

Abstract
Background
Regular colonoscopic surveillance for detection of dysplasia is recommended in longstanding inflammatory bowel disease (IBD), however, its sensitivity is disputed. Screening accuracy may increase by using a biomarker-based surveillance strategy.
Methods
A case-control study was performed to determine the prognostic value of DNA ploidy and p53 in IBD-related neoplasia. Cases with IBD-related colorectal cancer (CRC), detected in our surveillance program between 1985-2008, were selected and matched with two controls, for age, gender, disease characteristics, interval of follow-up, PSC, and previous surgery. Biopsies were assessed for DNA ploidy, p53, grade of inflammation and neoplasia. Progression to neoplasia was analyzed with Cox regression analysis, adjusting for potentially confounding variables.
Results
Adjusting for age, we found statistically significant Hazard ratios (HR) between development of CRC, and low grade dysplasia (HR5.5; 95%CI 2.6-11.5), abnormal DNA ploidy (DNA index (DI) 1.06-1.34, HR4.7; 95%CI 2.9-7.8 and DI>1.34, HR6.6; 95%CI 3.7-11.7) and p53 immunopositivity (HR3.0; 95%CI 1.9-4.7) over time. When adjusting for all confounders, abnormal DNA ploidy (DI 1.06-1.34, HR4.7; 95%CI 2.7-7.9 and DI>1.34, HR5.0; 95%CI 2.5-10.0) and p53 immunopositivity (HR1.7; 95%CI 1.0-3.1) remained statistically significant predictive of neoplasia.
Conclusion
In longstanding IBD, abnormal DNA ploidy and p53 immunopositivity are important risk factors of developing CRC. The yield of surveillance may potentially increase by adding these biomarkers to the routine assessment of biopsies.
Keywords: Inflammatory bowel disease, Colorectal cancer, Surveillance, Abnormal DNA ploidy, p53 immunopositivity
Introduction
Inflammatory bowel disease (IBD), which includes ulcerative colitis (UC) and Crohn’s disease (CD), is a chronic inflammatory disorder of the intestine. With approximately 4 million patients affected in the Western world, IBD is one of the most common chronic inflammatory disorders. Epidemiological evidence indicates that patients with IBD are at increased risk of developing colorectal cancer (CRC). The risk is considerable as it has been estimated that up to one in five patients will develop CRC within 30 years after an initial diagnosis of IBD [9].
Duration of the disease is a major clinical risk factor for IBD-related CRC, with a relative risk with long-standing colitis increasing to 6–18 [6, 7]. Most IBD-related CRC is seen in patients with pancolitis, whereas proctitis and left-sided colitis are associated with a small to intermediate cancer risk. Other risk factors for IBD-related CRC include young age at onset of IBD, presence of primary sclerosing cholangitis, presence of pseudopolyps, and a positive family history for CRC [1, 6, 7, 19].
Current guidelines from the American Gastroenterology Association (AGA) [2] and the British Society of Gastroenterology (BSG) [4, 8] recommend colonoscopic surveillance every 1 to 5 years for IBD-colitis patients starting 8 to 10 years after the initial diagnosis. This surveillance aims at early detection of neoplastic lesions, at a treatable stage [6]. Detection of dysplasia is generally accepted as the gold standard to predict cancer risk. Dysplasia as risk predictor for IBD-related cancer is however known to have significant shortcomings.
Dysplasia, which is commonly sub classified into two distinct morphological stages, i.e., low-grade dysplasia (LGD) and high-grade dysplasia (HGD), often arises multifocally throughout the colon of IBD patients. As dysplastic lesions are difficult to recognize by conventional endoscopy, the obtained biopsy specimens are prone to sampling error [18, 33]. In addition, misinterpretation of dysplasia occurs due to inter- and intraobserver variability between pathologists, and by the co-existent presence of variable degrees of inflammation [9, 26, 33].
In the search of better markers for the prediction of IBD-related CRC development, a variety of biomarkers have been studied, including DNA ploidy, and mutations of the APC gene, K-ras gene, DCC gene and p53 tumor suppressor gene [5, 18]. Of these, DNA ploidy and p53 gene mutations currently seem to be the most promising candidates [5, 16, 18, 25, 34]. Both markers are observed early during the cascade of CRC development and, in addition, they are assumed to be diffusely expressed thus decreasing the risk of sampling error [21, 22].
As the relative risk of abnormal DNA ploidy and p53 immunopositivity in developing IBD-related CRC is currently unknown, we investigated the prognostic value of these markers for neoplastic progression in a case–control study design by testing surveillance biopsies of high-risk IBD patients (biopsy specimens taken ≥8 years after the diagnosis of IBD), who either or not progressed towards CRC.
Materials and methods
Patients selection
This retrospective case–control study was conducted in the IBD patient cohort of the Erasmus MC, Rotterdam, the Netherlands. Cases were high-risk IBD patients with documented neoplastic progression (defined as patients with progression to CRC). Each case was preferentially matched with two controls. Controls were IBD-colitis patients without signs of neoplastic progression (defined as patients without HGD or CRC) during surveillance. Cases and controls were matched for age (with a maximum of 2 years difference), gender, colitis characteristics (i.e. extension and underlying UC or CD), age at onset of colitis symptoms, disease duration, interval of follow-up since the first colonoscopy (with a maximum of 1 year difference), presence of PSC, and previous surgery. Inclusion criteria for cases were (i) IBD history ≥8 years; (ii) confirmed IBD by colonoscopy and histology; and (iii) having undergone at least one surveillance colonoscopy with biopsy sampling prior to the development of neoplasia. An exclusion criterion was IBD with dysplasia or CRC at the first colonoscopy.
Biopsies specimens collected at routine surveillance colonoscopies or colectomy between 1985 and 2008 in our institute were retrieved and analyzed for histology, DNA ploidy and p53 as described below. They consist of 1,671 samples obtained from different locations of the colon and originated from 54 unrelated patients. Use of patient material was approved by the Medical Ethical Committee of the Erasmus MC.
Since 2001, a standardized biopsy protocol was used (four-quadrant biopsy specimens every 10 cm). Before 2001, the biopsy protocol was not standardized.
