

Abstract
CYP3A4, an important drug-metabolizing enzyme, is known to have genetic variants. We have previously reported that CYP3A4 variants such as CYP3A4.2, 7, 16, and 18 show different enzymatic kinetics from CYP3A4.1 (wild type). In this study, we quantitatively investigated the inhibition kinetics of two typical inhibitors, itraconazole (ITCZ) and cimetidine (CMD), on CYP3A4 variants and evaluated whether the genetic variation leads to interindividual differences in the extent of CYP3A4-mediated drug interactions. The inhibitory profiles of ITCZ and CMD on the metabolism of testosterone (TST) were analyzed by using recombinant CYP3A4 variants. The genetic variation of CYP3A4 significantly affected the inhibition profiles of the two inhibitors. In CYP3A4.7, the Ki value for ITCZ was 2.4-fold higher than that for the wild-type enzyme, whereas the Ki value for CMD was 0.64-fold lower. In CYP3A4.16, the Ki value for ITCZ was 0.54-fold lower than that for wild-type CYP3A4, whereas the Ki value for CMD was 3.2-fold higher. The influence of other genetic variations also differed between the two inhibitors. Docking simulations could explain the changes in the Ki values, based on the accessibility of TST and inhibitors to the heme moiety of the CYP3A4 molecule. In conclusion, the inhibitory effects of an inhibitor differ among CYP3A4 variants, suggesting that the genetic variation of CYP3A4 may contribute, at least in part, to interindividual differences in drug interactions mediated by CYP3A4 inhibition, and the pattern of the influences of genetic variation differs among inhibitors as well as substrates.
Introduction
CYP3A enzymes are expressed in the liver, kidney, and intestine in humans and are responsible for the metabolism of >50% of clinically used drugs (Shimada et al., 1994; Guengerich, 1999, 2008). Twenty genetic variants that lead to amino acid substitution have been identified in the CYP3A4 gene [The Human Cytochrome P450 (CYP) Allele Nomenclature Committee Web Site, http://www.cypalleles.ki.se/cyp3a4.htm].
In our previous study, we compared the enzymatic activities of major CYP3A4 variants from alleles with relatively higher allelic frequency, such as CYP3A4*1 (wild type),*2 (2.7% in whites) (Sata et al., 2000), *7 (1.4–3% in white) (Eiselt et al., 2001; Lamba et al., 2002), *16 (1.4–5% in Japanese) (Lamba et al., 2002; Fukushima-Uesaka et al., 2004), and *18 (1.3–2.8% in Japanese) (Fukushima-Uesaka et al., 2004) by using Escherichia coli expression systems (Miyazaki et al., 2008). CYP3A4.2 and CYP3A4.16 exhibited lower catalytic activity for 6β-hydroxylation of testosterone (TST), a probe substrate of CYP3A4, than CYP3A4.1 (wild type), whereas CYP3A4.18 had a higher activity, and CYP3A4.7 had comparable activity. Other researchers have also reported similar results; the TST activity was lower in CYP3A4.2, 7, and 16 variants than CYP3A4.1 (wild type) (Sata et al., 2000; Eiselt et al., 2001; Lamba et al., 2002; Murayama et al., 2002; Fukushima-Uesaka et al., 2004) and higher with CYP3A4.18 (Kang et al., 2009). Kang et al. (2009) have recently reported that the in vivo influence of CYP3A4*18 (i.e., subjects bearing the *18 allele) shows low bone mineral density ostensibly because of the enhanced turnover of both TST and estrogen. Taken together, genetic variation of CYP3A4 is quite likely to be a key factor in interindividual differences in responses to CYP3A4 substrates (Kang et al., 2009).
