

Abstract

Enantioselective syntheses of all of the named chiral members of the cleroindicin family (C-F) are reported. This effort demonstrates the synthetic utility of a 2,4-dihydroxybenzaldehyde as a starting material for natural product synthesis through the use sequential o-quinone methide chemistry and diastereoselective dearomatization. Natural cleroindicin F was shown to be nearly racemic, and an optically pure synthetic sample of cleroindicin F was found to racemize under slightly basic conditions. All other natural chiral cleroindicins are shown to be partially racemic.
Introduction
Extracts of the Clerodendrum genus of plants, found throughout tropical areas of Asia and Africa, exhibit many desirable medicinal properties.1 In an effort to discover the responsible bioactive agents, Sun et al. isolated several oxidized cyclohexanones from the indicum and related japonicum and bungei species.2 These compounds were designated as cleroindicins A-F (1-4, 6-7, Figure 1).
Figure 1.

The first named members of the cleroindicin family (blue) are shown along with their reported optical rotations (red). Compound 4 was first assigned as cleroindicin D.2b The ranges of optical rotations (black) for materials procured through prior “enantioselective” syntheses are also presented. Our optical rotations for our synthetic materials (green) suggest that all of the named chiral cleroindicins are partially racemic, and these finding may have profound ramifications on the speculated biosynthesis from cornoside.
The cleroindicins have been the focus of several synthetic efforts.3 However, few reports have described their enantioselective construction,4 and none in our opinion has fully resolved the issues of absolute stereochemistry and optical rotation for all constituents of the family of natural products. Like so many of the other 4-hydroxy-cyclohexanones that we had previously encountered,5 we began to suspect that the stereochemistry of one or more of these compounds was incorrectly assigned. Further investigation of the spectroscopic data ascribed to family members in the literature illuminated several other discrepancies.6 The assignment of (3S,4R) for the vicinal stereocenters in cleroindicin C (3) by Sun et al. had largely been based on the observation of a negative rotation in accordance with the octant rule for ketones.2b This assignment subsequently led to all future assignments of absolute stereochemistry for the remaining structures. However, from our perspective, this seemed rather dubious because cleroindicin D, which was initially assigned by Sun as 4, displays a positive optical rotation while cleroindicin F (7) was reported to display a small negative rotation. Moreover, Ogasawara had reported that oxidation of (−)-cleroindicin E (6), which was assumed to be the unnatural enantiomer, afforded (−)-cleroindicin C (3), the supposedly the natural enantiomer.3b Clearly, there was a problem with structural assignments and/or rotations.
This confusion over the stereochemistry, in turn, has clouded the understanding of the biosynthesis for these compounds.7 Most have presumed that the cleroindicins are biosynthesized by dearomatization of glycosylated p-(2-hydroxyethyl)phenol, which results in the achiral cyclohexadienone cornoside, (cf. Figure 1). Olefin reduction and deglycosylation provides cleroindicins A (1) and B (2). On the other hand, deglycosylation and olefin desymmetrization by 1,4-addition of the freed oxygen atom leads to cleroindicin F (7), whereupon further biomanipulations can be imagined as leading to the remaining optically active constituents of the cleroindicin family. Alternatively, cornoside might become desymmetrized by reduction of an olefin, and cleroindicin C (3) forms by deglycosylation and a successive 1,4-addition. Subsequent re-oxidations would then lead to compounds 4-5 and 7. We believed that syntheses of these materials could settle the lingering discrepancies and might resolve the biogenesis for the family. In addition, dearomatization of aromatic compounds has been of interest to us for some time,8 and the chiral cleroindicins (3-8) offered challenging tests for demonstration of the malleability inherent in our dissymmetric chiral cyclohexa-2,5-dienones building blocks.
Some time ago, we demonstrated that various cyclohexa-2,5-dienones could be accessed by a transformative sequence that employed an ortho-quinone methide (o-QM) for conjugate addition to provide various 4-alkylated resorcinols that were subsequently subjected to diastereoselective dearomatization.9 By application of this novel process to chiral members of the cleroindicin family, we hoped to resolve the lingering spectroscopic issues and further substantiate the utility of our process for addressing ornate cyclohexyl ring systems. Herein, we report our enantioselective strategy and method for addressing all of these chiral compounds, a pursuit that has unlocked much knowledge concerning the intrinsic malleability of bromo-cyclohexa-2,5-dienones and resolved some of the discrepancies surrounding the spectroscopic data and stereochemistry of these compounds as well as their speculated biosynthesis.
