

Abstract
In innate immune sensing, the detection of pathogen-associated molecular patterns by recognition receptors typically involve leucine-rich repeats (LRRs). We provide a categorization of 375 human LRR-containing proteins, almost half of which lack other identifiable functional domains. We clustered human LRR proteins by first assigning LRRs to LRR classes and then grouping the proteins based on these class assignments, revealing several of the resulting protein groups containing a large number of proteins with certain non-LRR functional domains. In particular, a statistically significant number of LRR proteins in the typical (T) and bacterial + typical (S+T) categories have transmembrane domains, whereas most of the LRR proteins in the cysteine-containing (CC) category contain an F-box domain (which mediates interactions with the E3 ubiquitin ligase complex). Furthermore, by examining the evolutionary profiles of the LRR proteins, we identified a subset of LRR proteins exhibiting strong conservation in fungi and an enrichment for “nucleic acid-binding” function. Expression analysis of LRR genes identifies a subset of pathogen-responsive genes in human primary macrophages infected with pathogenic bacteria. Using functional RNAi, we show that MFHAS1 regulates Toll-like receptor (TLR)–dependent signaling. By using protein interaction network analysis followed by functional RNAi, we identified LRSAM1 as a component of the antibacterial autophagic response.
Keywords: pathogen-response, anti-bacterial, inflammation, pathogen sensors, autophagy
Innate immunity is a conserved host response that entails the sensing of pathogen-associated molecular patterns through germline-encoded pattern recognition receptors (1), which initiate pathway-specific signaling networks, resulting in rapid responses that serve as the host's first line of defense. A striking feature among families of proteins functioning as sensors or effectors of innate immunity is the inclusion into their structure, various combinations of the following domains: Leucine-rich repeat (LRR), Toll/IL-1 receptor, Ig, nucleotide-binding site, pyrin, RIG-I–like receptor, caspase-recruitment, coiled-coil, immunoreceptor tyrosine-based activation motif, and C-type lectin (2).
The LRR domain is present in a large number of prokaryotic and eukaryotic proteins and is one of the most commonly occurring protein domains in proteins associated with innate immunity. This is especially so in invertebrates (e.g., sea urchin) and Cephalochordata (e.g., amphioxus) which have vastly expanded repertoires of LRR proteins (3) in the absence of adaptive immunity. In jawless vertebrates, combinatorial assembly of LRR gene segments in variable lymphocyte receptors generates the structural diversity for antigen recognition and forms the basis of an adaptive immune system (4, 5). Toll-like receptors (TLRs) and NOD-like receptors (NLRs) recognize via the LRR domain, molecular determinants from a structurally diverse collection of bacterial, fungal, viral, and parasite-derived components (6, 7). Mutations or polymorphisms in more than 30 LRR-containing proteins have been implicated in human diseases to date, notably polymorphisms in NOD2 in Crohn disease (8, 9), CIITA in rheumatoid arthritis and multiple sclerosis (10), and TLR5 in Legionnaire disease (11).
Most LRR domains consist of a chain of between 2 and 45 LRRs (12). Each repeat in turn is typically 20 to 30 residues long and can be divided into a highly conserved segment (HCS) followed by a variable segment (VS). The HCS usually consists of either the 11-residue sequence LxxLxLxxNxL or the 12-residue sequence LxxLxLxxCxxL, where L is Leu, Ile, Val, or Phe; N is Asn, Thr, Ser, or Cys; and C is Cys, Ser, or Asn (13, 14). Although these substitutions often preserve hydrophobicity or polarity, it is possible for the first and last leucines to be replaced by relatively hydrophilic residues (15). The function of many LRR domains is to provide a structural framework for protein–protein interactions (PPIs) (13). PDB structures for LRR-containing proteins show the LRR domains in an arc or horseshoe shape. The concave face (corresponding to the HCS of each LRR) consists of parallel β-strands, each usually three residues long, flanked by loops. Conversely, the convex face (corresponding to the VS of each LRR) is composed of a variety of secondary structures, which are often helical (13). Ligands can interact with either the convex or the concave face, although the latter is more typical (13, 16). The core of the arc is hydrophobic, typically shielded by caps at the N terminus of the first LRR and at the C terminus of the last LRR. In extracellular proteins or domains, these caps often contain two- or four-residue cysteine clusters (14).
Beyond innate immunity, extensive functional diversity occurs among LRR-containing proteins, which are involved in a variety of cellular processes including apoptosis, autophagy, ubiquitin-related processes, nuclear mRNA transport, and neuronal development. Although the presence of well characterized non-LRR domains can be used to classify LRR proteins along functional lines, such a classification scheme will place LRR proteins lacking any other identifiable functional domains into a single group unrelated to any specific function. To fully appreciate the signaling functions of LRR proteins, we devised semi-automated methods for grouping them on the basis of LRR classes. We identified the positions of individual LRR sequences within each LRR domain, and classified each repeat based upon consensus sequences and lengths of their VSs. Identified repeats were assigned into classes: bacterial (S), ribonuclease inhibitor–like (RI), cysteine-containing (CC), SDS22, plant-specific (PS), typical (T), and Treponema pallidum (Tp) (13, 17). By using this approach, we reasoned that functionally related LRR-only proteins would be placed together by virtue of their similar LRR domains. In addition, these LRR class-based clusters would contain all of the proteins bearing similar LRR repeat structures, with further subcategorization via the presence or absence of non-LRR domains.