On the other hand, inhibition of CYP3A4 is a major cause of drug-drug interactions (DDI) because CYP3A4 is responsible for the metabolism of many drugs that are widely used in the clinical settings (Zhou et al., 2007). Therefore, genetic variations of CYP3A4 that result in altered inhibitory kinetics might contribute to interindividual differences in the extent of CYP3A4-mediated DDI. However, the difference in the inhibition kinetics of CYP3A4 inhibitors among CYP3A4 genetic variants remains to be characterized. Moreover, no researchers have reported the clinical effect of genetic variation, i.e., CYP3A4*2, *7, *16, and *18, on the extent of DDI.
Characterization of the nature of binding of substrate and inhibitor to CYP3A4 may also aid in understanding differences in the inhibitory kinetics among variants. After the crystal structure of CYP3A4 was determined (Williams et al., 2004; Yano et al., 2004), several docking simulations have been carried out to identify the amino acid residues responsible for ligand binding (Kriegl et al., 2005; Park et al., 2005) and to reveal and predict their binding nature (Teixeira et al., 2010; Unwalla et al., 2010).
The aims of this study were to characterize the inhibition kinetics of two typical CYP3A4 inhibitors with distinctive structures, itraconazole (ITCZ) and cimetidine (CMD), on the TST 6β-hydroxylation activity of recombinant CYP3A4.1, 2, 7, 16, and 18 variants prepared from E. coli expression systems and to compare them with the results of docking simulation study for CYP3A4 variant molecules, substrates, and inhibitors.
Materials and Methods
Chemicals and Materials.
TST [(8R,9S,10R,13S,14S,17S)-17-hydroxy-10,13-dimethyl-1,2,6,7,8,9,11,12,14,15,16,17- dodecahydrocyclopenta[a]phenanthren-3-one] was purchased from Nacalai Tesque (Kyoto, Japan). 6β-Hydroxytestosterone (6β-OHT) and hydrocortisone were purchased from Spi-Bio, Bertin Group (Montigny le Bretonneux, France). ITCZ [(2R,4S)-rel-1-(butan-2-yl)-4-{4-[4-(4-{[(2R,4S)-2-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy}phenyl)piperazin-1-yl]phenyl}-4,5-dihydro-1H-1,2, 4-triazol-5-one] was purchased from Sigma-Aldrich (St. Louis, MO). CMD [2-cyano-1-methyl-3-(2-[(5-methyl-1H-imidazol-4-yl)methylthio]ethyl)guanidine] was purchased from Wako Pure Chemicals (Osaka, Japan). The membrane fractions for CYP3A4.1 (wild type), 2(S222P, 7(G56D, 16(T185S))), and 18(L293P) were prepared as described previously (Miyazaki et al., 2008). All other chemicals and reagents were of analytical or high-performance liquid chromatography (HPLC) grade and were obtained from commercial sources.
Inhibition of Enzymatic Activities of CYP3A4 Variants by ITCZ and CMD.
A 450-μl aliquot of 200 mM potassium phosphate buffer (pH 7.4) containing substrate (TST), inhibitor (ITCZ or CMD), 0.5 mM EDTA, and CYP3A4 membrane fraction (0.5 nmol P450/ml) was preincubated at 37°C for 10 min, and reactions were initiated with 50 μl of an NADPH-generating system (50 mM glucose 6-phosphate, 5 mM NADP+, 1 unit/ml glucose 6-phosphate dehydrogenase, and 30 mM MgCl2) to start reaction. The reaction mixtures (total of 500 μl) were incubated for 20 min, and the reactions were terminated by adding 450 μl of ice-cold acetonitrile, followed by spiking with 50 μl of hydrocortisone (0.5 mM) in methanol (internal standard). After centrifugation at 1200g for 20 min at 4°C, 6β-OHT (in the supernatant) was determined by the HPLC-UV method described below. TST and ITCZ were dissolved in ethanol and methanol, with the final concentrations of solvent in reaction mixture of ≤1 and ≤2.5% (v/v), respectively, which have been shown not to affect CYP3A4 activity (Chauret et al., 1998; Busby et al., 1999; Easterbrook et al., 2001). The enzymatic activity of CYP3A4 was evaluated by determining the production rate of 6β-OHT from TST.