Results and Discussion
Our drive towards cleroindins C-F began with the known benzaldehyde 9,9 which is available in two pots from 2,4-dihydroxy-benzaldehyde (Scheme 1). Treatment of compound 9 with [(2-(trimethylsilyl)ethoxy)methyl]lithium 1010 resulted in Boc migration, and afforded an intermediate phenoxide. This phenoxide succumbed to in situ β-elimination to form an o-quinone methide intermediate that underwent subsequent in situ 1,4-reduction with sodium borohydride to produce the phenol 11 in 68% yield. Our unusual one-pot cascade, which we have extensively investigated,11 is driven by relative anion stability. This particular example is noteworthy because addition of the lithium reagent superseded lithium halogen exchange. A standard Mitsunobu coupling of the amide 1212 afforded the inverted amide 13 in an 85% yield and >99% enantiomeric excess (ee) as confirmed by chiral HPLC analysis and comparison to a racemic standard prepared in a similar manner from racemic lactic acid.
Scheme 1.

Synthesis of the non-racemic cyclohexadienone 15a
a TMSE = CH2CH2SiMe3, DIAD = Diisopropyl azodicarboxylate
The remaining Boc residue smoothly cleaved upon exposure of 13 to aqueous lithium hydroxide to provide the phenol 14 in a 95% isolated yield, after work-up and chromatography. Oxidative dearomatization of 14 with PhI(OCOCF3)2 afforded a 76% yield of δ-lactone 15 as a single diastereomer. The structure of the syn diastereomer (−)-15 has been established by X-ray analysis of related intermediates13 and shows that the (R) configured 2-alkoxyethyl alkyl chain exists in a pseudo axial arrangement (15-3D, Figure 2) with regards to the 2-oxo-1,4-dioxane ring, whereas the (S) configured methyl residue that is adjacent to the lactone carbonyl occupies a pseudo equatorial arrangement. We believe that this rigid and locked conformation is what enables many of the subsequent chemoselective and diastereoselective manipulations of the core cyclohexadienone to occur.
Figure 2.
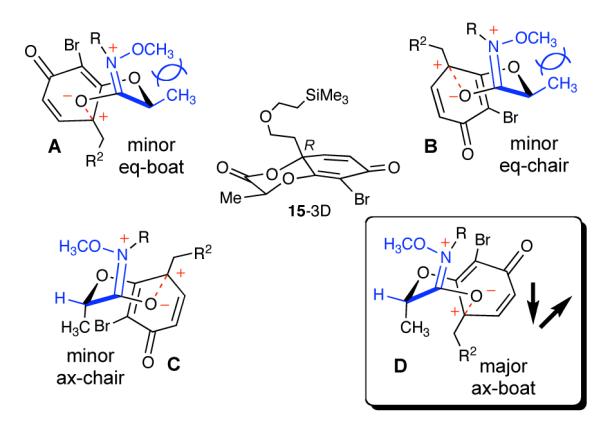
Structure 15 shown in three dimensions as well as the most plausible transition-state intermediates.
