

Abstract
Aim
Pretreatment with mineralocorticoid receptor (MR) antagonists is reported to reduce myocardial infarct size from ischemia/reperfusion. Here, we tested whether the MR antagonists potassium canrenoate and eplerenone could protect in the more clinically relevant schedule of administration at the end of ischemia.
Methods and results
In all models, hearts were subjected to 30 min regional ischemia followed by 120 min (rabbits 4 h) reperfusion. A bolus of canrenoate 5 min prior to reperfusion in open-chest mice decreased infarct size in a dose-dependent manner. Maximum protection was seen at 1 mg/kg where infarction was 18% of that in the control (p<0.001). Ecto-5′-nucleotidase (CD73), as well as adenosine A2b receptor knock-out mice could no longer be protected, suggesting a role for adenosine and the A2b receptor in the mechanism. A 1 mg/kg bolus of canrenoate prior reperfusion also reduced infarct size in open-chest rabbits. To explore the underlying mechanisms we studied isolated rat hearts. Eplerenone (10 μM) at the end of ischemia was similarly protective in the rat heart and the protection was abolished by co-treatment with inhibitors of the adenosine receptor, protein kinase C, PI3-kinase, and ERK. In addition eplerenone or canrenoate treatment increased phosphorylation of the pro-survival kinases Akt and ERK1/2 at reperfusion in the rat hearts.
Conclusion
Taken together, MR antagonists when given at the end of ischemia are highly effective and potent cardioprotective drugs with a signaling similar to that of ischemic preconditioning and, hence, could be a very promising candidate for the treatment of acute myocardial infarction in man.
Keywords: mineralocorticoid receptor antagonist, potassium canrenoate, eplerenone, cardioprotection, reperfusion
INTRODUCTION
Aldosterone causes sodium and water retention by a genomic action of the hormone on the kidney. These effects involve the translocation of the steroid-MR complex to the nucleus, where it acts as a transcriptional regulator. Several hours later the newly expressed proteins induce their effect. More recently, rapid, non-genomic effects of aldosterone occurring within minutes have also been described.1,2 Interestingly MR blockade also exerts rapid effects on calcium metabolism and the sodium hydrogen exchanger.3 Chai et al. reported that MR blockade prior to ischemia in the isolated rat heart reduced ischemic injury,4 suggesting that the MR antagonists might somehow cause pharmacological preconditioning.5 Because preconditioning is of little clinical value there has been an extensive search for a safe drug that reduces infarct size when given at reperfusion.6 Here, we test two selective MR blockers, potassium canrenoate, the i.v. compatible metabolite of spironolactone, and eplerenone, in several different animal models to see if either might also be protective at the end of ischemia. Finally, we test whether the protection from MR blockade might use any of the signaling pathways that are used by ischemic pre- and postconditioning.7 We tested the MR antagonists in three different species and a powerful anti-infarct effect was seen in all.
METHODS
The experiments in rats and mice were conducted in Greifswald, Germany, while rabbits were used in Maisons-Alfort, France. All experiments were conducted in accordance with The Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington, DC, 1996). The experimental protocols used in this study were either approved by the local authorities of the state of Mecklenburg-Vorpommern, Germany (rat and mouse), or according to French official regulations (rabbit).
Open-chest in situ mouse heart
We used the open-chest in situ mouse heart model described by Eckle et al.8 Briefly, mice were anesthetized with pentobarbital sodium (70 mg/kg i.p.) and additional anesthesia was administered as needed throughout the experiment. Animals were ventilated with room air supplemented with oxygen (peak inspiratory pressure of 10 mbar, positive end-expiratory pressure of 3 mbar). The ventilation frequency was set at 110 breaths/min and a tidal volume of 200–250 μl. To administer drugs a butterfly needle was placed in the tail vein. After a left thoracotomy a prominent branch of the left coronary artery was surrounded with a 7-0 nylon suture to form a snare. The mice were allowed to stabilize for 15min after surgery before the protocols were begun. In all cases the coronary branch was occluded for 30 min and reperfused for 2 h.
