammation.
A genetic ‘double hit’ to the host immune system has been proposed to be a determinant of the susceptibility and severity of IBD. 24 An example of a ‘double hit’ increasing the severity of inflammation is the amplification of colitis that occurs in mice when the loss of IL-10 signaling is combined with that of transforming growth factor β (TGFβ). The inflammatory process in the colons of these mice is far more severe than that in animals in which either type of signaling has been ablated individually. In fact, the submaximal inflammation associated with IL-10 deletion, is advantageous for the design of gain-of-function experiments to test the hypothesis that enhanced intestinal serotonergic signaling can act as a ‘double hit’ to increase the severity of the spontaneous inflammation arising in the setting of IL-10 ablation. In the current study, we tested and confirmed the hypothesis that knockout of SERT provides a second proinflammatory drive or ‘double hit’ that adds to the loss of the IL-10 modulation of the inflammatory response to commensal organisms (reviewed in 1). The proinflammatory effect of SERT knockout is underlined by our observation that the abundance of transcripts encoding the pro-inflammatory cytokines, IL-6 ( ~4.5-fold) and TNFα (~18-fold), is increased in IL-10−/−SERT−/− double knockout mice over IL-10−/−SERT+/+ mice and even more in wild-type animals. The result, as in the IL-10/TGFβ double knockout mice, is to increase the severity of inflammation to a degree that resembles the severe disease seen in human ulcerative colitis. 24 In the setting of IL-10 deficiency, adding a proinflammatory influence, such as SERT knockout-induced facilitation of serotonergic signaling, may exert an effect on inflammation that is similar to that of subtracting the anti-inflammatory influence of TGFβ.
It is of interest that SERT expression is increased in the inflamed colons of IL-10−/−SERT+/+ mice and even in IL-10−/−SERT+/− animals. In contrast, SERT expression is decreased in TNBS colitis; furthermore, colitis in IL-10 knockout mice is associated with a decrease in 5-HT-immunoreactive cells, while in TNBS-induced colitis, the number of 5-HT-immunoreactive cells is increased. 18–20,22,28 These seemingly contradictory effects need to be explained; however, whatever the reason for the differences, the disparate effects of TNBS-induced and IL-10 knockout-associated colitis on SERT and EC cells indicate that despite the common occurrence of inflammation, the two models are fundamentally different. It is possible that this difference is related to the use of an exogenous toxin, such as an alcoholic solution of TNBS, to induce an acute colitis. The initial toxic effects to enterocytes may cause SERT expression to fall because enterocytes are the major expressers of SERT in the mucosa (reviewed in [7]). Toxic damage to the surface epithelium may also lead to a compensatory increase in the generation of epithelial cells from stem cells in crypts and a more rapid turnover of enterocytes. Enhanced production of epithelial cells could underlie the increase in EC cells that is seen in TNBS-induced colitis. The T-cell mediated inflammation of the mucosa that occurs in celiac disease also causes an increase in epithelial cell turnover, an increase in EC cell numbers, decreased SERT expression, and increased bioavailability and release of 5-HT. 29 Increased epithelial turnover would be expected to reduce the time available for enterocyte differentiation, which might further contribute to their reduced SERT expression. Whatever the cause is, studies with SERT-deficient mice suggest that a reduction in expression of SERT would increase the severity of the later immunologically driven phase of TNBS-induced colitis. Tumor necrosis factor-α and interferon-γ reduce SERT expression in cultured Caco-2 cells and would be likely to keep levels of SERT low, because both are elevated during the later phase of TNBS-induced colitis. 30
In contrast to TNBS-induced colitis, the inflammation associated with the ablation of IL-10 is slowly developing and is not preceded by toxic damage to the GI epithelium. Inflammation thus is not being initiated in the setting of an already enhanced turnover of epithelial cells. The effects of inflammation, moreover, are more chronic and thus would allow compensatory mechanisms to oppose EC differentiation or enhanced activation of EC cells in the course of inflammation. A compensatory increase in SERT expression would also reduce the proinflammatory effects of 5-HT. In fact, these compensations occurring in IL-10 deficient mice may be why a ‘double hit’ is required to make the inflammation associated with IL-10 knockout severe.
The increased bioavailability of 5-HT due to SERT down-regulation would, in addition to enhancing the severity of inflammation, be likely to enhance symptoms associated with it. Potentiation of the 5-HT4 receptor-mediated increase in release of acetylcholine, which strengthens neurotransmission in prokinetic pathways, 31 would enhance propulsive and secretory reflexes and thereby contribute to inflammation-associated diarrhea. Potentiation of 5-HT3-mediated activation of visceral afferent nerves would increase discomfort and visceral hypersensitivity.