The ratio afforded by the dearomatization and lactonization depends on the size of the R2 substituent (Figure 2). Four transition states (A-D) define the energy surface leading to two plausible diastereomeric lactone products. The transition-states can be imagined as the ax-Boat D, the ax-Chair C and their corresponding ring-flips, the eq-Boat A and eq-Chair B. The latter two (A-B) contribute very little because steric interactions between the amide residue and eq-methyl group. Given the absolute stereochemistry of the major product, the ax-Boat D transition is preferred. However, the terms “boat” and “chair” apply loosely because five of the six contributing atoms in the six-membered transition state are in planar sp2 configuration. We speculate that transition-state D is preferred because the sum of the minor dipoles, which are comprised by the phenoxonium cation and the iminium zwitterion, is considerably smaller than for transition-state C. The expected correlation of diastereomeric effects and structural modifications to the chiral amide tether strongly support this reasoning.14
We now faced the challenge of chemoselective differentiation between the numerous functional groups within compound 15. This task mandated removal of the linkage that had directed the diastereoselective dearomatization, and which now served as the group protecting two of the oxygen substituents. Some time ago we developed several processes for removing related glycolic linkages that were employed in our early racemic studies of epoxysorbicillinol15 and rishirilide B.16 Unfortunately, neither sequence used for the achiral tether proved successful for cyclohexadienones derived from 12. Therefore, we considered another cleavage mode for the chiral directing group, a sequence that we had previously utilized with great success, which involved sequential reduction of the vinylogous ester and lactone, followed by ring-opening, a ring-flip, and a β-elimination of the remaining lactic acid linkage.9
To test this proposition, we treated the cyclohexadienone 15 (0.1 M in toluene, −78 °C) with Yamamoto’s Lewis acid (methylaluminium bis(2,6-di-tert-butyl-4-methylphenoxide, MAD, 2.5 equiv), followed by the addition of L-Selectride (1.05 equiv) (Scheme 2). As expected, the vinylogous ester reduced in a stereoselective anti fashion. Next, we formed the hydrofuran by submitting compound 16 (0.1 M in nitromethane) to zinc bromide (4 equiv), which caused the RO(CH2)2TMS functionality to cleave and thereby freed the oxygen atom to participate in an in situ 1,4-addition with the neighboring enone. The reaction smoothly resulted in the tricyclic compound 17 in 80% yield. At this juncture, we imagined that treatment of the α-bromo compound 17, or the corresponding amide 18, with zinc could furnish cleroindicin F (7) by formation of the corresponding zinc enolate followed by β-elimination. However, neither approach proved fruitful. We attribute this failure to an equatorially disposed C–O linkage, as treatment of 17 with zinc simply afforded the de-brominated tricycle 20 (Scheme 3, reaction not shown) without producing any of the desired β-elimination product. On the other hand, our attempts to open the δ-lactone via the aluminum amide of pyrrolidine and thereby facilitate a cyclohexyl ring-flip and further β-elimination also failed, and instead resulted in β-elimination of the opposite C–O bond followed by bromide displacement to afford the [3.3.1]-bicycle 19 in 55% yield.
Scheme 2a.

a First planned formation of cleroindicin F (7) via 17 fails. Compound 19 forms instead when opening 17 with the aluminum amide of pyrrolidine.
Scheme 3a.
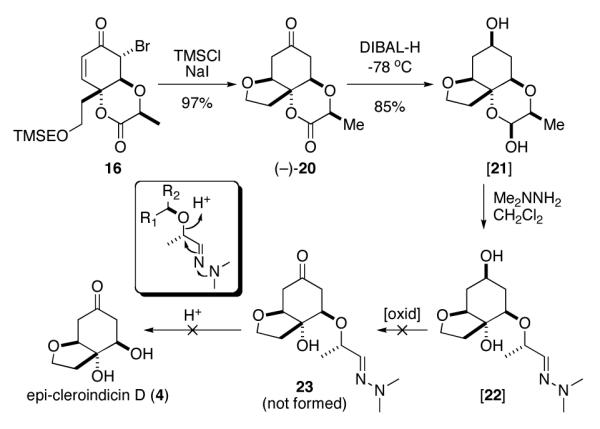
a Failed formation of epi-cleroindicin D (4) via intermediacy of 22.
With β-elimination as a method for removal of the chiral directing group no longer available to us, we considered our other options. Some time ago we reported a method for scission of an alpha C–O bond in aldehydes by sequential treatment of an aldehyde or its corresponding lactol with dimethylhydrazine followed by acid (inset, Scheme 3).5,14 Thus, we imagined that we could use this process to form 4, which at that time we still believed to be the structure of cleroindicin D, by severing the C–O bond in 23 (Scheme 3).
Treatment of 16 with TMSI caused cleavage of the bromide of the α-bromo-ketone and the RO(CH2)2TMS ether to afford the tricyclic compound (−)-20 in a respectable 80% yield for the two transformations (one-pot) (Scheme 3). The stability of the tricyclic compound to these notoriously harsh conditions is testimony to the sturdiness of the 2-oxo-1,4-dioxane ring as a diol protecting group.
Reduction of the two carbonyls in compound 20 with DIBAL-H (4.1 equiv) afforded a hemi-acetal that we tentatively assigned with the stereochemistry shown in 21. Further treatment of the lactol 21 with dimethyl hydrazine produced the chromatographically unstable hydrazone 22. Unfortunately, our plan, to oxidize the secondary alcohol and then to sever the C–O bond, also failed. Attempts aimed at oxidation of the secondary alcohol with Dess-Martin reagent led to either decomposition or returned the tricyclic lactone 20.