Analysis of 6β-OHT Using HPLC-UV Systems.
Concentrations of 6 β-OHT were measured by an HPLC-UV method reported previously (Miyazaki et al., 2008). In brief, the HPLC system consisted of a pump (LC-10AD; Shimadzu, Kyoto, Japan), a UV detector (SPD-10AV spectrophotometer; Shimadzu), and an octadecylsilane column (Cosmosil, 5C18-AR-2, 4.6 × 150 mm; Nacalai Tesque, Kyoto, Japan). The mobile phase consisted of methanol and water (58:42, v/v), pumped at a rate of 1.0 ml/min. The absorbance of 6β-OHT was measured at 254 nm, and the detection limit was 0.3 μM.
Analysis of Metabolism and Inhibition Kinetics.
The reaction rate (v) in the presence of a competitive inhibitor, such as ITCZ or CMD, is described by eq. 1.
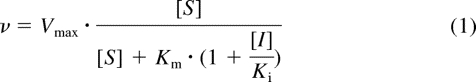 |
Equation 1 can be rearranged to give eq. 1′.
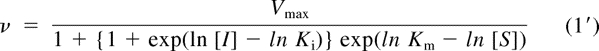 |
Equation 1′ was simultaneously fit to the observed reaction rate at varying concentrations of substrate ([S]) and inhibitor ([I]) using nonlinear least-squares fitting with software (MLAB; Civilized Software, Inc., Silver Spring, MD) to obtain parameters of enzyme kinetics, i.e., logarithm of Michaelis constant (ln Km), maximum reaction rate (Vmax), and logarithm of inhibitory constant (ln Ki), because the parameters Km and Ki conceivably follow log-normal distribution.
Docking Simulations of TST and Inhibitors to CYP3A4 Variants.
Molecular docking studies were performed on two CYP3A4-specific inhibitors with CYP3A4 variants, according to our previous reported method (Okada et al., 2009; Shimada et al., 2010). In brief, the CYP3A4 variant primary sequences were aligned with human CYP3A4 (Protein Data Bank code 1TQN), according to the reported amino acid substitution (Sata et al., 2000; Eiselt et al., 2001; Lamba et al., 2002; Murayama et al., 2002; Fukushima-Uesaka et al., 2004) using MOE software (version 2009.10; Chemical Computing Group, Montreal, QC, Canada) for modeling of a three-dimensional structure. Docking simulations were carried out for TST (substrate) and ITCZ or CMD (inhibitor) binding to CYP3A4 using the MMFF94x force field distributed in the MOE Dock software. The docking experiments were ranked according to total interaction energy (U value) (Okada et al., 2009).
Statistical Analysis.
Statistical differences between the Ki values of CYP3A4 variants were determined by analysis of variance, followed by Dunnett's multiple comparison tests using SPSS software for Windows (version 16; SPSS Inc., Chicago, IL). P values <0.05 were considered statistically significant.
Results
Comparison of Inhibition Kinetics of CYP3A4 Inhibition for CYP3A4 Variants.
Both ITCZ and CMD inhibited the TST 6β-hydroxylation activity of CYP3A4 variants in a concentration-dependent manner (Fig. 1). The Ki value of ITCZ for CYP3A4.7 was 179 μM and significantly (2.4-fold) higher than that for CYP3A4.1 (76 μM), whereas the Ki value of CMD was 230 μM and significantly (1.6-fold) lower than that for CYP3A4.1 (370 μM) (Table 1). In contrast, with regard to CYP3A4.16, the Ki value of ITCZ was 0.54-fold that for CYP3A4.1, and the Ki value of CMD was 3.2-fold, significantly increased in comparison with that for CYP3A4.1. For CYP3A4.18, the Ki value of CMD was significantly lower (0.39-fold), whereas for CYP3A4.2, the Ki value of ITCZ was lower (0.59-fold) than that for CYP3A4.1 (Table 1).
