

Abstract
Currently, some of the most effective treatments for heart failure target GPCRs such as the beta-adrenergic receptors (β1AR- β2AR) and angiotensin II type IA receptors (ATIaR). Ligands for these receptors not only function by blocking the deleterious G protein mediated pathway leading to heart failure, but also signal via G-protein independent pathways that involve receptor phosphorylation by G-protein receptor kinases (GRKs) leading to recruitment of the multifunctional protein, β-arrestin. Originally thought to play a role in GPCR desensitization and internalization, β-arrestin has recently been shown to mediate signaling independent of classical second messengers in a way that is often protective to the heart. The multi-functionality of β-arrestin makes it an intriguing molecule in the development of the next generation of drugs for cardiac diseases with the potential to simultaneously inhibit deleterious G-protein dependent pathways while activating beneficial β-arrestin-mediated signaling. In this review, we explore various facets of β-arrestin signaling and offer a perspective on its potential role as a key signaling molecule in the treatment of heart failure.
Keywords: β-arrestin, β-adrenergic receptor, Angiotensin II type 1a receptor, G protein coupled receptor kinase, extracellular-signal receptor kinase, epidermal growth factor receptor transactivation, signaling
Introduction
Heart failure is a major and growing public health problem affecting 1-6% of the US population [1]. Currently, some of the most effective treatments for heart failure target the beta-adrenergic receptors (β1AR- β2AR) and angiotensin II type IA receptors (ATIaR), which are both G-protein coupled receptors (GPCRs). GPCRs are seven transmembrane receptors that constitute the largest and most ubiquitous class of plasma membrane receptors that regulate a multitude of physiologic processes. Upon stimulation by circulating catecholamines and angiotensin II which are in excess in human heart failure [2], βARs and AT1aRs activate an associated G-protein (Gs for βARs and Gq for AT1aRs). This activation involves the exchange of bound GDP for GTP by the Gα subunit of the G protein, leading to dissociation of the heterotrimeric protein complex into Gα and Gβγ subunits and stimulation of downstream signaling via generation of second messengers [3]. Chronic Gs and Gq-protein mediated signaling is known to be deleterious by leading to increased myocyte size, apoptosis and contractile failure [4-6]. Indeed, antagonists for these receptors have positively impacted the morbidity and mortality of patients with heart failure. However, recent studies have shown that these antagonists not only function by blocking the deleterious G-protein mediated pathways that can lead to heart failure, but also signal via a G-protein independent pathway that appears to frequently use the multifunctional protein, β-arrestin [3, 7].
β-arrestins were originally identified through their ability to inhibit further G protein signaling through blockade of the GPCR-G protein interaction in a process called “desensitization” [3, 8]. However, investigations over the last few years have revealed diverse roles for the β-arrestins [3, 9]. In addition to being key regulators of GPCR endocytosis and trafficking[10], β-arrestins function as scaffold proteins linking GPCR activation to several downstream effectors such as the MAPK cascade (ERK [extracellular signal-regulated kinase] and JNK), Src, and the ubiquitin ligase Mdm2 [3, 9]. Furthermore, β-arrestins are now known to bind to calmodulin [11] and to CaMKIIδ [12, 13] which plays a key role in the progression of heart failure [14]. In this review we will explore the various facets of β-arrestin signaling in normal and disease conditions and its potential as a therapeutic target in human heart failure.
β-arrestin involvement in receptor desensitization
In vertebrates, members of the arrestin family (arrestin 1-4) were discovered by their ability to bind and inactivate signaling of rhodopsin [15] and the β2AR [8]. Expression of arrestins 1 and 4 are exclusive to the rods and cones of the eyes and are known as visual arrestins [3]. Arrestins 2 and 3, also known as β-arrestin 1 and 2 respectively, are ubiquitously expressed throughout mammalian tissues. Classic G-protein signaling occurs after ligand stimulation of GPCR and leads to generation of second messengers such as cyclic adenosine monophosphate (cAMP), diacylglycerol (DAG) and inositiol triphosphate (IP3) generated in response to Gs and Gq-coupled activation. This G protein mediated signaling can be terminated by a process known as “desensitization” whereby the receptor is uncoupled from its cognate G-protein by phosphorylation of residues within the C-terminal tail of the activated receptor by a family of kinases known as G protein-coupled receptor kinases (GRKs) [3, 16]. Phosphorylation of activated receptors by GRKs enhance their affinity for cytosolic β-arrestin, which in turn inhibits further G-protein signaling through: 1) blockade of the GPCR-G protein interaction [3], and 2) recruitment of enzymes that degrade second messenger molecules [17, 18].