In this article, we present a comprehensive categorization of human LRR-containing proteins by LRR class composition, functional associations, and involvement in host responses to infection stimuli. We demonstrate semi-automated methods that use LRR class composition to facilitate functional groupings. By integrating diverse datasets and using functional RNA interference, we experimentally place uncharacterized LRR proteins in innate immunity and autophagy.
Results
Annotation of LRRs.
We first compiled a list of 375 human proteins annotated as containing LRRs in InterPro (18), the Swiss-Prot section of UniProt (19), and LRRML (a conformational database and an extensible markup language description of LRRs) (17). As shown in Fig. S1, InterPro contained the largest number of LRR proteins: it included all 19 proteins found in LRRML and 303 of the 327 LRR proteins in Swiss-Prot.
To annotate the LRRs in the LRR proteins, we constructed hidden Markov models (HMMs) to represent the signatures of the seven LRR classes. That the resulting HMMs were constructed properly is evident from the strong similarities between the logos for the HMMs (Fig. S2) (20) and the corresponding consensus sequences described in the literature (13). Some differences were observed: for example, in the HMM for the S class, the first leucine is not as strongly conserved as in the consensus signature. Also, whereas the sixth residue of the consensus signature is valine, the most frequently occurring residue at the corresponding position of the HMM is cysteine, with valine as the third most common.
We next devised an algorithm that used the HMMs to identify the positions and class assignments of the LRRs. To fully capture the “irregular” LRRs with atypical amino acid sequences, we implemented a combination algorithm, using the HMMs to find regular LRRs, then pattern-matching to find adjacent, nonoverlapping matches to LRR amino acid sequences or predicted secondary structures (15, 21). This exploits the structural observation that LRRs occur in chains, thus leveraging the discriminatory power of HMMs to control otherwise promiscuous pattern matching.
By applying the annotation algorithm to the 375 proteins classified as LRR-containing, we found LRRs in almost all proteins classified by multiple databases but in very few of those classified by only a single database. There are 334 proteins in which at least one LRR can be identified. We provide a comprehensive map of these human LRR proteins, graphically displaying the LRR classes as well as non-LRR domains and their coordinates in each of these proteins (Dataset S1). In many of the proteins, most of the identified regular LRRs either belong to a single class (e.g., CC for FBXL2, PS for LRRC30; Fig. 1A) or are members of the S and T classes (e.g., ASPN and EPYC; Fig. 1B). This uniformity in the class membership of the LRRs has been observed (13, 15, 22). Nonetheless, several of the proteins contain LRRs from multiple classes (other than S and T). For instance, LRRC33 contains S, T, and SDS22 LRRs, whereas PS and S LRRs occur in MFHAS1 (Fig. 1C).
Fig. 1.

Examples of annotated LRR proteins. (A) Proteins containing LRRs from predominantly one class. (B) Proteins containing LRRs from predominantly the S and T classes. (C) Proteins containing LRRs from multiple classes. (D) Examples of proteins illustrating the occurrence of irregular LRRs both at the ends of LRR chains as well as within them. (E) Clustering of LRR proteins using LRR classes. A larger “zoomable” version is shown in Fig. S3. Cluster descriptions illustrate certain predominant features observed among representative LRR protein members in the respective clusters, e.g., F-box cluster consists predominantly of LRR proteins that also contain the F-box domain, whereas the NLR cluster consists mostly of NLR family members.
It is evident from Dataset S1 that most of the proteins contain one or more irregular LRR. As shown in Fig. 1D, these LRRs occur both at the ends of LRR chains (FBXL3) as well as within them (LRRC16A). By their very nature, irregular LRRs are more difficult to identify than regular LRRs, particularly at the ends of LRR chains. However, irregular LRRs that occur within LRR chains can be identified with greater confidence and are likely to be valid LRRs as they span the gaps between regular LRRs.
Clustering the LRR Proteins Using LRR Classes.
Based on the LRR class assignments, we grouped the LRR proteins in two different ways. For the first approach, we clustered the proteins based on the sequence similarity of their LRRs; each LRR in a given protein was allowed to match any LRR of the same class in another protein (the irregular LRRs were placed in their own class). Fig. 1E and Fig. S3 summarize the clustered results for human LRR proteins as a circular tree. As predicted, we observed functionally similar proteins clustering together e.g., members of the SLITRK, NLR (Fig. S3), and F-box families (Fig. S4). We were also able to observe LRR-only proteins being distributed among those containing non-LRR domains, fulfilling one of our goals in this effort: to cluster LRR proteins on the basis of LRR class composition.