Experimental protocol
Six groups were studied in control wild-type CD1 mice (Charles River, Kisslegg, Germany). Control mice had only the index occlusion followed by reperfusion. In drug-treated mice with potassium canrenoate was started i.v. 5 min before the onset of reperfusion. Canrenoate was given in different concentrations as a bolus. Control animals received the corresponding amount of saline. Two additional treatments (vehicle and 1 mg/kg BW canrenoate) were performed in CD73 knock-out and adenosine A2b receptor knock-out mice.9
Measurement of risk zone and infarct size
After completion of the protocol the coronary artery was reoccluded, and Evans blue was injected retrogradely through the aortic root to demarcate the ischemic zone (region at risk zone). Hearts were excised, perfused with 0.9% saline, weighed, frozen, and then cut into 1-mm-thick transverse slices. The slices were incubated in 1% triphenyltetrazolium chloride (TTC) in sodium phosphate buffer (pH 7.4) at 38°C for 20 min. TTC stains the non-infarcted myocardium brick-red indicating the presence of dehydrogenase enzymes. The slices were then immersed in 10% formalin to enhance the contrast between stained (viable) and unstained (necrotic) tissue. The areas of infarct and risk zone were determined by planimetry of each slice and volumes were calculated by multiplying each area by the slice thickness and summing the areas for each heart. Infarct size was expressed as a percentage of the risk zone.
Cardiac enzyme measurement
After removing the heart blood was collected from the abdominal aorta and centrifuged for measurement of cardiac troponin I (cTnI) in serum using a CTNI reagent kit and a Dimension Vista 1500, Integrated Analytics System (Siemens Healthcare Diagnostics, Deerfield IL).
Open chest in situ rabbit heart
Male New Zealand White rabbits (2.7–3.3 kg) were anesthetized with zolazepam and tiletamine (20–30 mg/kg i.v. each). Animals were ventilated with 100% oxygen. Anesthesia was thereafter maintained by i.v. pentobarbital as need to maintain a surgical plane. Arterial pressure was measured in a catheter in a marginal ear artery. An electrocardiogram was also recorded. A left thoracotomy was performed at the fourth intercostal space and a 3/0 Prolene suture was passed beneath an anterior branch of the left coronary artery to form a snare. Ischemia was confirmed by the occurrence of ST segment deviation in the electrocardiogram. Reperfusion was induced by releasing the snare. The chest was closed during reperfusion to prevent cardiac cooling.
Experimental protocol
All rabbits were subjected to a 30 min coronary artery occlusion. Five minutes before reperfusion, they randomly received an i.v. bolus administration of saline (control) or potassium canrenoate (1 mg/kg). In a first set of experiments, rabbits were euthanized after 4 h of reperfusion for infarct size assessment. In a second set of experiments, the open-chest surgery was done in sterile conditions and rabbits underwent 72 h reperfusion. During those 72 h, rabbits returned to animal room and received buprenorphine for analgesia (0.02 mg/kg/12 h sc).
Measurement of risk zone and infarct size
After completion of the 4 or 72 h of reperfusion, risk zone and infarct size were measured according to the above mouse protocol.
Isolated rat heart
Wistar rats were anesthetized with pentobarbital sodium (60 mg/kg ip) after which the heart was excised and perfused on a Langendorff apparatus with Krebs-Henseleit bicarbonate buffer containing (mM) 118.5 NaCl, 24.8 NaHCO3, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 2.5 CaCl2, and 10 glucose, and bubbled with 95% O2/5% CO2 to a pH of 7.35 – 7.45 at 38°C. A snare as above was passed around an epicardial coronary arterial branch.
Experimental protocol
Fourteen experimental groups were studied as in Fig. 1. The coronary branch was occluded for 30 min and reperfused for 2 h in all groups. In the control group no other treatment was given. In the eplerenone-treated groups 10 μM eplerenone was added to the perfusate starting 5 min prior to reperfusion. In the following inhibitor groups one of four inhibitors was co-infused with eplerenone including the adenosine receptor blocker 8-p-sulphophenyltheophylline (SPT, 100 μM), the PKC inhibitor chelerythrine (CHEL, 2.8 μM), the ERK inhibitor U0126 (500 nM), or a PI3-kinase inhibitor wortmannin (100 nM). Each inhibitor was tested alone to exclude independent effects of the blockers. In two additional groups, together with eplerenone, aldosterone was given in two different concentrations in order to compete with receptor binding. Aldosterone (500 nM) was finally given alone to exclude any direct effect. Finally, canrenoate was given (50 μM) to confirm its protective properties also in the isolated rat heart model. We delineated the risk zone with 2–9 μm green fluorescent microspheres and infarction with TTC staining as described above for in situ mouse hearts.
Fig. 1.