Histology
Formalin-fixed paraffin-embedded tissue samples were serially sectioned at 4 or 50 μm. The first and last section 4 μm were stained with hematoxylin & eosin stain and light microscopically evaluated. The degree of inflammation and dysplasia grade or presence of CRC was reevaluated by one expert gastrointestinal pathologist without knowledge of clinical and biomarker status, using the Geboes scoring system [10] and the International Classification of Dysplasia in Inflammatory Bowel Disease [29]. Patients were classified according to the most severe abnormality present in the biopsy specimens.
DNA ploidy by flow cytometry
DNA ploidy analysis was performed on formalin-fixed paraffin-embedded biopsy specimens using a pepsinization technique, which was modified from Hedley et al. [15]. For each sample, one 50-μm-thick section was obtained from the paraffin block. Following deparaffinization in xylene and subsequent rehydration. The tissue was suspended in phosphate-buffered-saline (PBS). Then, the specimens were incubated in 0.05% protease (Sigma-Aldrich, Zwijdrecht, The Netherlands) for 45 min at 37°C, minced mechanically with a syringe and a 60-μm needle, and filtered through a 50-μm nylon mesh. Subsequently, the suspension was centrifuged for 10 min at 900 g, the supernatant was discarded and the pellet was resuspended in PBS containing 0.01% RNAse (Simga-Aldrich). For DNA staining, 0.01% propidium iodide was added to the samples. At least 10,000–15,000 nuclei per specimen were analyzed by FACScan flow cytometry (Becton Dickinson, San Jose, CA). Data analysis was performed using CellQuest (Becton Dickinson) and Modfit 3.1 software packages (Verity Software House, Inc, Topsham, ME, USA). The DNA index (DI) was calculated as the ratio of the abnormal G0/G1 mean peak channel number to the normal diploid G0/G1 mean peak channel number. Histograms displaying a G0/G1 peak with a coefficient of variation (CV) of 1%≤CV≤10% were included in the analysis. Samples were considered diploid when the DNA index was 0.95≤DI≤1.05, near diploid in case of 1.06≤DI≤1.34, and aneuploid in case of a DI >1.34 [11, 32]. DNA histograms were classified without knowledge of the histological diagnosis.
p53 immunohistochemistry
Immunohistochemistry for p53 was performed on 4 μm formalin-fixed paraffin-embedded tissue slide. After deparaffinization in xylene and rehydration through graded ethanol, the sections were incubated with 3% H2O2 in methanol for 10 min. Then, the section were microwaved in monocitric acid buffer pH 6.0 for 20 min, washed, and blocked with 10% normal human plasma, 10% goat serum (Dako-cytomation, Heverlee, Belgium) and 5% bovine serum albumin (Sigma-Aldrich) in Tris–HCl pH 9.0. p53 immunostaining was accomplished using a mouse monoclonal antibody, i.e. clone DO-7 (1:100, Dako-cytomation). After overnight incubation with DO-7 at 4°C, p53 signals were amplified with Envision (Dako-cytomation). Subsequently, 3′-3′-diaminobenzidine (1 g/L, Sigma-Aldrich) was added to the slide to detect p53. The slides were counterstained in Harris’ hematoxylin, dehydrated and mounted. To determine the intraobserver variability between the different experiments, control slides (n = 3) of tissue that previously had shown to express p53 were included in the experiments. When the results of control slides differed from the results of previous staining, data were excluded from the analysis and the experiment was repeated. Substitution of the primary antibody by IgGb2 (Dako-cytomation) or Tris–HCl pH 9.0 in a matched serial section was used as a negative control. The intensity and percentage of positive cells were evaluated by two experts without knowledge of clinical and DNA ploidy status. Overexpression of p53 was defined as moderate and intense brown staining in >15% of the nuclei [14].
Statistical analysis
Statistical analyses were conducted using SPSS software (SPSS version 16.0, Chicago, Illinois, USA) and R software (R foundation for Statistical Computing, version 2.6.2).
Median and 25–75 percentiles were calculated for all continuous variables while proportions were calculated for all categorical variables.
At each follow-up visit (colonoscopies performed on regular basis ≥8 years after the diagnosis of IBD), histology, expression of the biomarkers, and grade of inflammation were determined in biopsy specimens taken from different locations of the colon of the same patient. For the analysis, we used from each time point the biopsy that was most different from normal (e.g. if for one patient at one time point one biopsy was aneuploid, one near diploid and three diploid, that time point was scored aneuploid).
To estimate the predictive value of histology, DNA ploidy, p53 and grade of inflammation we used Cox proportional hazard regression with time dependent covariates to take the multiple time points per patient into account. The relationship between these predictors and IBD-related neoplasia was expressed in hazard ratios (HR) with 95% confidence intervals (CI). We first adjusted for age, then for age and LGD and finally for the complete set of confounders (age, LGD, DNA ploidy, p53 immunopositivity and grade of inflammation).
Random effect regression analysis was used to calculate the statistical significance level of the correlation between outcome and the different predictors. This model takes the dependency between biopsy specimens per follow-up visit per patient into account. The data is presented by grade of neoplasia. P-values <0.05 were considered statistically significant. Smoothing splines were used to visualize trends over time for the biomarkers, DNA ploidy and p53 immunopositivity.
Results
Patient characteristics
In our cohort of IBD patients who underwent colonoscopic surveillance at the Erasmus MC—Rotterdam between 1985 and 2008, 36 patients developed CRC. Sixteen patients were excluded from the study because of an IBD history <8 years, HGD or CRC at their first surveillance colonoscopy, or lack of surveillance biopsies. For the twenty remaining IBD cases (CD, n = 8; UC, n = 12), 844 biopsy specimens, with a median (25–75 percentiles) of 6.0 (4.0–10.0) biopsies per colonoscopy or colectomy, were analyzed. 730 biopsy specimens had been taken prior to the development of HGD and/or CRC, and 171 specimens at the time of HGD and/or CRC. Cases were matched with 34 controls (CD, n = 14; UC, n = 20). From these, in total 827 biopsy specimens obtained during colonoscopic surveillance or colectomy were included. For six cases (CD, n = 2; UC, n = 4), no second matched control fulfilling all selection criteria could be selected from our patient cohort.
All patients’ characteristics are given in Table 1. No significant differences were observed between the cases and controls in age, gender, disease characteristics, interval of follow-up, treatment during surveillance, family history, PSC, and previous surgery.