Because the class annotations for the LRRs are not always optimal, the second method we used to group the LRR proteins was designed to be more robust to annotation errors. We first assigned all proteins containing fewer than five regular LRRs to the “unclassified” category. We then partitioned the remaining proteins into categories based on the class to which the majority of LRRs in each protein belong. We placed those proteins for which S and T LRRs together constitute the majority in a separate S+T category, whereas we classified as “mixed” those proteins for which each class is in the minority. Interestingly, more than 25% of the proteins are in the T or S+T categories and fewer than half contain fewer than five regular LRRs. A summary of the resulting category assignments is shown in Fig. 2A and Dataset S2A. Again, as expected, LRR-only proteins were found to be distributed among the categories and are thus grouped with proteins containing non-LRR domains.
Fig. 2.

Results from categorizing the LRR proteins by the majority LRR class in each protein. (A) Numbers of LRR proteins in each category; the proteins in each category are further subdivided into those that are LRR-only and those with non-LRR domains. (B) Distribution of the LRR proteins in each category between those with TM regions and those without; categories with a statistically significant association with the presence or absence of TM regions are indicated with an asterisk. (C) Percentages of the classified and unclassified LRR proteins containing an LRR of each class. (D) Numbers of classified and unclassified LRR proteins which contain each non-LRR InterPro domain.
As is evident in Fig. 2B, several of the class-based categories are associated with the presence or absence of transmembrane (TM) regions as predicted by Phobius (23) in the corresponding proteins. Specifically, a statistically significant number of proteins in the T and S+T categories contain TM regions, whereas a statistically significant number of proteins in the CC and unclassified categories do not. Several of the class-based categories are also associated with certain non-LRR domains. For example, at least 12 of the 15 proteins in the CC category contain an F-box domain (Fig. S4), whereas 17 of the 32 proteins in the RI category contain a NACHT NTPase domain (Dataset S1).
As noted earlier, almost half the LRR proteins are unclassified. Comparing these unclassified LRR proteins to those which are classified revealed several differences. In particular, all of the LRR proteins annotated as LRR-containing in only one database are unclassified. This observation is not surprising; one would expect that proteins with few LRRs would be less likely to be identified in a database as LRR-containing. Another difference between the classified and unclassified proteins is apparent in Fig. 2C: the T and S classes are the most commonly occurring regular classes in classified proteins but occur infrequently in unclassified proteins. Third, although several non-LRR domains occur in both classified and unclassified proteins (Fig. 2D), many non-LRR domains are unique to just the classified or unclassified proteins (Dataset S2B). Finally, when analyzing the sets of classified and unclassified proteins in terms of their molecular function and associated biological processes using the PANTHER classification system (24), we observed striking differences in key categories being over-represented (Dataset S2 C and D). Processes associated with receptor-mediated signaling, immunity and defense were significantly enriched among the classified LRR proteins. In contrast, nucleic acid–binding functions and cell structure–related processes were over-represented among the unclassified LRR proteins.
Because S and T are the most commonly occurring regular LRR classes in classified LRR proteins and such proteins are enriched for terms related to immunity and receptor-mediated signaling, we examined whether the subset of classified proteins with S or T LRRs would also exhibit such enrichments. Using the PANTHER classification system, a number of processes associated with receptor-mediated signaling were found to be statistically significant, including cytokine and chemokine mediated signaling and “immunity and defense” (FDR-adjusted P values, 0.02 and 0.05, respectively; Dataset S2E), suggesting that S and T LRRs might play a role in these processes.
Evolutionary Characteristics of the LRR Proteins.
We next examined the evolutionary profiles of the LRR proteins to determine whether the resulting patterns of conservation might yield any insights into the protein family. To generate this evolutionary profile, we clustered the LRR proteins so that proteins grouped together have orthologues in many of the same organisms. A heat map depicting this clustering is shown in Fig. 3. Fig. 3 also shows the heat maps generated by performing the same procedure for human proteins containing PDZ and SH2 domains. It is apparent that the evolutionary profiles for the PDZ and SH2 protein families are quite similar. Both protein families can be partitioned into two major groups: proteins with orthologues in all metazoans (group 4) and proteins with orthologues only in chordates (group 5). There are also some unique characteristics observed for a small number of SH2 proteins but not among PDZ proteins, such as those having orthologues primarily in mammals (group 6). Interestingly, none of the SH2 proteins have orthologues in eubacteria or archaea.
Fig. 3.

Heat maps indicating the species for which orthologues exist for human proteins containing LRR, PDZ, and SH2 domains. For each human LRR protein (row), each column corresponds to a species in which an orthologue of the protein exists. The species are partitioned into the taxonomically related groups indicated, and the species within each group are arranged from left to right in decreasing order of orthologue count. The specific human LRR proteins which are strongly conserved in fungi are highlighted. The protein subsets indicated by the numbers 1 through 6 are described in the text.