Fig. 1.

Inhibition kinetics of ITCZ or CMD for CYP3A4-mediated TST 6β-hydroxylation. A and D, CYP3A4.1 (wild type). B and E, CYP3A4.7. C and F, CYP3A4.16. A to C, ITCZ. D to F, CMD. The oxidation activities represent the mean ± S.D. from independent experiments. The substrate concentration ranged from 20 to 500 μM, and the ITCZ concentration ranged from 0 to 500 nM. Open circles, 0 nM ITCZ; light gray circles, 50 nM ITCZ; neutral gray circles, 100 nM ITCZ; dark gray circles, 250 nM ITCZ; and closed circles, 500 nM ITCZ. The CMD concentration ranged from 0 to 500 μM. Open circles, 0 μM CMD; light gray circles, 100 μM CMD; neutral gray circles, 250 μM CMD; dark gray circles, 500 μM CMD; and closed circles, 1000 μM CMD.
TABLE 1.
Kinetic parameters of testosterone 6β-hydroxylation in CYP3A4 wild type and variants
Geometric mean (−1 S.D. − +1 S.D.) for Vmax, Km, and Ki; arithmetic mean ± S.D. for Vmax/Km. Values are expressed to two significant digits. The Vmax/Km value represents an average for each set of each data.
| n | Vmax | Km | Vmax/Km |
Ki |
||
|---|---|---|---|---|---|---|
| ITCZ, nM | CMD, μM | |||||
| nmol per min/nmol P450 | μM | μl per min/nmol P450 | ||||
| CYP3A4.1 | 5 | 22 (18.6–25.1) | 290 (232–372) | 0.074 ± 0.0094 | 76 (45.1–129) | 370 (285–471) |
| CYP3A4.2 | 5 | 6.3 (4.96–7.63) | 220 (177–272) | 0.029 ± 0.0073 | 45 (30.7–65.7) | 360 (334–392) |
| CYP3A4.7 | 5 | 38 (24.9–51.5) | 400 (243–663) | 0.090 ± 0.018 | 180* (146–219) | 230* (192–286) |
| CYP3A4.16 | 5 | 4.2 (3.77–4.63) | 440 (338–555) | 0.0099 ± 0.0028 | 41 (17.1–99.8) | 1200* (784–1790) |
| CYP3A4.18 | 5 | 26 (22.0–29.3) | 140 (83.6–223) | 0.20 ± 0.096 | 82 (51.4–133) | 140* (104–196) |
n, the number of experiments.
P < 0.05 vs. CYP3A4.1.
Docking Simulations.
CYP3A4 variant crystal structures were generated in a homology model of CYP3A4 wild type using a MOE program. In the docking simulations with TST (substrate) and ITCZ (inhibitor) into CYP3A4 variants, the most stable energy states were in the positions shown in Fig. 2, A and B. Consistent with the results of enzymatic study, docking simulation shows that, in CYP3A4.1, ITCZ is docked so that its azole ring is located on the vertical center line of the heme ring, whereas, in CYP3A4.7, the position of ITCZ is far from the heme ring, and its orientation is also altered. Therefore, the access of ITCZ to the heme region, the reaction center for TST hydroxylation, of CYP3A4.7 was impaired in the presence of TST.
Fig. 2.

Docking simulation of TST, ITCZ, and CMD into P450 3A4 variants. The heme group of the P450 is shown at the lower part of each panel. In the figure, oxygen, nitrogen, sulfur, and iron atoms are colored with red, blue, yellow, and light blue, respectively. A and B were based on an orientation for models of P450 3A4.1 and 7 with ITCZ interaction energies (U value) of 18.0 and 187, respectively. C and D were based on an orientation for models of P450 3A4.1 and 16 with CMD interaction energies (U value) of −19.7 and −22.7, respectively.