β-arrestin mediated receptor trafficking: internalization, recycling and degradation
In addition to their role in desensitization, β-arrestins also play an important role in the recruitment of GPCRs into intracellular compartments, a process broadly known as receptor internalization. Internalization has been identified as a critical initial step in recycling of desensitized receptors [19] and also to activate key mitogenic pathways within the cell [20, 21]. The process of β-arrestin mediated receptor internalization occurs via interaction of the ligand bound receptor with proteins of the clathrin coated pit (CCP) machinery. β-arrestin binds to clathrin via the adaptor protein, AP2 [22] and is facilitated by the recruitment of phosphoinositide-3-kinase (PI3K) to the membrane [23] through its association with GRK2 [24-26]. On the plasma membrane PI3K generates 3, 4, 5-phosphtidylinositols (PIP3), which enhance recruitment of AP2 to the β-arrestin-clathrin receptor complex to subsequently promote receptor endocytosis [23-26].
After internalization, β-arrestin mediated GPCR trafficking is regulated by the location and variable binding affinity of different arrestin isoforms to GPCRs. Visual arrestin and β-arrestin1 are found in both the cytoplasm and the nucleus, while β-arrestin2 is localized only to the cytoplasm [27]. β-arrestins interact with GPCRs with differing affinities that have been classified according to the type of β-arrestin-GPCR association. Class A receptors, such as β1ARs, μ opioid receptors, and D1 dopamine receptors bind β-arrestin 2 with a greater affinity than β-arrestin 1 [27, 28] and their interaction is lost during internalization. Class B receptors, such as angiotensin II type 1A receptor, neurotensin receptor 1 and vasopressin V2 receptor, bind β-arrestin 1 and 2 with equal affinity and the interaction remains intact during internalization [27]. Internalized receptors are then sorted for degradation or recycling, a process known as trafficking. Receptors targeted for degradation traffic to lysosomes and are enzymatically degraded while receptors for recycling traffic to acidified vesicles where they are de-phosphorylated and recycle back to the plasma membrane [29].
β-arrestins in G-protein independent signaling
While β-arrestins play an important role in GPCR desensitization, internalization, and trafficking, recent data reveal that recruitment of β-arrestin to βARs and AT1aRs initiates a second wave of signaling independent of G-protein activation [30, 31] (Figure 1). Indeed, stimulation of the AT1aR in a β-arrestin-dependent, G protein-independent manner has revealed a phosphoproteome with over 1,500 phosphoproteins identified with connections to a diverse array of signaling pathways [32]. One widely studied signaling cascade is the mitogenic extracellular-signal receptor kinase (ERK) signaling pathway [33]. ERK signaling is classically activated by agonist binding to receptor tyrosine kinases (RTK) and many other GPCRs [34, 35] to initiate intracellular signaling promoting mitogenic and anti-apoptotic effects [33, 36]. These anti-apoptotic signals are mediated through both inhibition of caspase activity [37, 38] and enhancement of cellular proliferation by activation of proteins involved in nucleic acid synthesis [39], transcription [40], and translation [41].
Figure 1. GPCR signaling.