We therefore decided to retain the original carbonyl oxidation state by masking the ketone in (−)-20 as a ketal that could be easily deprotected at some later stage. The carbonyl functionality in 20 was readily transformed into the desired benzylidene ketal by treatment with the appropriate aryl diol in the presence of acid and azeotropic removal of water (Scheme 4). Subsequent cleavage of the 1,4-dioxa-2-one moiety trans-diol occurred over two pots. First, the lactone was reduced to the corresponding lactol with excess DIBAL-H in CH2Cl2. After work-up to remove aluminum salts, the crude lactol was submitted to dimethyl hydrazine and trace trifluoroacetic acid to break the C–O bond. The two-step process afforded the diol 25 in 75% yield, and this UV active compound displayed >99% ee by rigorous comparison to a racemic HPLC standard. Subsequent cleavage of the ketal by hydrogenolysis afforded (−)-epi-cleroindicin D (4), a structure previously misidentified as cleroindicin D, in nine steps (19.3% overall yield) from benzaldehyde 9. While Carreño recently reported clarification of the structure of cleroindicin D (5) by its racemic synthesis,18 it is interesting to note, that Yamasaki reported synthesizing a structure identified as (±)-57 in 1995, and reported data that closely matched that which was subsequently reported by Sun for cleroindicin D (5) and incorrectly assigned as the structure 4.
Scheme 4.

Synthesis of (−)-epi-cleroindicin D (4) and (±)-cleroindicin F (7) from the non-racemic tricycle 20
Since the synthetic compound 4 was evidently the undesired diastereomer epi-cleroindicin D, we decided to invert its secondary alcoholic stereocenter to prepare 5. We attempted to employ standard Mitsunobu conditions17 with 25 (Scheme 4). However, instead of the desired ester, we obtained the epoxide 26. We therefore targeted cleroindicin F (7), thinking that it might be reached by β-elimination. We independently converted the secondary alcohol into its corresponding acetate and mesylate, and then subjected each separately to hydrogenolysis of the respective benzylidene ketal. This two-pot sequence afforded the relevant acetate and mesylate (27, 28) in about 90% yield over the two steps. Next, we exposed these materials to base. Treatment of the acetate 27 with i-Pr2NEt or DBU had no effect. Both conditions simply returned the starting material unchanged. On the other hand, we found that treatment of the mesylate 28 with pyridine for 24 hours afforded optically enriched (−)-cleroindicin F (7) with 90% ee, as determined by chiral HPLC comparison with a racemic standard. Moreover, application of either i-Pr2NEt or DBU to mesylate 28 afforded nearly racemic cleroindicin F (7) in less than twenty-five minutes, presumably via the intermediacy of the achiral cyclohexadienone. This rapid racemization of (+)-7 was further confirmed by separate treatment of optically enriched 7 with either i-Pr2NEt or DBU. We therefore recognized that in order to prevent corruption of the optical activity, we would require non-basic, or perhaps nearly neutral conditions for the formation and subsequent reactions of 7. We therefore chose to install an even better leaving group and converted alcohol 25 into its corresponding tosylate 29 (Scheme 5).
Scheme 5.

Enantioselective synthesis of all remaining cleroindicins 3, 5-8
Hydrogenation of the ketal proceeded to the expected ketone bearing the β-tosylate. Upon silica gel chromatography, elimination proceeded to afford the synthetic enone (+)-7 ([α]D = +59.0°, c = 1.0, >99% ee). However, we again found that compound 7 can racemize under basic conditions [(0.1 M CH2Cl2, 1 equiv DBU, rt, 0% ee in 5 min) (0.1 M CH2Cl2, 1 equiv pyridine, rt, 90% ee after 12 h)]. A racemization might account for the very low rotation observed in the natural product 7, and other supposed enantioselective syntheses of 7.16b Moreover, this outcome suggested that we would have to be especially vigilant in the nucleophilic epoxidation of enone 7 to avoid loss of enantiomeric excess. After examining many conditions, we found that treatment of 7 with Ph3COOK, prepared from 4 equiv of Ph3COOH and 2 equiv of KH, afforded the epoxide 30 in greater than 99% ee. Further treatment of 30 with aluminum-mercury amalgam18 afforded synthetic cleroindicin D (5) ([α]D = −38.0°, c = 0.5), which proved identical in all other respects with natural cleroindicin D. Further hydrogenation of synthetic (+)-cleroindicin F (7) afforded synthetic (−)-cleroindicin C (3) ([α]D = −79.0°, c = 0.1), which when compared to the rotation of the natural material also suggests that natural cleroindicin C is partly racemic. Reduction of synthetic cleroindicin C (3) proceeded primarily on the convex face of the carbonyl to afford iso-cleroindicin E (8). On the other hand, reduction of 3 with samarium diiodide afforded a 2:1 ratio favoring cleroindicin E (6).