Table 1.
Characteristics of inflammatory bowel disease (IBD) patients
| Variables | Cases (n = 20 patients) | Controls (n = 34 patients) | p-value |
|---|---|---|---|
| Age at IBD diagnosis, y | 25.3 (19.2–38.4) | 25.8 (18.9–34.8) | 0.6 |
| Male, n | 20 (75.0%) | 24 (70.6%) | 0.7 |
| IBD history, y | 17.0 (12.0–23.8) | 18.5 (14.0–23.3) | 0.7 |
| Follow-up period, y | 8.0 (1.3–11.5) | 7.2 (2.2–12.6) | 0.8 |
| Colonoscopies/follow-up period, n | 4.4 (1.5–7.0)a | 4.3 (2.0–5.3) | 0.8 |
| Interval between follow-up colonoscopies, y | 1.6 (0.6–2.3)a | 2.0 (1.0–2.4) | 0.04 |
| Total number of follow-up visits | 116 | 148 | – |
| Type of colitis, n | 0.2 | ||
| Proctitis | 3 (15.0%) | 1 (2.9%) | |
| Left-sided colitis | 3(15.0%) | 12 (35.3%) | |
| Pancolitis | 14 (70.0%) | 20 (58.8%) | |
| Right-sided colitis | – | 1 (2.9%) | |
| Positive family history, n | 2 (10.0%) | 4/34 (11.8) | 0.8 |
| Primary sclerosing cholangitis, n | 6 (30.0%) | 6 (17.6%) | 0.3 |
| Medication use | 18 (90.0%)b | 34 (100%) | |
| Aminosalicylates, n | 15 (83%) | 31 (91%) | 0.4 |
| Corticosteroids, n | 11 (61%) | 21 (62%) | 1.0 |
| Immunosuppressives, n | 7 (38%) | 11 (32%) | 0.6 |
| Biologicals, n | 2 (11%) | 2 (6%) | 0.5 |
| Surgery during surveillance, n | 5 (25.0%) | 4 (11.4%) | 0.2 |
| Total number of biopsies, n | 844 | 827 | – |
| Biopsies per colonoscopy | 6.0 (4.0–10.0) | 5.6 (3.0–7.0) | 0.08 |
aPrior to the development HGD and/or CRC
bInformation on medication use of two patients was missing
y, years; n, number
Median (25–75 percentiles) given for continuous variables
During surveillance, ten colectomies were recommended for reasons of dysplasia (all cases), and six as a result of severe colitis (cases, n = 2; controls, n = 4). One patient with LGD refused to undergo surgery. In seven of the nine colectomies after a previous biopsy-based diagnosis of dysplasia, HGD and/or CRC lesions were detected by the pathologist. No sign of neoplasia were seen in the colectomies of the patients with severe colitis. All patients without histological conformation of HGD or CRC continued the surveillance program.
Histology
Grading surveillance biopsies according to the International Classification of Dysplasia in Inflammatory Bowel Disease [29] revealed that the majority of specimens (1123/1671; 67.2%) exhibited no dysplasia (ND). Biopsy specimens of one follow-up visit of the cases could not be graded due to severe architectural changes. When the specimens were analyzed per follow-up visit (Fig. 1a), ND was seen in 53.0% (61/115) of the follow-up visits in the cases, compared to 87.8% (130/148) in the controls. Indefinite dysplasia (IND) was found in 16.5% (19/115) and 11.5% (17/148), respectively. LGD was detected in 7.8% (9/115) of the follow-up visits in the cases, compared to 0.7% (1/148) in the controls. HGD and CRC were detected in 5.2% (6/115) and 17.4% (20/115) of the follow-up visits in cases, compared to none in controls.
Fig. 1.
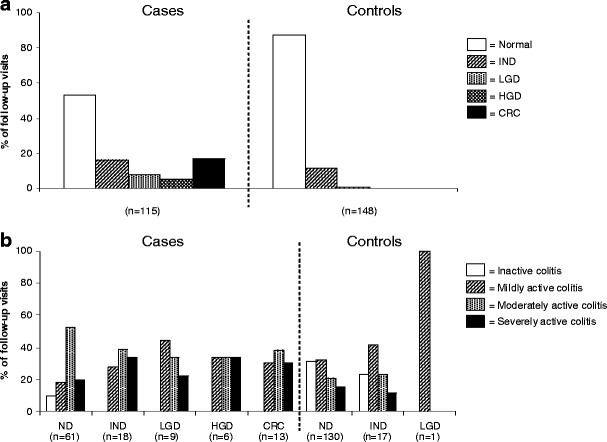
Prevalence of neoplasia and inflammatory activity in different IBD subgroups. a Percentages of follow-up visits with neoplasia graded according to the International Classification of Dysplasia in Inflammatory Bowel Disease [29] in the case and control group. Biopsy specimens of one follow-up visit of the cases could not be graded due to severe architectural changes. P-value from random effect regression analyses for difference between the cases and controls in grade of neoplasia is p = 0.7 for IND, p = 0.8 for LGD, p = 1.00 for HGD and p = 1.00. In the ND-group this analysis revealed p-value of p = 0.8 for IND. ND has been used as reference conditions. b Inflammatory activity graded according to the Geboes scoring system [10] in different subgroups of IBD patients. Biopsy specimens of eight follow-up visits of the cases could not be graded due to severe architectural changes or limited amount of tissue (IND n = 1, CRC n = 7). P-value from random effect regression analyses for difference between the cases and controls in inflammatory activity are p = 0.8 for mild colitis, p = 0.7 for moderate colitis and p = 0.7 for severe colitis. In the ND-group the same analysis revealed p-values of p = 0.9, p = 0.7 and p = 0.8, respectively. Inactive colitis has been used as reference conditions. Normal, ND, no dysplasia; IND, indefinite dysplasia; LGD, low grade dysplasia; HGD, high grade dysplasia; CRC, colorectal cancer (CRC). Mild colitis, neutrophil infiltration of <50% with no crypt destruction, erosion or ulceration; moderate colitis, neutrophil infiltration of >50% with no erosion or ulceration; severe colitis, erosion or ulceration
By endoscopy and macroscopy examination of the colon after colectomy, 80% (29/36) of the abnormal lesions were seen. Seventeen lesions were raised. Four lesions with dysplasia (LGD n = 3, HGD = 1) were missed by the endoscopists and were only detected histologically. For three lesions, the data on morphology of the lesion was missing.