For CMD, the most stable energy states in the position of TST and CMD are shown in Fig. 2, C and D. In CYP3A4.1, CMD was shown to dock vertically with its imidazole ring in interacting with the heme ring. However, in CYP3A4.16, CMD is docked into farther position with opposite orientation so that the access of the imidazole ring to the heme region is impaired in comparison with that in CYP3A4.1. These results are also consistent with the kinetic observations in our enzymatic study (vide supra).
Discussion
Consistent with our previous report (Miyazaki et al., 2008), the Km values for TST 6β-hydroxylation were increased in CYP3A4.7 and 16, decreased in CYP3A4.18, and comparable in CYP3A4.2 (in comparison with CYP3A4.1). The observed influences of genetic variation on the enzyme catalytic efficiency (Vmax/Km) were also consistent with previous in vitro findings (Sata et al., 2000; Eiselt et al., 2001; Lamba et al., 2002; Murayama et al., 2002; Fukushima-Uesaka et al., 2004). The absolute Km and Vmax values in the present study were not identical to those in our previous report (Miyazaki et al., 2008). Although the cause of this discrepancy remains uncertain, minor modification of the experimental condition, such as the composition of reaction buffer to solubilize both substrate and inhibitor in our present study, may have affected the absolute value of Vmax/Km. The Ki value of ITCZ for TST 6β-hydroxylation in recombinant CYP3A4.1 in this study was 76 nM (Table 1) and was comparable with Ki values previously estimated in human liver microsomes, i.e., 230 nM (corrected by fu,mic 0.056) (Galetin et al., 2005) and 10 to 27 nM (Zhou et al., 2007). The observed Ki value of for CMD was 370 μM (Table 1) and again consistent with the Ki value of 267 μM reported by Zhou et al. (2007). The Ki values for ITCZ and CMD varied 4- and >8-fold among CYP3A4 variants, respectively. Moreover, the influence of genetic variations was not parallel, e.g., the Ki value of ITCZ was increased in CYP3A4.7 and decreased in CYP3A4.16, whereas the Ki value of CMD was decreased in CYP3A4.7 and increased in CYP3A4.16. In other words, the influence of genetic variation on the inhibition kinetics is also dependent on the inhibitors.
Nonspecific binding of a substrate or inhibitor to the in vitro microsomal preparation may lead to underestimation of Km or Ki for an enzyme. Correction of a Ki value for an inhibitor requires consideration of the unbound fraction (fu,mic) in the microsomal preparation (Austin et al., 2002, 2005). ITCZ, used in this study as a typical inhibitor, is highly lipophilic, and its nonspecific binding to membrane fraction is 62 to 91% (Isoherranen et al., 2004) in Supersomes and 80 to 88% (Isoherranen et al., 2004) and 95% (Ishigam et al., 2001) in human liver microsomes. However, the aim of the present study was to investigate the relative influence of genetic variation but not to determine the absolute Ki values, so, accordingly, we did not measure fu,mic.
Perhaps the most plausible explanation for the differences among CYP3A4 variants is conformational changes caused by amino acid substitution that may lead altered accessibility of substrate and inhibitors to the heme iron of CYP3A4. We have previously carried out docking simulation studies for substrates and inhibitors of cytochrome P450 (P450) proteins, and many of the results are consistent with the enzymatic characteristics observed in the in vitro studies (Okada et al., 2009). In this study, we constructed a model to simulate the CYP3A4 variants with amino acid substitutions using a docking simulation program, MOE, and carried out simulation for the binding of substrates and inhibitors. As a result, CYP3A4.7 and CYP3A4.16 were shown to be structurally less suitable for access of ITCZ and CMD to the heme iron, respectively. These results are consistent with experimental observations of increased Ki values. It is likely that distinctive changes in the accessibility of inhibitors to the heme iron in each variant result in the distinctive changes in the affinity of each inhibitor. Apart from the accessibility, differences in the formation of coordinate bonds between CYP3A4 and inhibitors may also be responsible for differences in the influence of genetic variation among the inhibitors. With regard to the binding of the inhibitor metyrapone to CYP3A4, it has been shown that lone electron pair of the nitrogen atom of the pyrimidine ring forms a coordinate bond with the heme iron in CYP3A4 (Park et al., 2005). Both ITCZ and CMD have a negative charge moiety (imidazole and thiazole, respectively) that can form a coordinate bond with the CYP3A4 iron, and the differences in hydrogen bonding strength may affect the affinity of inhibitors to CYP3A4.