Conclusion
In summary, the enantioselective syntheses of the all the named chiral cleroindicins have been completed from 2,4-dihydroxybenzaldehyde. These synthetic materials were constructed in nearly enantiopure form. From the measured optical rotations of these synthetic materials, it appears that all of the named natural chiral cleroindicins are either partially or nearly racemic, as are many of the compounds obtained from prior ‘claimed’ enantioselective syntheses.19 Because all of the named chiral cleroindicins (C, D, E) are partially or nearly racemic, it would seem that each is derived from cleroindicin F, which racemizes during the biosynthesis. We further extrapolate from our data and the earlier data of Hase,7 that cornoside may be produced from cleroindicin F, and that an unnamed chiral cleroindicin ((3aS,6R,7aS)-2,3,3a,6,7,7a-hexahydrobenzofuran-3a,6-diol, 31, ([α]D = +102.0°)7 (see supporting information) may immediately precede cleroindicin F in the biosynthesis.
Experimental Section
2-Bromo-3-hydroxy-4-(2-(2-(trimethylsilyl)ethoxy)ethyl)phenyl tert-butyl carbonate (11)
n-BuLi (0.34 mL, 4.8 M, 1.05 equiv) was added in a dropwise fashion to a stirring solution of trimethyl(2-((tributylstannyl)methoxy)ethyl)silane (685 mg, 1.62 mmol, 1.05 equiv.) in THF (0.65 M) at −78 °C. After stirring for approximately 20 minutes, the Still alkoxy lithium solution was transferred via cannula to a solution of benzaldehyde 9 (645 mg, 1.55 mmol, 1 equiv) in THF (26 mL) at −78 °C. After 1 h, the reaction was quenched with water and the cold bath was removed. A solution of NaBH4 (469 mg, 12.4 mmol, 8 equiv) in cold water (5 mL) was added. The reaction was stirred for 24 h at room temperature, then quenched with 1 N HCl, extracted with Et2O four times. The combined organics were washed with brine, dried with Na2SO4, and concentrated in vacuo. Flash chromatography of the residue (5% EtOAc/hexanes, Rf = 0.15) yielded colorless oil 11 (457 mg, 1.05 mmol, 68% yield). 1H NMR (400 MHz, CDCl3) δ 9.17 (s, 1H), 7.00 (d, J = 8.4 Hz, 1H), 6.70 (d, J = 8 Hz, 1H), 3.70 (t, J = 4.8 Hz, 2H), 3.58 (t, J = 8.4 Hz, 2H), 2.92 (t, J = 4.8 Hz, 2H), 1.57 (s, 9H), 1.03 (t, J = 8.4 Hz, 2H), 0.02 (s, 9H); 13C NMR (100 MHz. CDCl3) δ 154.3, 151.3, 148.4, 129.8, 126.0, 114.3, 106.7, 84.1, 71.5, 69.3, 33.6, 27.8, 18.3, −1.3; IR (thin film) 2980, 2953, 2870, 1763, 1151 cm−1; HRMS (ESI) calcd for C18H29BrO5SiNa: 455.0865. Found 455.0872.