When comparing the different specimens obtained from a single patient at a single time point, we found that 41.6% (14/36) of the sample-sets displayed differences in the grading between the individual samples from that particular time point. In most of these cases (n = 9), HGD and CRC at one spot was accompanied by LGD or HGD at another spot. At five time points, biopsy specimens with neoplasia were found together with biopsy specimens without neoplasia (no LGD, HGD and CRC). As 66% (10/15) the colonoscopies were performed before the standardized biopsy protocol of 2001, data on the exact location of the biopsy is scarce.
We also graded the surveillance biopsies according to the Geboes scoring system [10]. Biopsy specimens of eight follow-up visits of the cases could not be graded due to severe architectural changes or limited amount of tissue (IND n = 1, CRC n = 7). Moderate colitis was more often seen in cases per follow-up visit than in controls, even before LGD, HGD and/or CRC occurred (Fig. 1b).
When comparing the sample-sets from a single patient at a single time point with neoplasia, differences in inflammatory activity between the individual samples was observed 16.6% (6/36) of the sample-sets. The highest inflammatory activity was always observed in samples obtained at the location with most severe histological abnormality of that particular time point.
DNA ploidy
Biopsy specimens of four follow-up visits (ND n = 3, LGD n = 1) of the cases and fourteen follow-up visits of the controls (ND n = 10, IND n = 4) could not be graded due to severity of inflammation or a limited amount of tissue. DNA aneuploidy (DI >1.34) in the cases was detected in 28.2% (33/110) of the follow-up visits. A near-diploid (DI 1.06 to 1.34) and diploid (DI 0.96 to 1.05) DNA content was seen respectively in 46.4% (51/110) and 25.5% (28/110) of the follow-up visits. For the controls, DNA aneuploidy was found in 2.2% (3/134) of the follow-up visits, and a near-diploid and diploid DNA content was observed in 3.0% (4/134) and 94.8% (127/134) of the follow-up visits, respectively.
When the cases were subdivided by grade of neoplasia, DNA aneuploidy was found in 50% (10/20) of the follow-up visits with CRC versus 16.7% (1/6) in those with HGD, 62.5% (5/8) in those with LGD, 31.6% (6/19) in those with IND, and 17.2% (10/58) in those with ND, while a near-diploid DNA content was seen in 45.0% (9/20), 33.3% (2/6), 12.5% (1/8), 52.6% (10/19), and 50.0% (29/58), respectively (Fig. 2a).
Fig. 2.
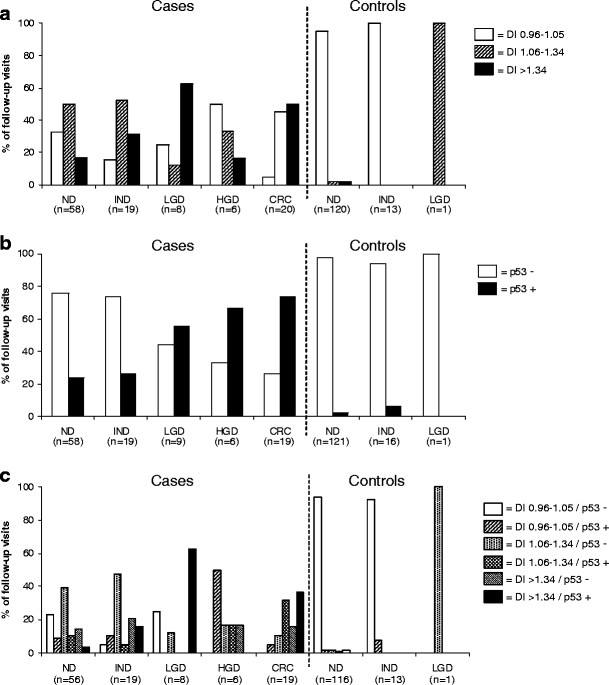
Prevalence of abnormal DNA content and p53 immunopositivity in different IBD subgroups. a Percentages of abnormal DNA content determined by flow cytometry in different subgroups of IBD patients. Biopsy specimens of four follow-up visits (ND n = 3, LGD n = 1) of the cases and fourteen follow-up visits of the controls (ND n = 10, IND n = 4) could not be graded due to severity of inflammation or a limited amount of tissue. P-values from random effect regression analyses for difference between the cases and controls in abnormal DNA content are p = 0.009 for DI 1.06–1.34 and p = 0.0.07 for DI >1.35. In the ND-group the same analysis revealed p-values of p = 0.006 and p = 0.03, respectively. b Percentages of p53 immunopositivity determined by immunohistochemistry in different subgroups of IBD patients. Biopsy specimens of four follow-up visits of the cases (ND n = 3, CRC n = 1) and ten follow-up visits of controls (ND n = 9, IND n = 1) could not be graded due to severity of inflammation or a limited amount of tissue. p53 overexpression is defined as moderate and intense brown staining in >15% of the nuclei. P-value from random effect regression analyses for difference between the cases and controls in p53 immunopositivity is p = 0.5. In the ND-group the p-value for p53 immunopositivity was p = 0.6. c Percentages of abnormal DNA content and p53 immunopositivity in different subgroups of IBD patients. P-values from random effect regression analyses for difference between the cases and controls in abnormal DNA content and p53 immunopositivity is p = 0.3 for DI 0.96–1.05/p53+, p = 0.1 for DI 1.06–1.34/p53-, p = 0.1 for DI 1.06–1.34/p53+, p = 0.04 for DI >1.34/p53-, p = 1.0 for DI >1.34/p53+. In the ND-group these p-values were p = 0.4, p = 0.2, p = 0.2, p = 0.03, p = 1.0 respectively. DI 0.96–1.05 and p53- have been used as reference conditions. ND, no dysplasia; LGD, low grade dysplasia; HGD, high grade dysplasia; CRC, colorectal cancer
In 27.7% (10/36) sample-sets obtained from single patients with neoplasia at a particular time point, differences in DI were seen between the individual biopsy specimens. The DI was the highest at the most severe histological abnormality present at that particular time point. In the sample-sets, where biopsy specimens with HGD and/or CRC were found together with specimens without neoplasia (n = 5, 3 sporadic lesions, 2 multifocal lesions), an abnormal DNA content was detected in specimens without neoplasia at a maximal distance of 65 cm from the lesion.