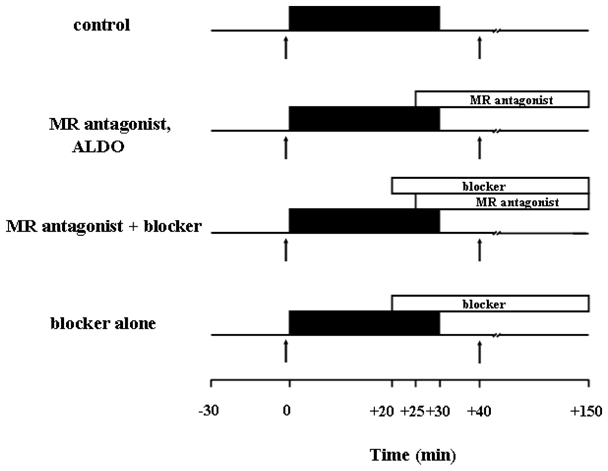
Experimental protocols of the isolated rat heart experiments. All hearts were stabilized for 30 min prior to experiments. Control hearts received 30 min of regional ischemia followed by 2 h of reperfusion. Mineralocorticoid receptor (MR) antagonists were given throughout reperfusion starting 5 min before reperfusion. The blockers were given either alone accordingly or 5 min before the MR antagonist treatment, while aldosterone (ALDO) was given together with the MR antagonist. The arrows indicate the times of the biopsies of the left ventricle. ■ = regional ischemia.
Biochemical studies
Isolated rat hearts were perfused as described above. Transmural biopsies of the left ventricle were obtained right before ischemia (baseline) and at 10 min of reperfusion following a 30 min period of global ischemia as indicated by the arrows in Fig.1. Seven groups were studied: control hearts without any further treatment, treatment with potassium canrenoate (50 μM) alone and in the presence of aldosterone, treatment with eplerenone (10 μM) alone and in the presence of either aldosterone or the PI3-kinase blocker wortmannin, and aldosterone alone. Finally, the level of phosphorylated Akt and phosphorylated ERK1/2 in the heart at reperfusion was determined by western blotting as described previously.10 Level of phosphorylation was measured as fold of total Akt and ERK1/2 resp., and normalized to the baseline sample (n = 6/group).
Materials
Eplerenone was provided by Pfizer (Karlsruhe, Germany). Polyclonal antibodies against Akt and its phosphorylated form (Ser473), ERK (p44/42 MAPK) and its phosphorylated form (Thr202/Tyr204), HRP-linked anti-rabbit IgG antibody used on Western blot analysis, and cell lysis buffer were purchased from Cell Signaling Technology (Beverly, MA), CHEL and U0126 were from Calbiochem (San Diego, CA), while all other chemicals were from Sigma-Aldrich Chemical Co. Eplerenone, wortmannin, and U0126 were dissolved in dimethyl sulfoxide (DMSO) before being diluted in Krebs-Henseleit buffer resulting in a DMSO concentration less than 0.01%. All other inhibitors were dissolved in the buffer directly.
Statistics
Data are presented as means ± SD. Differences in infarct size, troponin I levels, Akt and ERK1/2 phosphorylation amongst groups were compared by one-way ANOVA with Fisher LSD post hoc testing using SigmaStat 3.0 software. For the rabbit experiments, heart rate and mean blood pressure were compared between groups using a two-way ANOVA for repeated measures. A value of p<0.05 was considered significant.
RESULTS
Infarct size measurements in open-chest mice