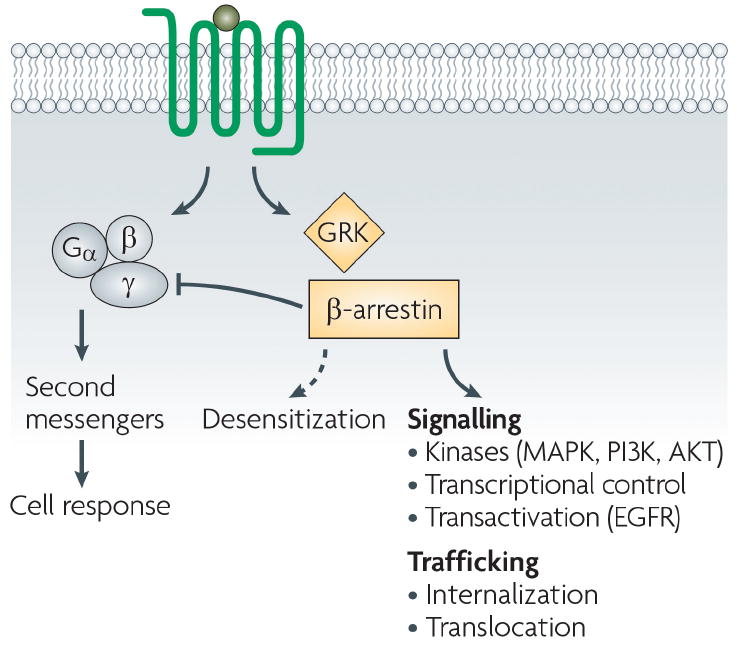
In response to ligand stimulation, GPCR signaling can occur via both G-protein mediated pathway (which leads to activation of second messengers) or via β-arrestin mediated pathway which further stimulate a plethora of downstream signaling pathways (MAPK, PI3K, AKT, EGFR etc.) (Reproduced with permission from Ref [31]).
Recent studies have shown that βARs and AT1aRs can initiate ERK signaling by both G-protein and β-arrestin mediated processes although with different kinetics and molecular consequences [30, 42]. For example, in cells overexpressing the AT1aR or the β2AR, G protein activation through the ligand bound receptor leads to peak ERK activity within two to five minutes [42, 43]. In contrast, β-arrestin-mediated signaling following angiotensin II (Ang II) stimulation of AT1aR or isoproterenol stimulation of β2AR has a slower and more prolonged pattern of ERK activation [42, 43]. Moreover, β-arrestin mediated signaling by the AT1aR and β1AR promotes cytoplasmic localization of phosphorylated ERK, while G protein mediated ERK activation results in both nuclear and cytoplasmic localization of phosphorylated ERK [42, 44-46] (Figure 2). Interestingly, a recent study by Cervantes et al. showed that β-arrestin can mediate cross talk between arrestin-dependent and G-protein dependent signaling pathways to direct the subcellular distribution of phosphorylated ERK [47]. They showed that β-arrestin acts as a coordinator to integrate signals from multiple GPCRs that lead to mitogenic signaling [47]. This distinct spatiotemporal profile of ERK activation induced by Gq-coupled receptors in the presence or absence of GPCR activation may have important implications in the maladaptive remodeling of different tissues in vivo.
Figure 2. β1AR-mediated transactivation of EGFR and direct ligand stimulation of EGF R differentially target ERK.

Wild type β1AR cells expressing EGFR-GFP and ERK2-RFP were stimulated for 20 min with Dobutamine or Epidermal Growth Factor (EGF). EGFR-GFP underwent internalization in response to Dobutamine or EGF (panels 3 and 5, arrowheads), which was blocked by the EGFR inhibitor AG1478 (panels 7 and 9). Only EGF induced nuclear translocation of ERK2-RFP (panel 6, arrow), which was prevented by AG1478 (panel 10). Reproduced with permission from Ref [45].