Five years prior to the development of CRC, an increase in the proportion of follow-up visits with DNA aneuploidy was found for the cases from 18 to 43%. In general, once DNA ploidy was positive it stayed positive over time. When a DI ≥1.06 was used as determinant, already ∼70% of cases were positive 10 years prior to the ultimate diagnosis of CRC, and this proportion remained stable over time. In the controls, the values were <5%, and no increase in the proportion of follow-up visits with an abnormal DNA content was seen over time (Fig. 3a).
Fig. 3.
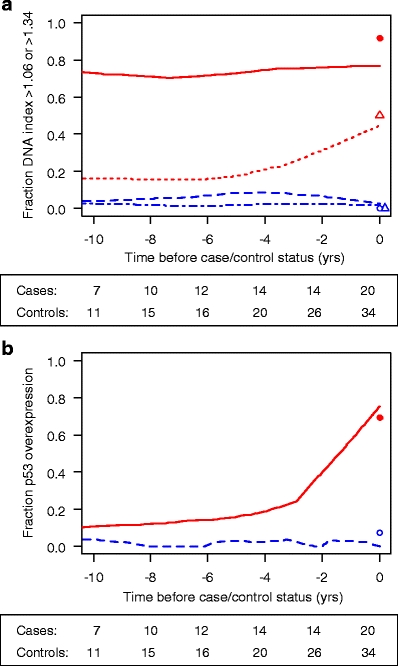
Fraction of biopsy specimens with abnormal DNA content/p53 immunopositivity over time prior to the development of colorectal cancer (CRC). a Fraction of abnormal DNA content. = cases, DI ≥1.06;
= cases, DI ≥1.06;  = cases, DI >1.34;
= cases, DI >1.34;  = controls, DI ≥1.06 and
= controls, DI ≥1.06 and  = controls, DI >1.34. b Fraction of p53 overexpression.
= controls, DI >1.34. b Fraction of p53 overexpression.  = cases;
= cases;  = controls. The numbers under each figure represent the number of IBD patients at that time point. The symbols at time point zero represent the fraction of patients at either time point with progression (cases n = 20) or at last follow-up visit without progression (controls n = 34)
= controls. The numbers under each figure represent the number of IBD patients at that time point. The symbols at time point zero represent the fraction of patients at either time point with progression (cases n = 20) or at last follow-up visit without progression (controls n = 34)
p53 expression
Biopsy specimens of four follow-up visits of the cases (ND n = 3, CRC n = 1) and ten follow-up visits of controls (ND n = 9, IND n = 1) could not be graded due to severity of inflammation or a limited amount of tissue. p53 immunopositivity in the cases was found in 37.3% (41/110) of the follow-up visits (Fig. 2b). By grade of neoplasia, these values were 73.7% (14/19) of the follow-up visits with CRC, 66.7% (4/6) with HGD, 55.6% (5/9) with LGD, 26.3% (5/19) with IND, and 24.1% (14/58) with ND. For the controls, only 2.9% (4/138; IND 1/16 and ND 3/121) of the follow-up visits were p53 positive.
In 16.6% (6/36) of the sample-sets obtained from single patients with neoplasia at a particular time point, differences in p53 immunopositivity were seen between the different biopsy specimens. p53 immunopositivity was found at the most severe histological abnormality present at that particular time point. In the sample-sets, where biopsy specimens with HGD and/or CRC were found together with specimens without neoplasia (n = 5), p53 immunopositivity was detected in specimens without neoplasia at a maximal distance of 55 cm from the lesion.
Prior to the development of CRC, an increase in the proportion of follow-up visits with p53 immunopositivity was seen in the cases from 10 to 75%. Once p53 was positive it remained positive over time. The increase in p53 immunopositivity was predominant at the time close to the development of CRC. In controls, p53 immunopositivity was detected in <5%, and no increase in proportion was detected over time (Fig. 3b).
When the p53 status of the cases was combined with the DNA ploidy data, only 14.8% (16/108) of the follow-up visits were p53 positive and had a DI >1.34 (aneuploid), in ten different patients, compared to none of the controls (Fig. 2c). When DNA ploidy with a DI of 1.06 to 1.34 (near-diploid) was considered as positive, 13.8% (13/108) of the follow-up visits in the cases were positive for both markers, in eight different patients, compared to one (0.8%, 1/130) follow-up visit of one control patient.
Prognostic value of the biomarkers
To compare the prognostic value of an abnormal DNA content, p53 immunopositivity and histological activity versus LGD for predicting neoplastic progression in subgroups of IBD patients, Cox regression with time dependent covariates was performed. With adjustment for age, significant associations with progression to advanced neoplasia were found for LGD, abnormal DNA ploidy, p53 immunopositivity, and moderate and severe colitis (Table 2).
Table 2.