The evolutionary profile heat map for the LRR proteins exhibits striking differences from those for the PDZ and SH2 proteins. For example, a significantly large number of the LRR proteins have orthologues primarily in mammals (group 1), whereas very few of the SH2 proteins and none of the PDZ proteins exhibit this property. Further, several of the LRR proteins that have orthologues only in eukaryotes do not have orthologues in fungi (group 2), whereas no such proteins are evident in the PDZ and SH2 heat maps. One particularly striking feature of the LRR heat map is the presence of a set of 21 proteins with strong conservation in fungi (group 3). Analyzing this set of proteins for over-represented PANTHER terms revealed an enrichment (with a P value adjusted for multiple testing of 1.277e-05) for the term “nucleic acid-binding” for the following seven proteins: CNOT6, CNOT6L, NXF1, NXF2, NXF2B, PDS5A, and SNRPA1.
To further stratify the clusters of LRR proteins shown in Fig. 3, we reclustered the proteins based on the relative degree of similarity between each protein and its orthologues. A heat map depicting the updated clustering is shown in Fig. S5A. For each species represented in the heat map, red denotes proteins that are more similar to their human orthologues than those depicted in green are to theirs. Because it was difficult to discern any clear patterns in the revised heat map, we rearranged the proteins so they would be grouped by their class-based categories (Fig. S5B). Doing so revealed that the SDS22, RI, and mixed categories contain very few proteins with high degrees of conservation (relative to the other LRRs). It is now also apparent that almost all the proteins in the T and S+T categories have orthologues only in metazoans.
Expression-Based Classification of LRR Genes Reveal Neuronal and Immune Clusters.
To build on the information derived from LRR class classification methods, we were interested to identify subsets of LRR genes that might have roles in processes associated with specialized tissue types. We examined the expression of LRR genes across 79 tissues in a human microarray panel (25). Of the 230 LRR genes for which probes were available, 39 showed higher-level expression across neuronal compared with other tissues (Wilcoxon test, P < 0.05; Fig. S6A), including genes known to be associated with the CNS such as NTRK2/SLIT and LRRTM family members, and functionally uncharacterized ones, e.g., C22orf36 and LRRC49 (Dataset S2H). Of particular interest are 93 LRR genes exhibiting elevated expression in immune tissues (Wilcoxon test, P < 0.05; Fig. S6A). These include, in addition to well studied pattern-recognition molecules such as TLRs and NLRs, others that are not presently known to be directly associated with immune function, such as SCRIB (a PDZ-domain–containing cell-junction protein) and genes involved with nuclear transport (NXF1, ANP32A, B, E; Dataset S2G). In addition, there is also a significant over-representation of genes encoding proteins associated with ubiquitin ligase complexes or activities (e.g., PPIL5, SKP2, LRSAM1, FBXL5, FBXL6, FBXO9, FBXO7, FBXO11, FBXW5; P = 4 × 10−15) in this immune cluster.
Pathogen-Responsive LRR Gene Expression in Human Primary Macrophages.
To uncover candidate LRR genes that might be involved in host-pathogen innate immune response but have not been previously identified to have immunologically defined roles, we examined the expression profiles of a subset of LRR genes by semiquantitative RT-PCR in primary human macrophages infected with Staphylococcus aureus, Mycobacterium tuberculosis, Listeria monocytogenes, enterohemorrhagic Escherichia coli (EHEC), and Salmonella enterica serovar typhi (S. typhi), across four time points (0 h, 1 h, 2 h, and 6 h after infection; Fig. 4A). The gene subset was selected as having possible innate immune involvement based on text mining, Gene Ontology annotations, or expression profiling. We observed a cluster of LRR genes (LRRC48, BPI, LRRK2, FLRT3, LRRTM3, TMOD1, NLRP11) that was strongly induced by L. monocytogenes and EHEC, showing early and sustained induction across the time periods examined (1 h to 6 h). Another group of genes (PHLPPL, SYNE2, SHOC2, LRRC28) displayed elevated expression when infected by each of the five pathogens but exhibited a particularly strong response at 6 h to EHEC. In contrast, the expression of FBXL11, LRCH4, and LMOD2 was decreased when infected with S. aureus, M. tuberculosis, L. monocytogenes, and EHEC. The expression of NLRP1, LRRTM2, LRRC59, and NXF1 was induced in response to M. tuberculosis and S. typhi, but appears to be refractory to the other infectious agents tested. A strong induction signal for MFHAS1 was observed with EHEC, L. monocytogenes, S.typhi, and M. tuberculosis (Fig. 4 A and B). Taken together, these findings suggest that these LRR proteins function as sensors or effectors of pathogen-mediated stress signals. The precise mechanisms by which microbial cues are sensed and regulated by these proteins remain to be determined.
Fig. 4.