Similar observations regarding differences in potency of inhibitors among genetic variants have been reported in CYP2C9. Kumar et al. (2006) have reported that the effect of an inhibitor varies among CYP2C9 variants and that the pattern of the influence of genetic variation is also distinctive for each inhibitor. A study with a single inhibitor is not sufficient to understand the effects of genetic variation on the potency of P450 inhibitors.
We estimated the clinical impact of CYP3A4 genetic variations on the DDI-induced increase in AUC by using the Ki values of inhibitors (Table 1). The ratio of the area under the curve (AUC) in the presence of an inhibitor (AUC′) to that without inhibitor (AUC), a measure of DDI, can be estimated using the following equation (Galetin et al., 2005): AUC′/AUC = 1 + [I]/Ki, where [I] represents the concentration of inhibitor. If it is assumed that the concentration of each inhibitor ([I]) is 4-fold of the respective Ki values in CYP3A4.1 (304 nM for ITCZ and 1480 μM for CMD), the AUC of a substrate is increased 5-fold by the inhibitor in a CYP3A4.1 homozygote (AUC ratio = 5.0). With the equivalent concentration of inhibitor, the AUC ratio by ITCZ is predicted to be 8.4 for a CYP3A4.16 homozygote (increased DDI), whereas it is predicted to be only 2.7 in a CYP3A4.7 homozygote. For CMD (with the same assumption that AUC in CYP3A4.1 is increased 5-fold by CMD), the AUC ratio is predicted to be 7.3 for CYP3A4.7, 2.2 for CYP3A4.16, and 12 for CYP3A4.18, suggesting that genetic variation leads to interindividual variations in DDI.
In conclusion, the potencies of typical inhibitors differed among wild-type CYP3A4 and CYP3A4 variants. The inhibitory potencies of ITCZ and CMD varied 4- and 8-fold, respectively, among these variants. The pattern for genetic variations also differed between ITCZ and CMD. These differences are hypothesized to result from the structural changes of CYP3A4 in each variant, which may affect the accessibility and affinity of an inhibitor to the catalytic site of CYP3A4. The genetic variation of CYP3A4 may possibly cause interindividual variations of DDI mediated by CYP3A4 inhibition.
This work was supported in part by the National Institutes of Health National Cancer Institute [Grant R37-CA090426] (to F.P.G.); the National Institutes of Health National Institute of Environmental Health Sciences [Grant P30-ES000267] (to F.P.G.); and a grant-in-aid for Scientific Research from the Open Research Center Project of Keio University [Grant 070010].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.036780.
ABBREVIATIONS:
- TST
- testosterone
- AUC
- area under the curve
- CMD
- cimetidine
- DDI
- drug-drug interaction
- HPLC
- high-performance liquid chromatography
- ITCZ
- itraconazole
- 6β-OHT
- 6β-hydroxytestosterone
- P450
- cytochrome P450.
Authorship Contributions
Participated in research design: Akiyoshi, Saito, Murayama, Yamazaki, Nakamura, Yamamoto, and Ohtani.
Conducted experiments: Akiyoshi, Saito, Murase, Miyazaki, Nakamura, Yamamoto, and Ohtani.
Contributed new reagents or analytic tools: Guengerich.
Performed data analysis: Murayama and Yamazaki.