We measured infarct size after 30 min regional ischemia and 2 h of reperfusion. Figure 2A reveals that treatment with a bolus of potassium canrenoate (CAN) caused a dose dependent reduction in infarct size with a peak effect at 1 mg/kg body weight (7.3±4.7% compared to 37.8±7.5% in control, p<0.001).
Fig. 2.
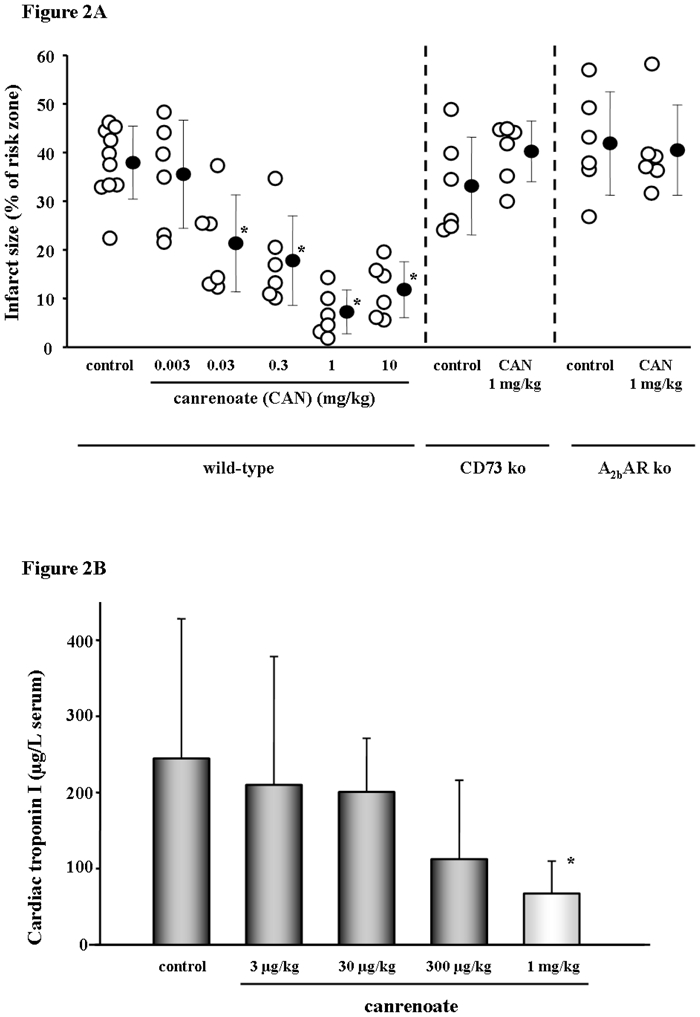
Results of the in situ open chest mouse experiments. A Potassium canrenoate (CAN) as a bolus given 5 min prior to reperfusion was protective with a concentration as low as 30 μg/kg, whereas 1 mg/kg showed very profound protection. The right panels depict experiments with the use of CAN either given to ecto-5′-nucleotidase (CD73) knock-out (ko) or A2b adenosine receptor (AR) ko mice. In both knock-out animals, CAN’s protection was blunted, suggesting a role for extracellular adenosine and the A2bAR in the protective signaling. Open symbols represent individual experiments while closed symbols are the means ± SD. *p<0.001 vs. control. B Plasma levels of cardiac troponin I (cTnI) at the end of the in situ open-chest mouse experiments. While a bolus of potassium canrenoate 1 mg/kg 5 min before reperfusion reduced cTnI release at the end of the reperfusion period, there was no significant decrease with lower canrenoate concentrations. *p=0.022 vs. control.
In order to test our hypothesis that the MR blockers protect through preconditioning-like signaling involving extracellular adenosine interacting with the A2b adenosine receptor (AR), we used cd73−/− or A2bAR−/− mice. It has previously been shown that CD73 an exonucleotidase is the source of preconditioning’s protective adenosine,11 which then presumably works through the A2bAR.9 As shown in Fig. 2A, the CD73 and A2bAR knock-out animals could no longer be protected with CAN.
The infarct size data were mirrored by the levels of cardiac troponin I (cTnI) measured in the serum at the end of reperfusion. Treatment with canrenoate significantly reduced the amount of cTnI release (single measurements expressed as means ± SD) again with a peak at 1 mg/kg (Fig. 2B).
Infarct size measurements in open-chest rabbits
We next tested 1 mg/kg canrenoate 5 min prior reperfusion in an in vivo rabbit model. Similarly, canrenoate was able to reduce infarct size in these hearts as shown in Fig. 3. Importantly, this beneficial effect was observed when reperfusion was extended up to either 4 h or 72 h. Canrenoate had no hemodynamic effect on heart rate or blood pressure (Table 1).
Fig. 3.
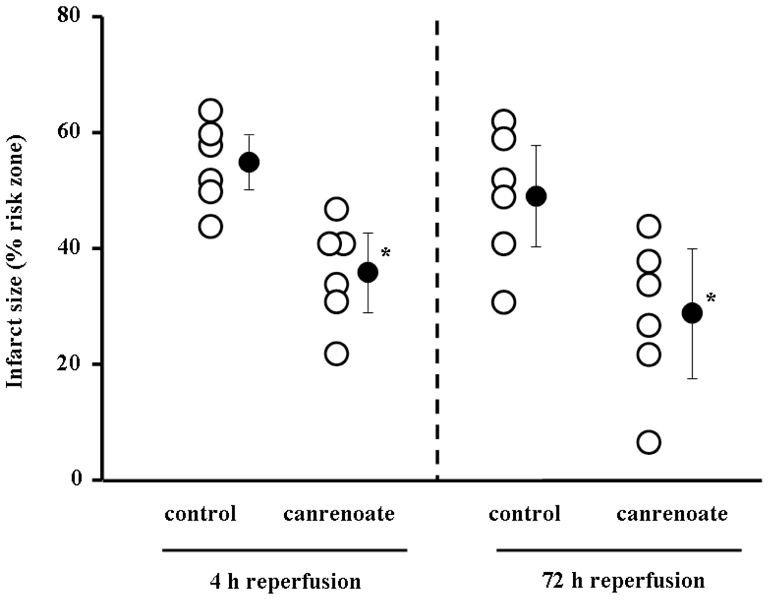
Results of the in situ rabbit heart experiments with infarct size assessment after 4 h of reperfusion (left panel) or 72 h (right panel). Canrenoate was applied as a 1 mg/kg bolus 5 min prior to reperfusion identical to the mouse experiments and clearly reduced infarct size presented at a percentage of the risk zone. *p<0.05 vs. control
Table 1.