Identifying genes encoding LRR proteins involved in immunity and antibacterial autophagy. (A) Heat map shows gene expression of LRR genes in human primary macrophages infected by the indicated pathogens at various time-points: 0 (uninfected) and 1, 2, and 6 h after infection (as examined by RT-PCR). GAPDH-normalized log2 expression values are expressed as signed difference ratios relative to the uninfected state and scaled by normalizing to the maximum absolute deviation of each gene's expression level from the uninfected control, so that all values lie between −1 and +1. (B) Further RT-PCR validation of a subset of M. tuberculosis–responsive genes from independent set of samples from human primary macrophages uninfected and infected with M. tuberculosis for 1 and 6 h after infection. Values are GAPDH-normalized log2 fold change relative to uninfected control, performed in duplicate. Error bars indicate ± SD. (C) Knockdown using siRNA directed against Mfhas1 in RAW 264.7 macrophages enhanced the level of secreted IL-6 production following LPS and (D) polyIC stimulation, as measured by ELISA. Data from three experiments is shown as mean ± SD. Asterisk indicates P < 0.05 as assessed using two-tailed t test. RT-PCR validation of siRNA knockdown is shown in Fig. S6B. (E) LRSAM1 is required for antibacterial autophagy. Knockdown of LRSAM1 results in loss of anti-Salmonella autophagy in HeLa cells. siRNA directed against FNBP1L or LRSAM1 resulted in loss of autophagic membranes surrounding internalized S. typhimurium. The anti-Salmonella autophagy rate was significantly lower for two of three LRSAM1 duplexes, correlating with effective knockdown at the RNA level (Fig. S6C). Data are shown as mean ± SE and are pooled from three independent experiments, each counting at least 50 infected cells per condition. Significance (P < 0.05) was assessed using two-tailed t tests with Bonferroni correction.
LRR-Containing Protein MFHAS1 Regulates TLR-Dependent Signal Transduction.
We next investigated whether LRR proteins, especially MFHAS1 (malignant fibrous histiocytoma amplified sequence 1), were essential for the activation of TLR-dependent pathways. For this, we down-regulated MFHAS1 expression in RAW macrophages using siRNA. siRNA knockdown of MFHAS1 in RAW 264.7 macrophages strongly enhanced IL-6 production following LPS and polyI:C stimulation (Fig. 4 C and D), suggesting a potential immune modulatory role for MFHAS1. Under similar experimental conditions, knockdown of CNOT6L did not alter LPS- and polyIC-mediated TLR activation. Apart from its LRR domain, MFHAS1 contains one other identifiable and recently described Roco (Ras-like GTPase and C-terminal of Roc) domain (26), which is also present in a group of LRR proteins in Dictyostelium and prokaryotes. Although there is currently no reported involvement of MFHAS1 in host defense processes, the strong induction of MFHAS1 expression observed following pathogen challenge (Fig. 4 A and B) is consistent with a role in immune regulation.
Network Analysis Identifies a Potential Autophagy-Related Role for the LRR Protein LRSAM1.
Previously we identified FNBP1L as a protein required for anti-bacterial autophagy, based on PPI data (27). Using a similar approach, we constructed first-order PPI networks for human LRR proteins to explore whether LRR proteins interacted with components of the core autophagy “machinery.” Interestingly, WDFY3 was the only human LRR protein that has been previously linked to autophagy (28). From network analysis, we identified LRSAM1 (leucine-rich repeat and sterile α-motif containing 1) as a potential interactor with GABARAPL2, which belongs to the MAP1 LC3/ATG8 family of proteins. RNAi directed against LRSAM1 resulted in good knockdown for two duplexes (siRNA1 and 2) and a modest reduction in RNA level for the third (siRNA3; Fig. S6C). The level of LRSAM1 knockdown correlated with the level of anti-Salmonella autophagy observed in HeLa cells, with higher knockdown resulting in lower rates of successful autophagy of intracellular bacteria. This result was confirmed in three separate experiments and the data pooled (Fig. 4E). Statistically significant reductions in anti-Salmonella autophagy were observed in FNBP1L and LRSAM1 (siRNA 1 and 2), but not LRSAM1 siRNA3, compared with control siRNA duplex transfection. Thus, LRSAM1 has an essential role in antibacterial autophagy.
Discussion
In this report, we have described two approaches for grouping the human LRR-containing proteins using predicted class assignments for the individual LRRs. For the first approach, we clustered the proteins based on the sequence similarity of the LRRs belonging to each class. As illustrated by the NLR and SLITRK clusters in Fig. S3 and F-box–containing proteins in Fig. S4, this method was able to group together proteins with similar function while distributing the LRR-only proteins among those containing non-LRR domains, thereby allowing the assignment of putative functional extensions to LRR-only proteins within these clusters. This is necessary because, based on the annotations in InterPro, almost half the 375 human LRR proteins do not contain non-LRR domains. The absence of these domains from which functional insights could usually be gleaned, limits their characterization. Hence, a majority of these LRR proteins have no known function. On the basis of very similar LRR class composition, we found CEP72 [an LRR-only protein that was recently implicated in ulcerative colitis from genome-wide association studies (29)] clustered with LRRC36 (Fig. S3), an RORγ-binding protein, suggesting a gene-regulatory connection that requires additional functional studies to validate.
For the second approach, we partitioned the LRR proteins into categories based on the majority LRR class of each protein. As demonstrated in Fig. 2A, this method yielded categories in which LRR-only proteins are grouped with non-LRR domains. These categories also have the potential to yield insights into the functions of uncharacterized proteins. In particular, as shown in Fig. 2B, several of the categories are associated with the presence or absence of TM regions. Further, most of the proteins in the CC category contain an F-box domain, whereas half the proteins in the RI category contain a NACHT NTPase domain. Finally, although the unclassified category contains almost half the LRR proteins, we still found statistically significant associations with several ontology terms when we compared the proteins in this category to those in the remaining categories. Specifically, the unclassified LRRs exhibit an over-representation of terms related to nucleic acid binding and cell structure, whereas the classified LRR proteins are enriched for terms related to immunity and receptor-mediated signaling. We also observed that this latter enrichment is valid for the subset of classified proteins which contain S or T LRRs. As classified LRRs all contain five or more LRRs, this observation suggests a possible link between receptor-mediated signaling and LRR proteins with an S or T LRR and at least four other LRRs.