Wrote or contributed to the writing of the manuscript: Akiyoshi, Saito, Murayama, Yamazaki, Guengerich, and Ohtani.
References
- Austin RP, Barton P, Cockroft SL, Wenlock MC, Riley RJ. (2002) The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its prediction from physicochemical properties. Drug Metab Dispos 30:1497–1503 [DOI] [PubMed] [Google Scholar]
- Austin RP, Barton P, Mohmed S, Riley RJ. (2005) The binding of drugs to hepatocytes and its relationship to physicochemical properties. Drug Metab Dispos 33:419–425 [DOI] [PubMed] [Google Scholar]
- Busby WF, Jr, Ackermann JM, Crespi CL. (1999) Effect of methanol, ethanol, dimethyl sulfoxide, and acetonitrile on in vitro activities of cDNA-expressed human cytochromes P-450. Drug Metab Dispos 27:246–249 [PubMed] [Google Scholar]
- Chauret N, Gauthier A, Nicoll-Griffith DA. (1998) Effect of common organic solvents on in vitro cytochrome P450-mediated metabolic activities in human liver microsomes. Drug Metab Dispos 26:1–4 [PubMed] [Google Scholar]
- Easterbrook J, Lu C, Sakai Y, Li AP. (2001) Effects of organic solvents on the activities of cytochrome P450 isoforms, UDP-dependent glucuronyl transferase, and phenol sulfotransferase in human hepatocytes. Drug Metab Dispos 29:141–144 [PubMed] [Google Scholar]
- Eiselt R, Domanski TL, Zibat A, Mueller R, Presecan-Siedel E, Hustert E, Zanger UM, Brockmoller J, Klenk HP, Meyer UA, et al. (2001) Identification and functional characterization of eight CYP3A4 protein variants. Pharmacogenetics 11:447–458 [DOI] [PubMed] [Google Scholar]
- Fukushima-Uesaka H, Saito Y, Watanabe H, Shiseki K, Saeki M, Nakamura T, Kurose K, Sai K, Komamura K, Ueno K, et al. (2004) Haplotypes of CYP3A4 and their close linkage with CYP3A5 haplotypes in a Japanese population. Hum Mutat 23:100. [DOI] [PubMed] [Google Scholar]
- Galetin A, Ito K, Hallifax D, Houston JB. (2005) CYP3A4 substrate selection and substitution in the prediction of potential drug-drug interactions. J Pharmacol Exp Ther 314:180–190 [DOI] [PubMed] [Google Scholar]
- Guengerich FP. (1999) Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol 39:1–17 [DOI] [PubMed] [Google Scholar]
- Guengerich FP. (2008) Cytochrome P450 and chemical toxicology. Chem Res Toxicol 21:70–83 [DOI] [PubMed] [Google Scholar]
- Ishigam M, Uchiyama M, Kondo T, Iwabuchi H, Inoue S, Takasaki W, Ikeda T, Komai T, Ito K, Sugiyama Y. (2001) Inhibition of in vitro metabolism of simvastatin by itraconazole in humans and prediction of in vivo drug-drug interactions. Pharm Res 18:622–631 [DOI] [PubMed] [Google Scholar]
- Isoherranen N, Kunze KL, Allen KE, Nelson WL, Thummel KE. (2004) Role of itraconazole metabolites in CYP3A4 inhibition. Drug Metab Dispos 32:1121–1131 [DOI] [PubMed] [Google Scholar]
- Kang YS, Park SY, Yim CH, Kwak HS, Gajendrarao P, Krishnamoorthy N, Yun SC, Lee KW, Han KO. (2009) The CYP3A4*18 genotype in the cytochrome P450 3A4 gene, a rapid metabolizer of sex steroids, is associated with low bone mineral density. Clin Pharmacol Ther 85:312–318 [DOI] [PubMed] [Google Scholar]
- Kriegl JM, Arnhold T, Beck B, Fox T. (2005) A support vector machine approach to classify human cytochrome P450 3A4 inhibitors. J Comput Aided Mol Des 19:189–201 [DOI] [PubMed] [Google Scholar]