Prognostic value of LGD, abnormal DNA ploidy, p53 immunopositivity and inflammatory activity for neoplastic progression in high-risk IBD patients according to Cox regression analysis with time-dependent covariates
| Variable | Adjusted for age | Adjusted for age and LGD | Adjusted for all other variables | |||
|---|---|---|---|---|---|---|
| HR (95%CI) | p | HR (95%CI) | p | HR (95%CI) | p | |
| LGD | 5.5 (2.6–11.5) | <0.0001 | 2.0 (0.8–4.9) | 0.2 | ||
| DNA ploidy, 1.06≤DI≤1.34a | 4.7 (2.9–7.8) | <0.0001 | 3.6 (2.2–5.9) | <0.0001 | 4.3 (2.5–7.2) | <0.0001 |
| DNA ploidy, DI >1.34 | 6.6 (3.7–11.7) | <0.0001 | 5.3 (3.3–8.3) | <0.0001 | 4.8 (2.3–9.6) | <0.0001 |
| p53 immunopositivityb | 3.0 (1.9–4.7) | <0.0001 | 2.2 (1.3–3.7) | 0.002 | 1.7 (1.0–3.1) | 0.04 |
| Mildly active inflammation | 2.3 (0.9–5.3) | 0.07 | 2.1 (0.8–5.4) | 0.1 | 1.9 (0.7–4.9) | 0.2 |
| Moderately active inflammation | 4.1 (1.8–9.1) | 0.001 | 4.0 (1.7–9.6) | 0.002 | 3.0 (1.2–7.2) | 0.02 |
| Severely active inflammation | 3.4 (1.4–8.2) | 0.005 | 4.7 (2.2–9.9) | <0.0001 | 2.3 (0.9–5.9) | 0.1 |
aDI, DNA index, calculated as ratio of the abnormal G0/G1 mean peak channel number to the normal diploid G0/G1 mean peak channel number
bp53 immunopositivity, defined as moderate and intense brown staining in >15% of the nuclei [14]
LGD, low grade dysplasia
Considering DNA ploidy with a DI 1.06–1.34 (near-diploid) over the length of surveillance as positive, a 4.7-fold increased risk for neoplastic progression was calculated. When DNA ploidy marked as DI >1.34 (aneuploid) was used as criterion, the risk of advanced neoplasia was 6.6-fold increased. For p53 immunopositivity, a 3.0-fold increased risk for advanced neoplasia was observed over time. For mild, moderate and severe colitis, the age adjusted analyses revealed respectively a 2.3, 4.1, and 3.4-fold increased risk for advanced neoplasia. For LGD, the risk for neoplastic progression was established at 5.5-fold.
When the risk values were adjusted for both age and presence of LGD, abnormal DNA ploidy, p53 immunopositivity, and moderate and severe colitis remained statistically significant predictive of neoplasia (Table 2). The risks for advanced neoplasia were established at 3.6-fold for a DI 1.06–1.34, 5.3-fold for a DI >1.34, 1.7-fold for p53 immunopositivity, and 4.0 and 4.7-fold for moderate and severe colitis.
Adjustment for all confounders revealed significant relationships between DNA ploidy status, p53 immunopositivity and moderate colitis, and the risk for later development of advanced neoplasia (Table 2). The increased risks for advanced neoplasia was calculated at 4.3-fold for a DI 1.06–1.34, and 4.8-fold for a DI >1.34. For p53 immunopositivity and moderate colitis, the risk was established at 1.7 and 3.0-fold, respectively.
Discussion
Many centers perform colonoscopic surveillance in patients with longstanding IBD, but despite these programs, CRC remains an important cause of IBD-related mortality [20]. Particularly for patients with PSC and/or extensive colitis with a duration of >8 years, conventional colonoscopic surveillance with random biopsy sampling every 1–3 year seems to be insufficient [6, 23, 30]. Several recent studies have focused on the value of adding specific biomarkers to predict the development of IBD-related CRC [16, 22, 25, 34].
At present, DNA ploidy and p53 are recognized as promising candidates for prediction of IBD-related CRC. Their obvious advantage is that they are both diffusely expressed and present at an early stage in the IBD related dysplasia-carcinoma sequence (Fig. 2, and ref. [17, 18, 22, 26]). However, the relative risk of abnormal DNA ploidy and p53 immunopositivity in developing IBD-related CRC has never been calculated. In the present study, a case–control study design based on our IBD surveillance database was performed to determine the risk of advanced neoplasia of both markers in subgroups of IBD patients. Potential biases in the study population were avoided by matching for age, gender, colitis characteristics, age of onset, duration of the disease, interval of follow-up, presence of PSC, and previous surgery.
In our study population, in age adjusted analyses significant relationships were observed between development of advanced neoplasia and prior presence of LGD (HR5.5), abnormal DNA ploidy (DI 1.06–1.34, HR4.7, and DI >1.34, HR6.6), and p53 immunopositivity (HR3.0) over time. When histological results were taken into account, i.e. the presence of LGD, both DNA ploidy and p53 immunopositivity maintained statistical significant predictors for development of advanced neoplasia (Table 2). Moreover, after adjustment for more confounders, abnormal DNA ploidy and p53 immunopositivity remained statistically significant predictive of neoplasia, with increasing risk for abnormal DNA ploidy depending on higher DI levels (Table 2).
Although risk stratification based on severity of histological signs of inflammation in IBD-related CRC was not the primary goal of this paper, we observed a correlation between severity of histological inflammation and increasing risk of neoplasia (Table 2). Evaluation of surveillance biopsy specimens showed that cases more often had histological signs of moderate colitis compared to the controls, in the years prior to the development of advanced neoplasia (HR 3.0). This observation is in agreement with recently published data [13, 31], and supports the concept that severity of inflammation in IBD patients is a risk factor for IBD-related neoplasia [13].
Despite the advantage of our case–control study design, there are several potential limitations to this report. Firstly, all patient material was tested retrospectively. Unfortunately, a prospective trial analogous to our study will encounter large difficulties and costs because of required patient numbers, years of follow-up, and surveillance colonoscopies needed [6, 7]. Secondly, our study was based on immunohistochemistry, which has the intrinsic shortcoming that the amount of staining is not linearly related to the amount of protein present, and may vary with the kit used.
Albeit most colonoscopies were performed during remission of the disease, some biopsy specimens have been taken when remission could not be achieved. The presence of histological signs of inflammation, and the regional or patchy presence of dysplasia, abnormal DNA ploidy and p53 immunopositivity [16, 21] which may even change over time [3, 24], might be a third putative drawback of the study. However, we tried to minimize these problems by using multiple biopsy specimens taken from different locations of the colon, validated scoring systems and two experts to evaluate histology and p53 immunopositivity. In order to calculate the hazard ratios we had to assume that the biopsy results at each location and time point accurately represented the grade of neoplasia, DNA ploidy status and p53 immunoreactivity in the colon at that interval. Obviously, this may not be correct, but given the number of specimens tested per patient, we expect that this bias was minimal.
Although we studied patients with UC as well as CD, no subdivision was made between the patient groups. This decision to interpret all results in the same analysis was based on the finding that no clear differences were seen between histology, DNA ploidy status and p53 immunoreactivity within the two different patient groups (data not shown).