Further investigation of β-arrestin mediated signaling by the β1AR has led to the discovery of epidermal growth factor receptor (EGFR) transactivation as a mechanism for β-arrestin mediated ERK signaling following catecholamine stimulation [48]. Transactivation of the EGFR following catecholamine stimulation of the β1AR involves Src-dependent matrix-metalloproteinase mediated release of heparin-binding epidermal growth factor (HB-EGF) to stimulate EGFR phosphorylation [45, 48]. HB-EGF acts as a ligand for the EGFR leading to the internalization of EGFR and initiation of ERK signaling [49-51]. Similarly, for the β2AR, the ligand ICI118551, which is an inverse agonist for Gs-stimulated adenylyl cyclase, can induce ERK phosphorylation that depends on β-arrestin2 expression [52]. More recently, a β-arrestin dependent mechanism for transactivation of EGFR by endogenous AT1aRs has been reported in vascular smooth muscle cells [53]. These data further support the concept of G protein independent signaling and β-arrestin independent signaling pathways and that some ligands classified as inverse agonists rely β-arrestin to activate signaling.
In addition to activation of the extracellular signal-regulated kinase (ERK) MAPK cascade, several 7TMRs have been also shown to activate the PI3K-AKT pathways in a β-arrestin-dependent manner [30]. Stimulation of 7TM protease-activated receptors with thrombin has been shown to activate AKT, a downstream target of PI3K, in a β-arrestin1-dependent manner [54]. Likewise, stimulation of the AT1R activates AKT in several cell types including vascular smooth muscle cells (VSMCs) [55, 56], although in these studies the role of β-arrestin was not tested. Ahn et al. examined the role of β-arrestin 2 in AT1R-regulated apoptosis and determined the downstream pathways mediating this regulation in rat VSMCs [57]. They showed that receptor stimulation leads to activation of two pathways, ERK/p90RSK and PI3K/AKT, which converge to phosphorylate and inactivate the pro-apoptotic protein BAD (BCL2-associated agonist of cell death) [57]. Since apoptosis of VSMCs plays a significant role in vascular remodeling in vascular diseases such as atherosclerosis and neointimal responses after injury, it is possible that β-arrestin may play an important role in these processes [58-60].
The interaction of β-arrestin with an activated receptor and subsequent signaling is regulated by a family of GRKs (1-7) [16]. GRK 1 and 7 are exclusively expressed in the retina, whereas the rest are ubiquitously expressed in mammalian tissues. GRKs 2 and 3 interact with Gβγ subunits and reside in the cytoplasm, whereas GRK 4, 5, and 6 are membrane bound [16]. Different isoforms of GRKs play important roles in the phosphorylation of the receptor to initiate signaling via the alternative β-arrestin biased pathway [61]. For example, inhibition of GRK 5 or 6 using siRNA attenuates β-arrestin mediated ERK activation via the β1AR and AT1aR while it remains unaffected (or may even increase) when siRNA is used to knockdown GRK 2 or 3 expression [48, 62, 63]. β-arrestin dependent ERK signaling is also initiated by the ligand-bound AT1aR or β2AR when GRK 5 or 6 are overexpressed in the cell [43, 63]. Interestingly, the β-arrestin component of the ERK response is reciprocally diminished by GRK 2 overexpression [43, 63]. These data implicate that receptor phosphorylation by the different GRKs together with β-arrestin binding is a critical factor in directing receptor signaling toward G-protein dependent and G-protein independent pathways.
β-arrestin signaling and cardioprotection
Physiologically, the β1AR is a Gs-coupled receptor that regulates cardiac inotropy and chronotropy via activation of cAMP-mediated protein kinase A (PKA) [2, 64]. Long-term chronic catecholamine stimulation of the β 1AR, as occurs in heart failure, promotes receptor downregulation and detrimental cardiac remodeling [65, 66]. Experimental evidence now shows that transactivation of EGFR through a β-arrestin-mediated mechanism promotes cardioprotective signaling in response to chronic catecholamine stress [48] or mechanical stress [67].
For the β1AR, the physiologic relevance of this pathway was demonstrated using transgenic mice overexpressing wild-type β1ARs or mutant β1ARs lacking GRK (β1AR GRK-) phosphorylation sites. Under conditions of chronic catecholamine stress, transgenic mice overexpressing β1AR GRK- are unable to induce β -arrestin mediated EGFR transactivation and show marked myocyte apoptosis and left ventricular dilatation compared to mice expressing wild type β1ARs or mutant β1ARs lacking PKA phosphorylation sites [48]. Consistent with an EGFR transactivation mechanism, wild-type mice chronically stimulated with catecholamines show the same deterioration in cardiac function when pre-treated with the selective EGFR inhibitor, erlotinib [48].