(3S,8aR)-5-Bromo-3-methyl-8a-(2-(2-(trimethylsilyl)ethoxy)ethyl)benzo[b][1,4]dioxine-2,6(3H,8aH)-dione (15)
Phenol 14 (0.605 g, 1.35 mmol) was dissolved in CH3NO2 (13.5 mL) and cooled to −10 °C. [Bis(trifluoroacetoxy)iodo]benzene (0.871 g, 2.03 mmol, 1.50 equiv) was added in one portion. The reaction mixture was stirred at −10 °C for 15 min, and gradually turned light yellow. When residual water was present in the phenol, a darker reaction mixture was observed, and a lower yield was obtained. To quench the reaction, deionized water (20 mL) was added, immediately followed by CH2Cl2 (20 mL) and it was stirred vigorously at 0 °C for 15 min. Upon initial addition of water, the solution turned dark green then faded to yellow after several minutes. The aqueous layer was extracted with CH2Cl2 (4 × 20 mL). The combined organics were washed with brine (10 mL), dried (Na2SO4), and concentrated. The crude product was purified by flash chromatography with 10% EtOAc/hexanes (Rf = 0.15) to afford 15 (0.414 g, 1.03 mmol, 76% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.85 (d, J = 10 Hz, 1H), 6.37 (d, J = 10 Hz, 1H), 4.95 (q, J = 7.2 Hz, 1H), 3.35 (m, 4H), 2.35 (m, 2H), 1.84 (d, J = 7.2 Hz, 3H), 0.83 (t, J = 9.2 Hz, 2H), −0.04 (s, 9H); 13C NMR (100 MHz. CDCl3) δ 179.5, 166.4, 161.4, 142.1, 127.5, 108.0, 78.8, 74.4, 68.6, 64.2, 42.2, 19.2, 18.1, −1.3; IR (thin film) 2950, 2866, 1762, 1670, 1250 cm−1; HRMS (ESI) calcd for C16H23BrO5SiNa: 425.0396. Found 425.0409. [α]D25 = −52.9 (CHCl3, c = 0.9).
Cleroindicin F (7), non-racemized
To a solution of diol 25 (7.0 mg, 0.024 mmol) in pyridine (0.3 mL) was added TsCl (14.0 mg, 0.072 mmol, 3.0 equiv). After 18 h of stirring, the reaction mixture was diluted with CH2Cl2, washed with CuSO4 (aq), extracted with CH2Cl2 (4x), washed with water and brine. The organic phase was dried over Na2SO4 and concentrated under reduced pressure. The resulting mono-tosylate 29 was purified by flash chromatography (40% EtOAc/hexanes, Rf = 0.2).
A solution of the above mono-tosylate 29 in THF/MeOH/CH2Cl2 (0.16 mL/0.16 mL/0.16mL) containing 5% Pd/C (3 mg) was stirred under 1 atm of hydrogen at room temperature for 12 h. The reaction mixture was filtered through a pad of Celite, and concentrated under reduced pressure. The crude product was purified by flash chromatography (70% EtOAc/hexanes, Rf = 0.15) to provide cleroindicin F (7) as colorless oil (6.3 mg, 0.041 mmol, 72 % yield over 2 steps, >99% ee). Note: Base-induced elimination, or treatment of 7 with base (pyridine or DBU), yielded partially or fully racemized cleroindicin F (7).19 The enantiomeric ratio was determined by HPLC (Chiralcel AD-H, 4% IPA/Hexanes, tR1 = 48.78 min, t = 57.30 min) for each reaction. 1H NMR (500 MHz, CDCl3) δ 6.77 (d, J = 10 Hz, 1H), 6.04 (d, J = 10 Hz, 1H), 4.25 (t, J = 6.0 Hz, 1H), 4.08 (dd, J = 8.5 Hz, J = 15 Hz, 1H), 3.96 (dd, J = 8.5 Hz, J = 15 Hz, 1H), 3.50 (bs, 1H), 2.80 (dd, J = 4.5 Hz, J = 17 Hz, 1H), 2.63 (dd, J = 6 Hz, J = 17 Hz, 1H), 2.32 (m, 1H), 2.24 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 196.7, 147.8, 129.2, 81.9, 76.0, 66.5, 40.5, 39.8; IR (thin film) 3418, 2920, 2851, 1666 cm−1; HRMS (ESI) calcd for C8H10O3: 154.0630. Found 154.0625. [α]D25 = +59.0 (MeOH, c = 1.0). 1H and 13C NMR, IR, and HRMS data were consistent with that reported in the literature, though our rotation is significantly higher.2b, 7
Supplementary Material
Acknowledgement
Continued research support for exploration of this cyclohexadienone chemistry from the National Institutes of Health (GM-64831) is greatly appreciated.