Cox regression analysis with time-dependent covariates and random effect regression analyses in the ND-group revealed that patients positive for DNA ploidy and/or p53 are at high-risk of developing CRC. Therefore, extending the conventional colonoscopic surveillance with DNA ploidy and p53 could permit assigning IBD patients to either a low-risk (negative for dysplasia, DNA ploidy and p53), intermediate-risk (positive for dysplasia, DNA ploidy or p53) or high-risk (positive for dysplasia and DNA ploidy/p53) group. This refinement of risk stratification is of interest, since it would allow to allocate the resources more appropriate by reducing the number of unnecessary endoscopies for patients at a low-risk of developing CRC, while enabling more intensive surveillance endosocopies for those at high-risk of developing CRC. The results of this study suggest that the interval of colonoscopic surveillance in low-risk patients can be deferred up to 5 years. This has to be evaluated in future prospective studies. The surveillance in intermediate- and high-risk patients should however be intensified to every year and every 3 months, respectively. In the patients at risk, novel endoscopy techniques, such as high magnification endoscopy, chromoendoscopy, narrow band imaging, or autofluorescence endoscopy may enhance the detection of suspicious lesions [12, 27, 28, 35].
In conclusion, the data from our study support the notion that the use of DNA ploidy and p53 enhance the sensitivity of correctly selecting patients at an increased risk of IBD-related CRC development. Given the retrospective nature of our study and its limited power, the potential utility of DNA ploidy and p53 immunohistochemistry into clinical management of patients with longstanding IBD warrants further investigation.
Acknowledgement
This project was financially supported by the Erasmus MC by a grant to M.M. Gerrits (Mrace - Doelmatigheid & Zorg; project no. DRP/REJ/271190).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Monique M. Gerrits and Min Chen contributed equally to this work.
References
- 1.Ahmadi A, Polyak S, Draganov PV. Colorectal cancer surveillance in inflammatory bowel disease: the search continues. World J. Gastroenterol. 2009;15:61–66. doi: 10.3748/wjg.15.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.F.A. Farraye, R.D. Odze, J. Eaden, S.H. Itzkowitz. AGA medical position statement on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology 138, 738–745 (2010) [DOI] [PubMed]
- 3.Befrits R, Hammarberg C, Rubio C, Jaramillo E, Tribukait B. DNA aneuploidy and histologic dysplasia in long-standing ulcerative colitis. A 10-year follow-up study. Dis. Colon Rectum. 1994;37:313–319. doi: 10.1007/BF02053590. [DOI] [PubMed] [Google Scholar]
- 4.Cairns SR, Scholefield JH, Steele RJ, Dunlop MG, Thomas HJ, Evans GD, Eaden JA, Rutter MD, Atkin WP, Saunders BP, Lucassen A, Jenkins P, Fairclough PD, Woodhouse CR. Guidelines for colorectal cancer screening and surveillance in moderate and high risk groups (update from 2002) Gut. 2010;59:666–689. doi: 10.1136/gut.2009.179804. [DOI] [PubMed] [Google Scholar]
- 5.Claessen MM, Schipper ME, Oldenburg B, Siersema PD, Offerhaus GJ, Vleggaar FP. WNT-pathway activation in IBD-associated colorectal carcinogenesis: potential biomarkers for colonic surveillance. Cell. Oncol. 2010;32:303–310. doi: 10.3233/CLO-2009-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.P.D. Collins, C. Mpofu, A.J. Watson, J.M. Rhodes, Strategies for detecting colon cancer and/or dysplasia in patients with inflammatory bowel disease. Cochrane Database Syst. Rev. (2006):CD000279 [DOI] [PubMed]
- 7.Eaden J. Review article: colorectal carcinoma and inflammatory bowel disease. Aliment. Pharmacol. Ther. 2004;20:24–30. doi: 10.1111/j.1365-2036.2004.02046.x. [DOI] [PubMed] [Google Scholar]
- 8.Eaden JA, Mayberry JF. Guidelines for screening and surveillance of asymptomatic colorectal cancer in patients with inflammatory bowel disease. Gut. 2002;51:V10–V12. doi: 10.1136/gut.51.suppl_5.v10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eaden J, Abrams K, McKay H, Denley H, Mayberry J. Inter-observer variation between general and specialist gastrointestinal pathologists when grading dysplasia in ulcerative colitis. J. Pathol. 2001;194:152–157. doi: 10.1002/path.876. [DOI] [PubMed] [Google Scholar]
- 10.Geboes K, Riddell R, Ost A, Jensfelt B, Persson T, Lofberg R. A reproducible grading scale for histological assessment of inflammation in ulcerative colitis. Gut. 2000;47:404–409. doi: 10.1136/gut.47.3.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giaretti W, Molinu S, Ceccarelli J, Prevosto C. Chromosomal instability, aneuploidy, and gene mutations in human sporadic colorectal adenomas. Cell. Oncol. 2004;26:301–305. doi: 10.1155/2004/816591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goetz M, Neurath MF. Imaging techniques in inflammatory bowel disease: recent trends, questions and answers. Gastroentérol. Clin. Biol. 2009;33:S174–S182. doi: 10.1016/S0399-8320(09)73152-5. [DOI] [PubMed] [Google Scholar]
- 13.Gupta RB, Harpaz N, Itzkowitz S, Hossain S, Matula S, Kornbluth A, Bodian C, Ullman T. Histologic inflammation is a risk factor for progression to colorectal neoplasia in ulcerative colitis: a cohort study. Gastroenterology. 2007;133:1099–1105. doi: 10.1053/j.gastro.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hage M, Siersema PD, Vissers KJ, Steyerberg EW, Haringsma J, Kuipers EJ, van Dekken H. Molecular evaluation of ablative therapy of Barrett’s oesophagus. J. Pathol. 2005;205:57–64. doi: 10.1002/path.1685. [DOI] [PubMed] [Google Scholar]