To obtain further insight into function, we also examined the evolutionary profiles of the LRR proteins. We observed striking differences in the profile for human LRR proteins when compared with those of PDZ and SH2 proteins. In particular, a small subset of the LRR proteins exhibits strong conservation in fungi and is enriched for the PANTHER ontology term “nucleic acid binding.” One of these, NXF1, a nuclear RNA-export factor, was recently identified in two genome-wide siRNA screens as a host-dependency factor for influenza virus (30, 31). We also uncovered an intriguing connection between the class-based LRR categories and the degrees to which the categories’ members are conserved. As illustrated in Fig. S5B, we found that the SDS22, RI, and mixed categories contain very few proteins with high degrees of conservation (relative to the other LRRs).
Next, we were interested in identifying a subset of LRR genes in immunity and host defense. To identify bacterial-responsive LRR genes, we examined expression profiles of LRR genes in primary human macrophages infected with five pathogenic bacteria and across multiple time points using RT-PCR. We identified MFHAS1 as a candidate gene for further investigation as a result of its induction response to four of the five pathogens examined. Validation by siRNA knockdown in RAW macrophages places MFHAS1 in the signaling network downstream of TLR signaling and highlights its potential role as a modulator of the inflammatory response.
We experimentally validated candidate LRR genes identified through integrating information from diverse sources including ontology annotations, literature co-citations, PPI data, and gene expression microarray data, with the aim of gaining additional insights for functional exploration. From literature mining, we note that the LRR protein WDFY3 has been previously implicated to play a role in autophagy by binding to phosphatidylinositol-3-phosphate (Ptdlns3P), which regulates endocytic and autophagic membrane traffic (28). Extending this search by using PPI network analysis, we identified LRSAM1 as the only human LRR protein in the network that potentially interacted with GABARAPL2, a member of the MAP1 LC3/ATG8 family of proteins, in the core autophagy apparatus. LRSAM1, also known as Tal, has a SAM domain and a RING (E3 ubiquitin-protein ligase) domain, which mediates monoubiquitination of TSG101 at multiple sites and regulates receptor endocytosis by inactivating the ability of TSG101 to sort endocytic and exocytic (as observed with HIV-1 viral protein) cargos (32). Given this role in cargo sorting and membrane packaging of cargos, as well as the known role for bacterial ubiquitination in triggering antibacterial autophagy (33), we postulated that LRSAM1 was a plausible candidate to operate within the anti-Salmonella autophagy pathway. Using a functional siRNA approach, knockdown of LRSAM1 resulted in reductions in anti-Salmonella autophagy.
We provide a resource and framework to facilitate the functional understanding of 375 human LRR proteins, many of which are currently uncharacterized. The results of this study have contributed to our understanding of how LRR class composition might be used to facilitate the functional classification of less well studied LRR proteins alongside those of known function. Using an integrative approach, we elucidated functions of two LRR genes in regulating responses to pathogens. First, via expression profiling of pathogen-responsive LRR genes, followed by functional siRNA, we show that MFHAS1 regulates TLR signaling. Finally, by using PPI data, we identified LRSAM1 as a component of the anti-bacterial autophagic response. Together these findings provide insights into the function and molecular pathways associated with LRR proteins.
Materials and Methods
Construction of the LRR HMMs.
We extracted for each LRR class the associated set of LRR sequences in the LRRML database. As LRRML contained no sequences for the TpLRR class and very few for the PS class, we added to these classes’ sets sequences obtained from two publications (34, 35), respectively. After removing the duplicate sequences in each set, we used the remaining sequences to construct a “seed” HMM for each LRR class. To construct each HMM, we first iteratively aligned and filtered the associated sequences using Clustalw2 (36) (version 2.09, default options) until no positions in the resulting alignment were associated with gaps in 95% or more of the sequences. We then used HMMER (37) (version 2.3.2, default options) to build and calibrate an HMM from the alignment. As the HMMs were to be used to annotate the LRRs only in human proteins but the sequences used to construct the HMMs were not specific to humans, we modified each HMM by repeating the following steps three times. (i) With HMMER, all human UniProt sequences were scanned for matches to the HMMs using a domain E-value cutoff of 0.1 and a global E-value cutoff of 1 × 1020. (ii) Each matching sequence segment is assigned to the class for which it has the smallest E-value. (iii) For each class, the new sequences were added to those already used to construct the class's HMM, removing any duplicates. (iv) As described earlier, we iteratively aligned and filtered the sequences for each class. The final alignments were used to construct new HMMs.
Annotation of the LRRs.