For the AT1aR, Rakesh et al. showed that mechanical stretch triggered a conformational change in the AT1aR leading to β-arrestin recruitment to the receptor in the absence of ligand [67]. Mechanical stretch of the receptor in a cellular system and an ex vivo murine heart model led to activation of β-arrestin mediated, antiapoptotic ERK signaling in the absence of detectable G protein activation [67]. Hearts from mice lacking β-arrestin or AT1aRs failed to induce a cardioprotective response to mechanical stretch as shown by blunted ERK and Akt activation, impaired transactivation of the EGFR, and enhanced myocyte apoptosis [67] (Figure 3). These data show that the heart responds to acute increases in mechanical stress by activating β-arrestin–mediated cell survival signals [67]. Consistent with these data transgenic mice with cardiac-specific over-expression of AT1aRs that are unable to couple with G protein show less myocardial apoptosis and fibrosis, and enhanced cardiac hypertrophy, after chronic stimulation with angiotensin II [68].
Figure 3. Effect of mechanical stress on the heart.
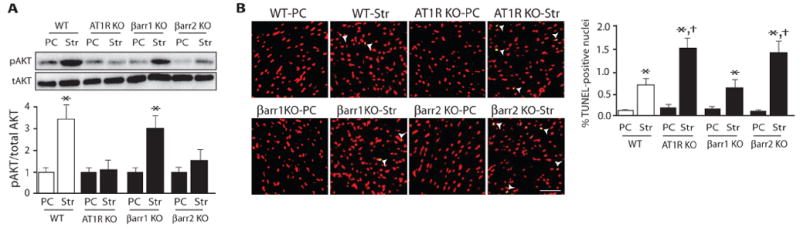
Mechanical stress activates prosurvival antiapoptotic signaling in an AT1aR- and β-arrestin2–dependent manner. Mechanical stretch of wild-type hearts led to Akt phosphorylation, which was absent in hearts from AT1aR KO and β-arrestin2 KO mice (panel A). The rate of apoptotic cell death as measured by TUNEL [terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate (dUTP) nick end labeling] staining was significantly increased in hearts from β-arrestin2 KO or AT1aR KO mice compared to those from wild-type (panel B) (Reproduced with permission from Ref [67]).
Biased agonism at adrenergic and angiotensin receptors
Based on the studies mentioned for both AT1aR and the β1AR, it appears that chronic G-protein signaling is detrimental in heart failure, where as β-arrestin dependent signaling by these receptors is cardioprotective. Many of the currently available therapies to treat heart failure, such as angiotensin-receptor blockers (ARBs) and β adrenergic receptor blockers (beta-blockers) target signaling at these receptors but block both G-protein and β-arrestin mediated signaling. The emerging concept of β-arrestin mediated signaling suggests the possibility of developing ligands that activate a receptor to preferentially signal through one pathway. This gives rise to the paradigm of “biased agonism” which is a property of the ligand-receptor complex, whereby a ligand or a receptor may be biased towards a particular signaling pathway. Thus “biased ligands” or “biased receptors” are those that preferentially exploit either β-arrestin or G-protein signaling pathways [31] (Figure 4). This concept, also described as “functional selectivity” or “collateral/pluridimensional efficacy” has major implications for pharmacological therapeutics targeting GPCRs including the adrenergic and angiotensin receptors [69-72].
Figure 4. Biased ligands and biased receptors.