- 15.Hedley DW, Friedlander ML, Taylor IW, Rugg CA, Musgrove EA. Method for analysis of cellular DNA content of paraffin-embedded pathological material using flow cytometry. J. Histochem. Cytochem. 1983;31:1333–1335. doi: 10.1177/31.11.6619538. [DOI] [PubMed] [Google Scholar]
- 16.Holzmann K, Weis-Klemm M, Klump B, Hsieh CJ, Borchard F, Gregor M, Porschen R. Comparison of flow cytometry and histology with mutational screening for p53 and Ki-ras mutations in surveillance of patients with long-standing ulcerative colitis. Scand. J. Gastroenterol. 2001;36:1320–1326. doi: 10.1080/003655201317097191. [DOI] [PubMed] [Google Scholar]
- 17.Hussain SP, Amstad P, Raja K, Ambs S, Nagashima M, Bennett WP, Shields PG, Ham AJ, Swenberg JA, Marrogi AJ, Harris CC. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res. 2000;60:3333–3337. [PubMed] [Google Scholar]
- 18.Itzkowitz S. Colon carcinogenesis in inflammatory bowel disease: applying molecular genetics to clinical practice. J. Clin. Gastroenterol. 2003;36:S70–S74. doi: 10.1097/00004836-200305001-00012. [DOI] [PubMed] [Google Scholar]
- 19.Itzkowitz SH, Harpaz N. Diagnosis and management of dysplasia in patients with inflammatory bowel diseases. Gastroenterology. 2004;126:1634–1648. doi: 10.1053/j.gastro.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 20.Karlen P, Lofberg R, Brostrom O, Leijonmarck CE, Hellers G, Persson PG. Increased risk of cancer in ulcerative colitis: a population-based cohort study. Am. J. Gastroenterol. 1999;94:1047–1052. doi: 10.1111/j.1572-0241.1999.01012.x. [DOI] [PubMed] [Google Scholar]
- 21.Klump B, Holzmann K, Kuhn A, Borchard F, Sarbia M, Gregor M, Porschen R. Distribution of cell populations with DNA aneuploidy and p53 protein expression in ulcerative colitis. Eur. J. Gastroenterol. Hepatol. 1997;9:789–794. doi: 10.1097/00042737-199708000-00010. [DOI] [PubMed] [Google Scholar]
- 22.Lashner BA, Bauer WM, Rybicki LA, Goldblum JR. Abnormal p53 immunohistochemistry is associated with an increased colorectal cancer-related mortality in patients with ulcerative colitis. Am. J. Gastroenterol. 2003;98:1423–1427. doi: 10.1111/j.1572-0241.2003.07573.x. [DOI] [PubMed] [Google Scholar]
- 23.Lim CH, Dixon MF, Vail A, Forman D, Lynch DA, Axon AT. Ten year follow up of ulcerative colitis patients with and without low grade dysplasia. Gut. 2003;52:1127–1132. doi: 10.1136/gut.52.8.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lofberg R, Brostrom O, Karlen P, Ost A, Tribukait B. DNA aneuploidy in ulcerative colitis: reproducibility, topographic distribution, and relation to dysplasia. Gastroenterology. 1992;102:1149–1154. [PubMed] [Google Scholar]
- 25.Nathanson JW, Yadron NE, Farnan J, Kinnear S, Hart J, Rubin DT. p53 mutations are associated with dysplasia and progression of dysplasia in patients with Crohn’s disease. Dig. Dis. Sci. 2008;53:474–480. doi: 10.1007/s10620-007-9886-1. [DOI] [PubMed] [Google Scholar]
- 26.Pohl C, Hombach A, Kruis W. Chronic inflammatory bowel disease and cancer. Hepatogastroenterology. 2000;47:57–70. [PubMed] [Google Scholar]
- 27.Ramsoekh D, van Leerdam ME, van Ballegooijen M, Habbema JD, Kuipers EJ. Population screening for colorectal cancer: faeces, endoscopes or X-rays? Cell. Oncol. 2007;29:185–194. doi: 10.1155/2007/610496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramsoekh D, Haringsma J, Poley J, van Putten P, van Dekken H, Steyerberg EW, van Leerdam ME, Kuipers EJ. A back-to-back comparison of white light video endoscopy with autofluorescence endoscopy for adenoma detection in high-risk subjects. Gut. 2010;59:785–793. doi: 10.1136/gut.2008.151589. [DOI] [PubMed] [Google Scholar]
- 29.Riddell RH, Goldman H, Ransohoff DF, Appelman HD, Fenoglio CM, Haggitt RC, Ahren C, Correa P, Hamilton SR, Morson BC, et al. Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical applications. Hum. Pathol. 1983;14:931–968. doi: 10.1016/S0046-8177(83)80175-0. [DOI] [PubMed] [Google Scholar]
- 30.Robertson DJ, Greenberg ER, Beach M, Sandler RS, Ahnen D, Haile RW, Burke CA, Snover DC, Bresalier RS, McKeown-Eyssen G, Mandel JS, Bond JH, Van Stolk RU, Summers RW, Rothstein R, Church TR, Cole BF, Byers T, Mott L, Baron JA. Colorectal cancer in patients under close colonoscopic surveillance. Gastroenterology. 2005;129:34–41. doi: 10.1053/j.gastro.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 31.Rutter M, Saunders B, Wilkinson K, Rumbles S, Schofield G, Kamm M, Williams C, Price A, Talbot I, Forbes A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology. 2004;126:451–459. doi: 10.1053/j.gastro.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 32.Shankey TV, Rabinovitch PS, Bagwell B, Bauer KD, Duque RE, Hedley DW, Mayall BH, Wheeless L, Cox C. Guidelines for implementation of clinical DNA cytometry. International Society for Analytical Cytology. Cytometry. 1993;14:472–477. doi: 10.1002/cyto.990140503. [DOI] [PubMed] [Google Scholar]
- 33.Sjoqvist U. Dysplasia in ulcerative colitis–clinical consequences? Langenbecks Arch. Surg. 2004;389:354–360. doi: 10.1007/s00423-003-0455-6. [DOI] [PubMed] [Google Scholar]
- 34.Sjoqvist U, Befrits R, Soderlund S, Ost A, Karlen P, Tribukait B, Rubio C, Rutgeerts P, Geboes K, Lofberg R. Colorectal cancer in colonic Crohn’s disease–high frequency of DNA-aneuploidy. Anticancer Res. 2005;25:4393–4397. [PubMed] [Google Scholar]
- 35.Stallmach A, Bielecki C, Schmidt C. Malignant transformation in inflammatory bowel disease—surveillance guide. Dig. Dis. 2009;27:584–590. doi: 10.1159/000233302. [DOI] [PubMed] [Google Scholar]