We first scanned every LRR protein using the LRR HMMs and compiled a list of all matches with positive scores. We then identified and scored potential irregular LRRs according to the criteria detailed in SI Materials and Methods. For a given LRR arrangement and regular LRR class assignment for a protein, we defined the total score for the protein to be the sum of the LRR scores adjusted for the inter-LRR regions. We next used an HMM to simultaneously identify the LRR arrangement and regular LRR class assignment, which maximized the total score for each protein. Each identified regular and irregular LRRs were rescored using the LRR HMMs to fine-tune the associated class assignment. LRRs with negative HMM scores were annotated as irregular. A graphical representation of LRR proteins is provided in Dataset S1. For clarity of display, we merged any overlapping, non-LRR InterPro domains using dependency or relational terms.
Clustering LRR Proteins Using LRR Sequence Similarity.
Sequences of the annotated LRRs in LRR proteins were extracted and placed into groups based on LRR classes. The irregular LRRs constituted a separate class and were placed in their own group. Similarity scores were then computed for every pair of LRRs belonging to the same class. For BLAST E-values greater than 1, the similarity score was defined as 0, whereas for E-values of 0 or lower, the similarity score was defined as the negative log10 of the E-value. Using these scores, we next identified for each LRR the best matching LRR in every protein. For each LRR protein, a similarity score was also computed for every other LRR protein by summing the scores of the best-matching LRRs in that protein. To summarize this formally: for each protein i, we then calculated for every protein j a similarity score si,j equal to the sum of the scores of the best-matching LRRs in protein j or −∞ if no matching LRRs existed (in the latter case, we also set sj,i equal to −∞). Using the resulting similarity scores, we constructed a matrix D = [di,j], where for all i and j, di,j represented the normalized distance between proteins i and j and is defined by the following formula:
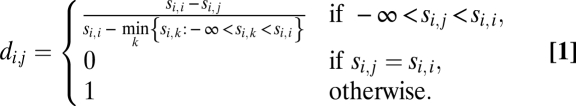 |
Next, a symmetric matrix E = (D + DT) / 2 was created. We then used E to hierarchically cluster the LRR proteins with the method of McQuitty (38) and the hclust function in the R programming language. The clustering result was combined with positional information for each LRR class in each LRR protein using a Perl program and visualized using the representation scheme described previously (39).
Determining Association Between LRR Categories and TM Regions.
For each LRR category, we computed the P value for the two-sided Fisher exact test, implemented in the R programming language. A P value of 0.01 was used to determine statistical significance.
Identification of Enriched PANTHER Ontology Terms.
Enrichment of ontology terms from the PANTHER classification system (24) was computed using one-sided Fisher exact test implemented in the R programming language. P values were corrected for multiple-hypothesis testing using the method of Benjamini and Hochberg (40).
Generation of Evolutionary Profile Heat Maps.
Genes for the 375 LRR proteins were mapped to Ensembl protein IDs. The OrthoMCL database (41) was queried for each protein to identify orthologues and the organisms in which orthologues are present. Vectors containing strings of 0s and 1s denoting the extent of orthologue representation across taxonomic groupings were constructed and used to hierarchically cluster the LRR genes with Cluster 3.0 (42). The clustered results were represented using a heat map. To generate a heat map depicting the relative degrees of conservation of the LRRs, we also incorporated distances (percent divergences) computed using Clustalw2 (36) between LRR proteins and their orthologues (details in SI Materials and Methods).
Supplementary Material
Acknowledgments
We thank Mark Daly and Fred Ausubel for helpful discussions. This work was supported by National Institutes of Health Grants AI062773, DK83756, DK060049, and DK060049 (to R.J.X., R.J.W.H., and J.E.) and a Research Fellowship Award from the Crohn's and Colitis Foundation of America (to A.C.Y.N.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Microbes and Health,” held November 2–3, 2009, at the Arnold and Mabel Beckman Center of the National Academies of Sciences and Engineering in Irvine, CA. The complete program and audio files of most presentations are available on the NAS Web site at http://www.nasonline.org/SACKLER_Microbes_and_Health.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1000093107/-/DCSupplemental.