Hemodynamic parameters
| baseline | ischemia 5 min | ischemia 15 min | ischemia 29 min | reperfusion 30 min | |
|---|---|---|---|---|---|
| Heart rate (beats/min) | |||||
| Experiments with 30 min ischemia and 4 h reperfusion | |||||
| Control | 248±40 | 243±22 | 247±22 | 239±29 | 235±16 |
| Canrenoate | 256±23 | 250±24 | 246±26 | 242±13 | 222±29 |
| Experiments with 30 min ischemia and 72 h reperfusion | |||||
| Control | 242±26 | 240±22 | 239±40 | 233±26 | 218±29 |
| Canrenoate | 248±22 | 234±29 | 234±24 | 231±24 | 222±24 |
| Mean blood pressure (mmHg) | |||||
| Experiments with 30 min ischemia and 4 h reperfusion | |||||
| Control | 70±11 | 62±13 | 62±13 | 61±13 | 62±11 |
| Canrenoate | 74±15 | 61±4 | 60±2 | 69±7 | 66±7 |
| Experiments with 30 min ischemia and 72 h reperfusion | |||||
| Control | 76±13 | 69±9 | 65±9 | 64±9 | 61±4 |
| Canrenoate | 75±13 | 69±11 | 63±11 | 63±9 | 66±7 |
Infarct size measurements in isolated rat hearts
We next performed mechanistic studies in isolated rat hearts subjected to 30 min regional ischemia. As seen in Fig. 4, eplerenone at 10 μM throughout reperfusion reduced infarct size from 40.8±5.3% in the control group to 10.9 ± 7.2% (p<0001), while only 1 μM eplerenone had no effect (data not shown).
Fig. 4.
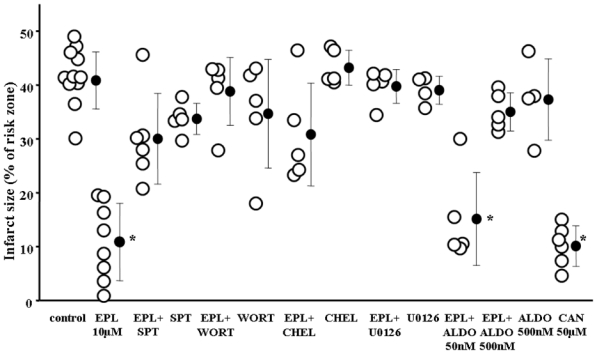
Results of the isolated rat hearts experiments. Drugs were given as depicted in Fig. 1. Eplerenone (EPL) resulted in a significant reduction in infarct size, which could be abolished with the co-treatment of pharmacological inhibitors of known protective signaling elements. [8p-sulfophenyladenosine (SPT) = adenosine receptor blocker, wortmannin (WORT) = PI3 kinase inhibitor, chelerythrine (CHEL) = PKC inhibitor, and U0126 = ERK blocker]. While a low aldosterone (ALDO) concentration did not overcome the eplerenone’s protection, a higher ALDO concentration did. All blockers and ALDO alone had no effect on infarct size. Canrenoate also showed protection in this model. *p<0.001 vs. control
Pharmacological inhibition of the adenosine receptor (SPT), PKC (CHEL), PI3-kinase (WORT), or ERK (U0126) each abolished eplerenone’s protection, indicating that protection depended on the same signaling elements as preconditioning. The inhibitors alone had no effect.
Eplerenone is a competitive inhibitor of aldosterone. To rule out any non-MR mechanism, aldosterone was given simultaneously with the eplerenone treatment to repopulate MR. Co-infusion of 50 nM aldosterone blunted eplerenone’s protection and 500 nM aldosterone abolished it. Aldosterone alone had no effect on infarct size.
Canrenoate (50 μM) similarily worked in the isolated rat heart model and reduced infarct size from 40.8±5.3% in the control group to 10.2±3.8% (p<0001), while only 10μM had no effect (data not shown).
Phosphorylation of Akt and ERK1/2
Akt and ERK1/2 activation by phosphorylation at reperfusion are central to protection by preconditioning. We found a marked increase in Akt and ERK1/2 phosphorylation following MR blockade at reperfusion (Figs. 5A, 5B, and 5C). Canrenoate and eplerenone, both increased Akt and ERK1/2 phosphorylation and that was abolished with co-treatment with aldosterone. Aldosterone alone had no effect, and the PI3-kinase inhibitor wortmannin totally blocked eplerenone-induced phosphorylation.