Footnotes
Supporting Information Available: Additional procedures and spectral data for all new compounds are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Cheng H-H, Wang H-K, Ito J, Bastow KFTY, Nakanishi Y, Xu Z, Luo T-Y, Lee K-H. J. Nat. Prod. 2001;64:915–919. doi: 10.1021/np000595b. [DOI] [PubMed] [Google Scholar]
- (2) (a).Tian J, Shao Q-S, Lin ZW, Sun H-D. Chin. Chem. Lett. 1996;7:279–282. [Google Scholar]; (b) Tian J, Zhao Q-S, Zhang H-J, Lin ZW, Sun H-D. J. Nat. Prod. 1997;60:766–769. [Google Scholar]; (c) Tian J, Zhao Q-S, Zhang H-J, Lin Z-W, Sun H-D. Chin. Chem. Lett. 1997;8:129–132. [Google Scholar]; (d) Yang H, Hou A-J, Mei S-X, Sun H-D, Che C-T. J. Asian Nat. Prod. Res. 2002;4:165–169. doi: 10.1080/1028602021000000053. [DOI] [PubMed] [Google Scholar]
- (3) (a).Chen J, Tian J, Wu FE, Kawabe N, Tokuda M. Chinese Chem. Lett. 2001;12:771–774. [Google Scholar]; (b) Honzumi M, Kamikubo T, Ogasawara K. Synlett. 1998:1001–1003. [Google Scholar]
- (4) (a).See ref. 3b and Canto M, de March P, Figueredo M, Font J, Rodriguez S, Alarez-Larena A, Piniella JF. Tetrahedron: Asymmetry. 2002;13:455–459.
- (5).Hoarau C, Pettus TRR. Org. Lett. 2006;8:2843–2846. doi: 10.1021/ol061000s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6) (a).Racemic cleroindicin F (7) has also been called rengyolone, Endo K, Hikino H. Can. J. Chem. 1984;62:2011–2014. and halleridone, Messana I, Sperandei M, Mulatari G, Galeffi C, Bettolo G. B. Marini. Phytochemistry. 1984;23:2617–2619.
- (7).Hase T, Kawamoto Y, Kasai R, Yamasaki K, Picheansoonthon C. Phytochemistry. 1995;39:235–241. [Google Scholar]
- (8).Magdziak D, Meek SJ, Pettus TRR. Chem. Rev. 2004;104:1383–1429. doi: 10.1021/cr0306900. [DOI] [PubMed] [Google Scholar]
- (9).Mejorado L, Hoarau C, Pettus TRR. Org. Lett. 2004;6:1535–1538. doi: 10.1021/ol0498592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10) (a).Kozikowski AP, Okita M, Kobayashi M, Floss HG. J. Org. Chem. 1988;53:863–869. [Google Scholar]; (b) Still WC. J. Am. Chem. Soc. 1978;100:1481–1487. [Google Scholar]
- (11) (a).Van De Water R, Magdziak D, Chau J, Pettus TRR. J. Am. Chem. Soc. 2000;122:6502–6503. [Google Scholar]; (b) Jones RM, Van de Water RW, Lindsey CC, Pettus TRR. J. Org. Chem. 2001;66:3435–3441. doi: 10.1021/jo001752e. [DOI] [PubMed] [Google Scholar]
- (12) (a).Patterson I, Wallace DJ, Cowden CJ. Synthesis. 1998:639–652. [Google Scholar]; (b) Less SL, Leadlay PF, Dutton CJ, Staunton J. Tetrahedron Lett. 1996;37:3519–3520. [Google Scholar]
- (13).For an example, please see: Wenderski TA, Huang S, Pettus TRR. Communication to the Cambridge Structural Database, deposition number CCDC 726677. 2009.
- (14).Mejorado L, Pettus TRR. J. Am. Chem. Soc. 2006;128:15625–15631. doi: 10.1021/ja062987w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pettus LH, Van de Water RW, Pettus TRR. Org. Lett. 2001;3:905–908. doi: 10.1021/ol0155438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16) (a).Wang J, Pettus LH, Pettus TRR. Tetrahedron Lett. 2004;45:1793–1796. [Google Scholar]; (b) Wang JH, Pettus TRR. Tetrahedron Lett. 2004;45:5895–5899. [Google Scholar]
- (17).Martin SF, Dodge JA. Tetrahedron Lett. 1991;32:3017–3020. [Google Scholar]
- (18).Barradas S, González-López M. Carreño, Latorre A, Urbano A. Org. Lett. 2007;9:5019–5022. doi: 10.1021/ol702236e. [DOI] [PubMed] [Google Scholar]
- (19).You Z, Hoveyda AH, Snapper ML. Angew. Chem. Int. Ed. 2009;48:547–550. doi: 10.1002/anie.200805338.; consider the supporting information for cleroindicin F.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.