In summary, studies of the knockout of SERT imply that the function of SERT-mediated 5-HT uptake in the colon is not limited to terminating serotonergic neurotransmission or the paracrine signaling of 5-HT released from EC cells that initiates peristaltic and secretory reflexes. Serotonin reuptake transporter evidently also plays a significant role in modulating the proinflammatory effects of 5-HT. During the early period of inflammation, when the colonic epithelium is intact, it is possible that upregulation of SERT in enterocytes can increase 5-HT uptake and dampen the proinflammatory effects of released 5-HT. This upregulation occurs in the epithelium of IL-10 knockout mice and may be a compensatory change that limits the damage due the lack of adequate regulation of immunity. During later phases of inflammation when substantial epithelial damage or destruction exists, SERT expression is decreased. The loss of SERT would amplify the proinflammatory drive due to 5-HT release, which might then exert a catalytic effect on inflammation, worsening its severity. The reduction of SERT expression in the epithelium of patients with ulcerative colitis is thus likely to contribute both to the severity of the inflammatory process and to the mediation of symptoms associated with it. Given the importance of SERT and the potential of 5-HT to act as a proinflammatory mediator, it is reasonable to suggest that antidepressants and selective serotonin reuptake inhibitors, which inhibit SERT, be used with caution in patients in whom intestinal inflammation exists or may develop.
Acknowledgments
This work was supported by the Columbia University (Ludwig-Schaefer-Award to S.C.B.), by the Land Baden-Württemberg (Landesforschungsschwerpunkt ‘Immune and neurologic regulation in the gastrointestinal tract’, coordinated by S.C.B.), and by the Zentrum für Ernährungsmedizin of the Universities of Hohenheim and Tübingen, Germany.
Abbreviations
- 5-HT
5-hydroxytryptamine = serotonin
- EC cell
enterochromaffin cell
- IBD
inflammatory bowel disease
- IBS
irritable bowel syndrome
- IL
interleukin
- SERT
serotonin reuptake transporter
- TGFβ
transforming growth factor β
- TNBS
2,4,6-trinitrobenzene sulfonic acid
- TNFα
tumor necrosis factor alpha
Footnotes
Competing interests: the authors have no competing interests.
References
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–34. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.De Schepper HU, De Man JG, Moreels TG, Pelckmans PA, De Winter BY. Review article: gastrointestinal sensory and motor disturbances in inflammatory bowel disease - clinical relevance and pathophysiological mechanisms. Aliment Pharmacol Ther. 2008;27:621–37. doi: 10.1111/j.1365-2036.2008.03624.x. [DOI] [PubMed] [Google Scholar]
- 3.Kraneveld AD, Rijnierse A, Nijkamp FP, Garssen J. Neuro-immune interactions in inflammatory bowel disease and irritable bowel syndrome: future therapeutic targets. Eur J Pharmacol. 2008;585:361–74. doi: 10.1016/j.ejphar.2008.02.095. [DOI] [PubMed] [Google Scholar]
- 4.Askenase PW, Herzog WR, Millet I, et al. Serotonin initiation of delayed-type hypersensitivity: mediation by a primitive Thy-1+ antigen-specific clone or by specific monoclonal IgE antibody. Skin Pharmacol. 1991;4(Suppl 1):25–42. doi: 10.1159/000210981. [DOI] [PubMed] [Google Scholar]
- 5.Leon-Ponte M, Ahern GP, O’Connell PJ. Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-HT7 receptor. Blood. 2007;109:3139–46. doi: 10.1182/blood-2006-10-052787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young MR, Matthews JP. Serotonin regulation of T-cell subpopulations and of macrophage accessory function. Immunology. 1995;84:148–52. [PMC free article] [PubMed] [Google Scholar]
- 7.Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. 2007;132:397–414. doi: 10.1053/j.gastro.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Bogunovic M, Dave SH, Tilstra JS, et al. Enteroendocrine cells express functional Toll-like receptors. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1770–83. doi: 10.1152/ajpgi.00249.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Idzko M, Panther E, Stratz C, et al. The serotoninergic receptors of human dendritic cells: identification and coupling to cytokine release. J Immunol. 2004;172:6011–9. doi: 10.4049/jimmunol.172.10.6011. [DOI] [PubMed] [Google Scholar]
- 10.Fiebich BL, Akundi RS, Seidel M, et al. Expression of 5-HT3A receptors in cells of the immune system. Scand J Rheumatol Suppl. 2004;119:9–11. [PubMed] [Google Scholar]
- 11.Yang GB, Qiu CL, Zhao H, Liu Q, Shao Y. Expression of mRNA for multiple serotonin (5-HT) receptor types/subtypes by the peripheral blood mononuclear cells of rhesus macaques. J Neuroimmunol. 2006;178:24–9. doi: 10.1016/j.jneuroim.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 12.Yin J, Albert RH, Tretiakova AP, Jameson BA. 5-HT(1B) receptors play a prominent role in the proliferation of T-lymphocytes. J Neuroimmunol. 2006;181:68–81. doi: 10.1016/j.jneuroim.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 13.McDuffie JE, Motley ED, Limbird LE, Maleque MA. 5-hydroxytryptamine stimulates phosphorylation of p44/p42 mitogen-activated protein kinase activation in bovine aortic endothelial cell cultures. J Cardiovasc Pharmacol. 2000;35:398–402. doi: 10.1097/00005344-200003000-00008. [DOI] [PubMed] [Google Scholar]