Fig. 5.
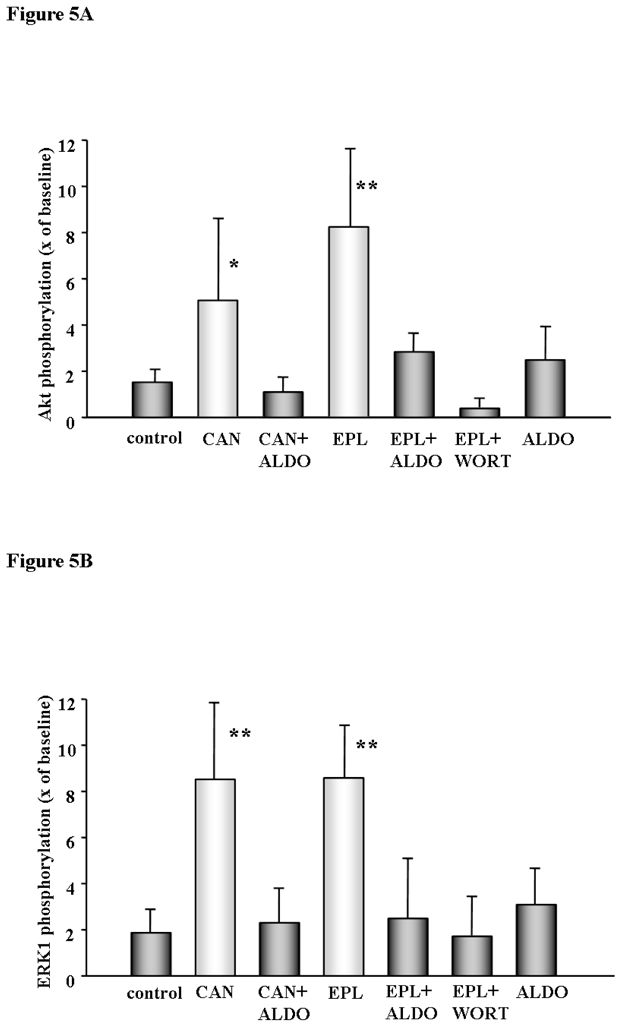
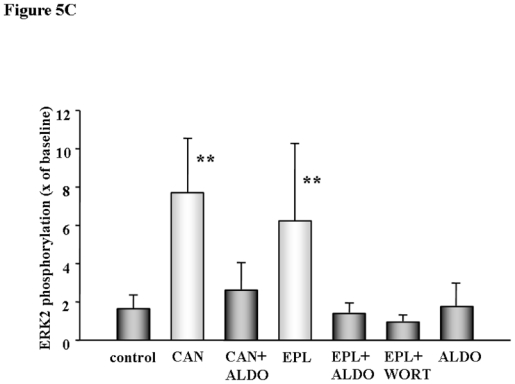
Fig. 5A, 5B, and 5C Akt and ERK1/2 phosphorylation in isolated rat heart. Myocardial samples were obtained from transmural biopsies of isolated rat hearts of the left ventricle right before ischemia (baseline) and at 10 min of reperfusion following a 30 min period of global ischemia as indicated by the arrows in Fig. 1. Phosphorylation of all tested kinases was clearly increased at reperfusion compared to untreated control when either canrenoate (CAN) or eplerenone (EPL) were present. Protection was abolished with co-infusion of aldosterone (ALDO) or wortmanin (WORT), while ALDO had no effect on its own. Results represent the mean±SD of 6 independent experiments, *p<0.01, **p<0.001 vs. control.
DISCUSSION
The mineralocorticoid receptor antagonists potassium canrenoate and eplerenone are both very cardioprotective against infarction when administered prior to reperfusion. Their very rapid action suggests a non-genomic effect of MR blockade. Further, we could show that the observed infarct-reducing properties of MR blockade involve key elements of the signaling pathway used by pre- and postconditioning. Finally, similar protection was demonstrated in three species.
Ischemia-induced infarction can be reduced either with preconditioning, an intervention prior to ischemia, or by postconditioning, an intervention at the onset of reperfusion. While Chai et al. recently reported that MR blockade was cardioprotective when applied as a preconditioning stimulus,4 we found that MR antagonists are also protective in the more clinically relevant setting of administration at the end of ischemia. Both phenomena, ischemic pre- and postconditioning share a common signaling pathway that includes PKC, adenosine receptors, PI3-kinase/Akt, and ERK.12 This signaling is thought to protect by suppressing the opening of mitochondrial permeability transition pores at reperfusion. In the present study, we showed that protection from MR blockade prior to reperfusion depends on these same signaling elements suggesting a common mechanism.