References
- 1.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 2.Pålsson-McDermott EM, O'Neill LA. Building an immune system from nine domains. Biochem Soc Trans. 2007;35:1437–1444. doi: 10.1042/BST0351437. [DOI] [PubMed] [Google Scholar]
- 3.Huang S, et al. Genomic analysis of the immune gene repertoire of amphioxus reveals extraordinary innate complexity and diversity. Genome Res. 2008;18:1112–1126. doi: 10.1101/gr.069674.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagawa F, et al. Antigen-receptor genes of the agnathan lamprey are assembled by a process involving copy choice. Nat Immunol. 2007;8:206–213. doi: 10.1038/ni1419. [DOI] [PubMed] [Google Scholar]
- 5.Alder MN, et al. Diversity and function of adaptive immune receptors in a jawless vertebrate. Science. 2005;310:1970–1973. doi: 10.1126/science.1119420. [DOI] [PubMed] [Google Scholar]
- 6.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 7.Peter ME, Kubarenko AV, Weber AN, Dalpke AH. Identification of an N-terminal recognition site in TLR9 that contributes to CpG-DNA-mediated receptor activation. J Immunol. 2009;182:7690–7697. doi: 10.4049/jimmunol.0900819. [DOI] [PubMed] [Google Scholar]
- 8.Hugot JP, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 9.Ogura Y, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 10.Swanberg M, et al. MHC2TA is associated with differential MHC molecule expression and susceptibility to rheumatoid arthritis, multiple sclerosis and myocardial infarction. Nat Genet. 2005;37:486–494. doi: 10.1038/ng1544. [DOI] [PubMed] [Google Scholar]
- 11.Hawn TR, et al. A common dominant TLR5 stop codon polymorphism abolishes flagellin signaling and is associated with susceptibility to Legionnaires’ disease. J Exp Med. 2003;198:1563–1572. doi: 10.1084/jem.20031220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enkhbayar P, Kamiya M, Osaki M, Matsumoto T, Matsushima N. Structural principles of leucine-rich repeat (LRR) proteins. Proteins. 2004;54:394–403. doi: 10.1002/prot.10605. [DOI] [PubMed] [Google Scholar]
- 13.Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol. 2001;11:725–732. doi: 10.1016/s0959-440x(01)00266-4. [DOI] [PubMed] [Google Scholar]
- 14.Kajava AV. Structural diversity of leucine-rich repeat proteins. J Mol Biol. 1998;277:519–527. doi: 10.1006/jmbi.1998.1643. [DOI] [PubMed] [Google Scholar]
- 15.Matsushima N, et al. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genomics. 2007;8:124. doi: 10.1186/1471-2164-8-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brodsky I, Medzhitov R. Two modes of ligand recognition by TLRs. Cell. 2007;130:979–981. doi: 10.1016/j.cell.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 17.Wei T, et al. LRRML: A conformational database and an XML description of leucine-rich repeats (LRRs) BMC Struct Biol. 2008;8:47. doi: 10.1186/1472-6807-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunter S, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37(database issue):D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The UniProt Consortium The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38(database issue):D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuster-Böckler B, Schultz J, Rahmann S. HMM Logos for visualization of protein families. BMC Bioinformatics. 2004;5:7. doi: 10.1186/1471-2105-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dolan J, et al. The extracellular leucine-rich repeat superfamily; a comparative survey and analysis of evolutionary relationships and expression patterns. BMC Genomics. 2007;8:320. doi: 10.1186/1471-2164-8-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsushima N, Ohyanagi T, Tanaka T, Kretsinger RH. Super-motifs and evolution of tandem leucine-rich repeats within the small proteoglycans—biglycan, decorin, lumican, fibromodulin, PRELP, keratocan, osteoadherin, epiphycan, and osteoglycin. Proteins. 2000;38:210–225. doi: 10.1002/(sici)1097-0134(20000201)38:2<210::aid-prot9>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 23.Käll L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Mi H, et al. PANTHER version 7: Improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010;38(database issue):D204–D210. doi: 10.1093/nar/gkp1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su AI, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marín I, van Egmond WN, van Haastert PJ. The Roco protein family: A functional perspective. FASEB J. 2008;22:3103–3110. doi: 10.1096/fj.08-111310. [DOI] [PubMed] [Google Scholar]
- 27.Huett A, et al. A novel hybrid yeast-human network analysis reveals an essential role for FNBP1L in antibacterial autophagy. J Immunol. 2009;182:4917–4930. doi: 10.4049/jimmunol.0803050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simonsen A, et al. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J Cell Sci. 2004;117:4239–4251. doi: 10.1242/jcs.01287. [DOI] [PubMed] [Google Scholar]
- 29.McGovern DP, et al. NIDDK IBD Genetics Consortium Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 2010;42:332–337. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brass AL, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hao L, et al. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008;454:890–893. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amit I, et al. Tal, a Tsg101-specific E3 ubiquitin ligase, regulates receptor endocytosis and retrovirus budding. Genes Dev. 2004;18:1737–1752. doi: 10.1101/gad.294904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng YT, et al. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–5916. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 34.Shevchenko DV, et al. Molecular characterization and cellular localization of TpLRR, a processed leucine-rich repeat protein of Treponema pallidum, the syphilis spirochete. J Bacteriol. 1997;179:3188–3195. doi: 10.1128/jb.179.10.3188-3195.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang XS, Choi JH, Heinz J, Chetty CS. Domain-specific positive selection contributes to the evolution of Arabidopsis leucine-rich repeat receptor-like kinase (LRR RLK) genes. J Mol Evol. 2006;63:612–621. doi: 10.1007/s00239-005-0187-z. [DOI] [PubMed] [Google Scholar]
- 36.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 37.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 38.McQuitty LL. Similarity analysis by reciprocal pairs for discrete and continuous data. Educ Psychol Meas. 1966;26:825–831. [Google Scholar]
- 39.Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- 40.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 41.Chen F, Mackey AJ, Stoeckert CJ, Jr, Roos DS. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006;34(database issue):D363–D368. doi: 10.1093/nar/gkj123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.