- Kumar V, Wahlstrom JL, Rock DA, Warren CJ, Gorman LA, Tracy TS. (2006) CYP2C9 inhibition: impact of probe selection and pharmacogenetics on in vitro inhibition profiles. Drug Metab Dispos 34:1966–1975 [DOI] [PubMed] [Google Scholar]
- Lamba JK, Lin YS, Thummel K, Daly A, Watkins PB, Strom S, Zhang J, Schuetz EG. (2002) Common allelic variants of cytochrome P4503A4 and their prevalence in different populations. Pharmacogenetics 12:121–132 [DOI] [PubMed] [Google Scholar]
- Miyazaki M, Nakamura K, Fujita Y, Guengerich FP, Horiuchi R, Yamamoto K. (2008) Defective activity of recombinant cytochromes P450 3A4.2 and 3A4.16 in oxidation of midazolam, nifedipine, and testosterone. Drug Metab Dispos 36:2287–2291 [DOI] [PubMed] [Google Scholar]
- Murayama N, Nakamura T, Saeki M, Soyama A, Saito Y, Sai K, Ishida S, Nakajima O, Itoda M, Ohno Y, et al. (2002) CYP3A4 gene polymorphisms influence testosterone 6β-hydroxylation. Drug Metab Pharmacokinet 17:150–156 [DOI] [PubMed] [Google Scholar]
- Okada Y, Murayama N, Yanagida C, Shimizu M, Guengerich FP, Yamazaki H. (2009) Drug interactions of thalidomide with midazolam and cyclosporine A: heterotropic cooperativity of human cytochrome P450 3A5. Drug Metab Dispos 37:18–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Lee S, Suh J. (2005) Structural and dynamical basis of broad substrate specificity, catalytic mechanism, and inhibition of cytochrome P450 3A4. J Am Chem Soc 127:13634–13642 [DOI] [PubMed] [Google Scholar]
- Sata F, Sapone A, Elizondo G, Stocker P, Miller VP, Zheng W, Raunio H, Crespi CL, Gonzalez FJ. (2000) CYP3A4 allelic variants with amino acid substitutions in exons 7 and 12: evidence for an allelic variant with altered catalytic activity. Clin Pharmacol Ther 67:48–56 [DOI] [PubMed] [Google Scholar]
- Shimada T, Tanaka K, Takenaka S, Murayama N, Martin MV, Foroozesh MK, Yamazaki H, Guengerich FP, Komori M. (2010) Structure-function relationships of inhibition of human cytochromes P450 1A1, 1A2, 1B1, 2C9, and 3A4 by 33 flavonoid derivatives. Chem Res Toxicol 23:1921–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. (1994) Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 270:414–423 [PubMed] [Google Scholar]
- Teixeira VH, Ribeiro V, Martel PJ. (2010) Analysis of binding modes of ligands to multiple conformations of CYP3A4. Biochim Biophys Acta 1804:2036–2045 [DOI] [PubMed] [Google Scholar]
- Unwalla RJ, Cross JB, Salaniwal S, Shilling AD, Leung L, Kao J, Humblet C. (2010) Using a homology model of cytochrome P450 2D6 to predict substrate site of metabolism. J Comput Aided Mol Des 24:237–256 [DOI] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. (2004) Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 305:683–686 [DOI] [PubMed] [Google Scholar]
- Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. (2004) The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J Biol Chem 279:38091–38094 [DOI] [PubMed] [Google Scholar]
- Zhou SF, Xue CC, Yu XQ, Li C, Wang G. (2007) Clinically important drug interactions potentially involving mechanism-based inhibition of cytochrome P450 3A4 and the role of therapeutic drug monitoring. Ther Drug Monit 29:687–710 [DOI] [PubMed] [Google Scholar]