Fujita et al. demonstrated that aldosterone worsens injury from ischemia by a rapid, non-genomic effect.13 Here, we could not see any difference between the untreated control group and aldosterone-treated isolated rat hearts. In contrast to our Langendorff-perfused heart model, Fujita et al. used an in situ dog model which could explain these differences.
Most postconditioning agents have a very narrow therapeutic window, and delaying the intervention for only a few minutes after the onset of reperfusion eliminates the protection.14,15 Canrenoate-induced protection was also absent when the bolus was given within 1 min or 15 min after onset of reperfusion (data not shown), confirming a non-genomic mechanism due to the rapidity of the protection with MR blockers.
In large-scale clinical trials (RALES and EPHESUS resp.)16,17 treatment of post-myocardial infarction patients with MR antagonists reduced mortality most likely via the prevention of remodeling. In these trials, MR antagonists were given long after the onset of reperfusion, and, therefore, it is not likely that the observed benefits were due to infarct size reduction. Since mortality after AMI is directly correlated with infarct size, the anti-infarct effect of immediate MR blockade shown in the present study could have additional beneficial effects in patients with AMI.
The protection could either be due to the inhibition of detrimental effects of aldosterone itself, to a unique receptor response to antagonist binding, or an MR-independent nonspecific effect. Since we could overcome the cardioprotective effect of the competitive MR inhibitors eplerenone and canrenoate with a high concentration of aldosterone, it is most likely that elimination of aldosterone’s effect at the MR was involved. Nevertheless, any additional nonspecific drug effects of canrenoate or eplerenone cannot be fully excluded. Since ischemia itself leads to a release of aldosterone by the heart,18 we can speculate that occupying MR may inhibit the cell’s natural protection against ischemia. If the inhibition were suppressed with MR blockade or overcome by pre- or postconditioning, the endogenous protective signaling could then promote cell survival.
The powerful cardioprotection of pre- and postconditioning must be made available to patients with ST-segment elevation AMI (STEMI). Several forms of postconditioning, either repetitive short ischemic episodes at reperfusion19 or pharmacological preconditioning with either cyclosporine A20 or atrial natriuretic peptide,21 have been tested in STEMI patients and the results were encouraging. Unfortunately, none of these interventions is ideally suited for routine clinical use. Our demonstration of good protection prior to reperfusion across species lines encourages us that potassium canrenoate is a good candidate for a clinical trial in patients with STEMI. Canrenoate is routinely used clinically as a diuretic with an initial dose of 200 mg. Canrenoate caused a profound infarct size reduction in animal models of AMI in mice and rabbits at a much lower, single bolus, dose of ~1 mg/kg.
Acknowledgments
The CD73 knock-out mice were kindly provided by Dr. Thompson, Oklahoma Medical Research Foundation, Oklahoma City, OK, and the A2bAR knock-out mice by Dr. Eltzschig, Department of Anesthesiology, University of Colorado, Denver CO. The study was supported by funds from Pfizer Pharma GmbH, Karlsruhe, Germany
The authors thank Dr. Downey for the critical review of the manuscript.
References
- 1.Alzamora R, Marusic ET, Gonzalez M, Michea L. Nongenomic effect of aldosterone on Na+,K+-adenosine triphosphatase in arterial vessels. Endocrinology. 2003;144:1266–1272. doi: 10.1210/en.2002-220950. [DOI] [PubMed] [Google Scholar]
- 2.Lösel RM, Feuring M, Falkenstein E, Wehling M. Nongenomic effects of aldosterone: cellular aspects and clinical implications. Steroids. 2002;67:493–498. doi: 10.1016/s0039-128x(01)00176-3. [DOI] [PubMed] [Google Scholar]
- 3.Michea L, Delpiano AM, Hitschfeld C, Lobos L, Lavandero S, Marusic ET. Eplerenone blocks nongenomic effects of aldosterone on the Na+/H+ exchanger, intracellular Ca2+ levels, and vasoconstriction in mesenteric resistance vessels. Endocrinology. 2005;146:973–980. doi: 10.1210/en.2004-1130. [DOI] [PubMed] [Google Scholar]