- 14.Camilleri M, Andrews CN, Bharucha AE, et al. Alterations in expression of p11 and SERT in mucosal biopsy specimens of patients with irritable bowel syndrome. Gastroenterology. 2007;132:17–25. doi: 10.1053/j.gastro.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen JJ, Li Z, Pan H, et al. Maintenance of serotonin in the intestinal mucosa and ganglia of mice that lack the high-affinity serotonin transporter: abnormal intestinal motility and the expression of cation transporters. J Neurosci. 2001;21:6348–61. doi: 10.1523/JNEUROSCI.21-16-06348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coates MD, Johnson AC, Greenwood-Van MB, Mawe GM. Effects of serotonin transporter inhibition on gastrointestinal motility and colonic sensitivity in the mouse. Neurogastroenterol Motil. 2006;18:464–71. doi: 10.1111/j.1365-2982.2006.00792.x. [DOI] [PubMed] [Google Scholar]
- 17.Bian X, Patel B, Dai X, Galligan JJ, Swain G. High mucosal serotonin availability in neonatal guinea pig ileum is associated with low serotonin transporter expression. Gastroenterology. 2007;132:2438–47. doi: 10.1053/j.gastro.2007.03.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linden DR, Chen JX, Gershon MD, Sharkey KA, Mawe GM. Serotonin availability is increased in mucosa of guinea pigs with TNBS-induced colitis. Am J Physiol Gastrointest Liver Physiol. 2003;285:G207–16. doi: 10.1152/ajpgi.00488.2002. [DOI] [PubMed] [Google Scholar]
- 19.Linden DR, Foley KF, McQuoid C, Simpson J, Sharkey KA, Mawe GM. Serotonin transporter function and expression are reduced in mice with TNBS-induced colitis. Neurogastroenterol Motil. 2005;17:565–74. doi: 10.1111/j.1365-2982.2005.00673.x. [DOI] [PubMed] [Google Scholar]
- 20.O’Hara JR, Ho W, Linden DR, Mawe GM, Sharkey KA. Enteroendocrine cells and 5-HT availability are altered in mucosa of guinea pigs with TNBS ileitis. Am J Physiol Gastrointest Liver Physiol. 2004;287:G998–1007. doi: 10.1152/ajpgi.00090.2004. [DOI] [PubMed] [Google Scholar]
- 21.Coates MD, Mahoney CR, Linden DR, et al. Molecular defects in mucosal serotonin content and decreased serotonin reuptake transporter in ulcerative colitis and irritable bowel syndrome. Gastroenterology. 2004;126:1657–64. doi: 10.1053/j.gastro.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 22.Bischoff SC, Mailer R, Pabst O, et al. Role of serotonin in intestinal inflammation: knockout of serotonin reuptake transporter exacerbates 2,4,6-trinitrobenzene sulfonic acid colitis in mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G685–95. doi: 10.1152/ajpgi.90685.2008. [DOI] [PubMed] [Google Scholar]
- 23.Ghia JE, Li N, Wang H, et al. Serotonin has a key role in pathogenesis of experimental colitis. Gastroenterology. 2009;137:1649–60. doi: 10.1053/j.gastro.2009.08.041. [DOI] [PubMed] [Google Scholar]
- 24.Kang SS, Bloom SM, Norian LA, et al. An antibiotic-responsive mouse model of fulminant ulcerative colitis. PLoS Med. 2008;5:e41. doi: 10.1371/journal.pmed.0050041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Atkinson W, Lockhart S, Whorwell PJ, Keevil B, Houghton LA. Altered 5-hydroxytryptamine signaling in patients with constipation- and diarrhea-predominant irritable bowel syndrome. Gastroenterology. 2006;130:34–43. doi: 10.1053/j.gastro.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 26.Spiller R. Serotonin and GI clinical disorders. Neuropharmacology. 2008;55:1072–80. doi: 10.1016/j.neuropharm.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 27.Scheiffele F, Fuss IJ. Induction of TNBS colitis in mice. Curr Protoc Immunol. 2002;Chapter 15(Unit 15.19) doi: 10.1002/0471142735.im1519s49. [DOI] [PubMed] [Google Scholar]
- 28.O’Hara JR, Lomax AE, Mawe GM, Sharkey KA. Ileitis alters neuronal and enteroendocrine signalling in guinea pig distal colon. Gut. 2007;56:186–94. doi: 10.1136/gut.2006.102780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coleman NS, Foley S, Dunlop SP, et al. Abnormalities of serotonin metabolism and their relation to symptoms in untreated celiac disease. Clin Gastroenterol Hepatol. 2006;4:874–81. doi: 10.1016/j.cgh.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 30.Te Velde AA, De KF, Sterrenburg E, et al. Comparative analysis of colonic gene expression of three experimental colitis models mimicking inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:325–30. doi: 10.1002/ibd.20079. [DOI] [PubMed] [Google Scholar]
- 31.Liu M, Geddis MS, Wen Y, Setlik W, Gershon MD. Expression and function of 5-HT4 receptors in the mouse enteric nervous system. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1148–63. doi: 10.1152/ajpgi.00245.2005. [DOI] [PubMed] [Google Scholar]