- 4.Chai W, Garrelds IM, de Vries R, Danser AH. Cardioprotective effects of eplerenone in the rat heart: interaction with locally synthesized or blood-derived aldosterone? Hypertension. 2006;47:665–670. doi: 10.1161/01.HYP.0000205831.39339.a5. [DOI] [PubMed] [Google Scholar]
- 5.Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1113–1151. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- 6.Hausenloy DJ, Yellon DM. Clinical translation of cardioprotective strategies : Report and recommendations of the Hatter Institute 5th International Workshop on Cardioprotection. Basic Res Cardiol. 2008;103:493–500. doi: 10.1007/s00395-008-0736-x. [DOI] [PubMed] [Google Scholar]
- 7.Davidson SM, Hausenloy D, Duchen MR, Yellon DM. Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int J Biochem Cell Biol. 2006;38:414–419. doi: 10.1016/j.biocel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 8.Eckle T, Grenz A, Köhler D, Redel A, Falk M, Rolauffs B, Osswald H, Kehl F, Eltzschig HK. Systematic evaluation of a novel model for cardiac ischemic preconditioning in mice. Am J Physiol Heart Circ Physiol. 2006;291:H2533–H2540. doi: 10.1152/ajpheart.00472.2006. [DOI] [PubMed] [Google Scholar]
- 9.Eckle T, Krahn T, Grenz A, Köhler D, Mittelbronn M, Ledent C, Jacobson MA, Osswald H, Thompson LF, Unertl K, Eltzschig HK. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- 10.Krieg T, Qin Q, McIntosh EC, Cohen MV, Downey JM. ACh and adenosine activate PI3-kinase in rabbit hearts through transactivation of receptor tyrosine kinases. Am J Physiol Heart Circ Physiol. 2002;283:H2322–H2330. doi: 10.1152/ajpheart.00474.2002. [DOI] [PubMed] [Google Scholar]
- 11.Kitakaze M, Minamino T, Node K, Komamura K, Hori M. Activation of ecto-5′-nucleotidase and cardioprotection by ischemic preconditioning. Basic Res Cardiol. 1996;91:23–26. doi: 10.1007/BF00788856. [DOI] [PubMed] [Google Scholar]
- 12.Downey JM, Krieg T, Cohen MV. Mapping preconditioning’s signaling pathways: an engineering approach. Ann N Y Acad Sci. 2008;1123:187–196. doi: 10.1196/annals.1420.022. [DOI] [PubMed] [Google Scholar]
- 13.Fujita M, Minamino T, Asanuma H, Sanada S, Hirata A, Wakeno M, Myoishi M, Okuda H, Ogai A, Okada K, Tsukamoto O, Koyama H, Hori M, Kitakaze M. Aldosterone nongenomically worsens ischemia via protein kinase C-dependent pathways in hypoperfused canine hearts. Hypertension. 2005;46:113–117. doi: 10.1161/01.HYP.0000171184.84077.80. [DOI] [PubMed] [Google Scholar]
- 14.Yang X-M, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV. Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol. 2004;44:1103–1110. doi: 10.1016/j.jacc.2004.05.060. [DOI] [PubMed] [Google Scholar]
- 15.Kin H, Zhao Z-Q, Sun H-Y, Wang N-P, Corvera JS, Halkos ME, Kerendi F, Guyton RA, Vinten-Johansen J. Postconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res. 2004;62:74–85. doi: 10.1016/j.cardiores.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 17.Pitt B, White H, Nicolau J, Martinez F, Gheorghiade M, Aschermann M, van Veldhuisen DJ, Zannad F, Krum H, Mukherjee R, Vincent J. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol. 2005;46:425–431. doi: 10.1016/j.jacc.2005.04.038. [DOI] [PubMed] [Google Scholar]
- 18.Silvestre J-S, Heymes C, Oubénaïssa A, Robert V, Aupetit-Faisant B, Carayon A, Swynghedauw B, Delcayre C. Activation of cardiac aldosterone production in rat myocardial infarction: effect of angiotensin II receptor blockade and role in cardiac fibrosis. Circulation. 1999;99:2694–2701. doi: 10.1161/01.cir.99.20.2694. [DOI] [PubMed] [Google Scholar]
- 19.Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L’Huillier I, Aupetit JF, Bonnefoy E, Finet G, André-Fouët X, Ovize M. Postconditioning the human heart. Circulation. 2005;112:2143–2148. doi: 10.1161/CIRCULATIONAHA.105.558122. [DOI] [PubMed] [Google Scholar]
- 20.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, André-Fouët X, Revel D, Kirkorian G, Monassier JP, Derumeaux G, Ovize M. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 21.Kitakaze M, Asakura M, Kim J, Shintani Y, Asanuma H, Hamasaki T, Seguchi O, Myoishi M, Minamino T, Ohara T, Nagai Y, Nanto S, Watanabe K, Fukuzawa S, Hirayama A, Nakamura N, Kimura K, Fujii K, Ishihara M, Saito Y, Tomoike H, Kitamura S. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet. 2007;370:1483–1493. doi: 10.1016/S0140-6736(07)61634-1. [DOI] [PubMed] [Google Scholar]