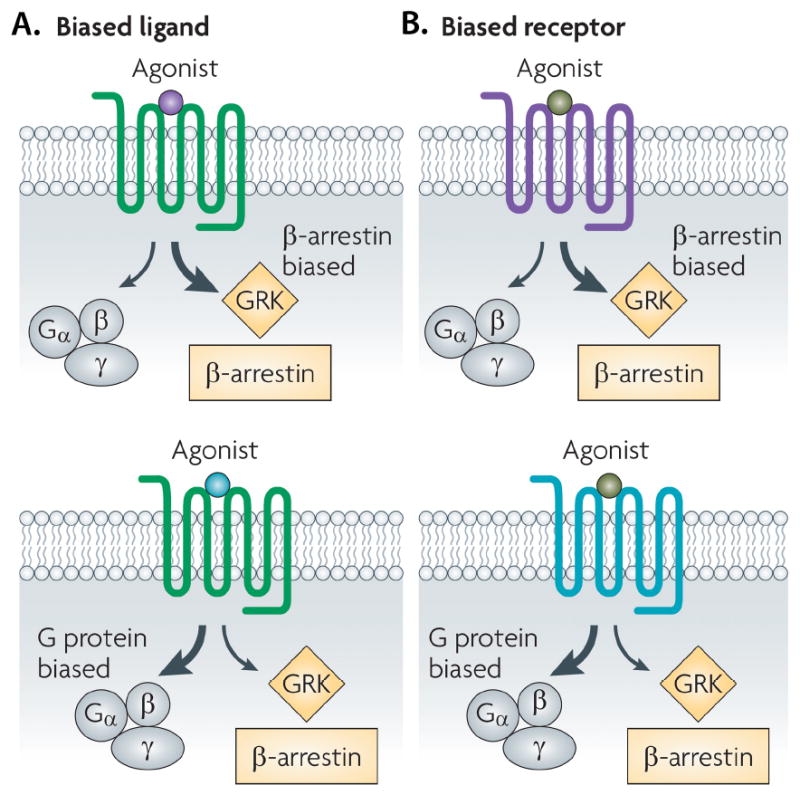
GPCR can preferentially activate various signaling mechanisms following ligand stimulation. A) Binding of a biased ligand (purple: β arrestin bias; blue: G-protein bias), to an unbiased receptor results in a biased response activating either G-protein mediated or β-arrestin pathway. B) Similarly, binding of an unbiased ligand to the biased receptor (β-arrestin-biased in purple or G protein-biased in blue) also results in a biased response (Reproduced with permission from Ref [31]).
Based on this concept of “biased agonism”, a variety of ligands targeting the βAR have been screened to evaluate their ability to signal through multiple pathways (a measure of their functional selectivity or bias). Using isoprotereonol as a reference compound, Baker et al. showed the agonist and inverse agonist (defined as a decrease in basal activity after ligand binding) response to β2AR activation with a series of ligands [73]. For certain ligands, such as the antagonist propranolol, there was increased cAMP response element (CRE)-mediated transcription and MAP kinase activation despite a significant decrease in cAMP production (a traditional antagonist or inverse agonist effect) [73]. Galandrin et al. have identified complex “efficacy profiles” for β1- and β2ARs after stimulation with a variety of traditional antagonists [69]. Antagonists such as carvedilol, bucindolol and labetolol were shown to activate adenylyl cyclase and ERK pathways for both β1ARs and β2ARs with variable efficacies. Among all the traditional “β-blockers”, carvedilol induced the greatest ERK signaling [69]. Further investigation has examined the ability of ligands to recruit β-arrestin and activate ERK in a “β-arrestin-biased” fashion [62, 74]. Of the many β-blockers tested, carvedilol, was shown to be a β-arrestin-biased ligand for both the β1AR [62] and β2AR [74].
The AT1aR also demonstrates biased signaling upon ligand stimulation using a synthetically generated biased Ang II analog known as SII ([sarcosine1,Ile4,Ile8]AngII) [75]. Upon AT1aR stimulation, SII recruits β-arrestin causing an ERK signaling response without inducing any G protein activity or G protein mediated protein kinase C (PKC) activation [42, 75]. In vivo treatment with SII also leads to ERK signal transduction and enhanced myocyte contractility [76, 77]. Interestingly, mechanical stretch of the heart activates ligand-independent β-arrestin signaling of the AT1aR that appears to be an endogenous stimulus analogous to that of a biased ligand [67].
Natural occurring polymorphisms of the human β1AR show variable responses to agonist stimulation and to different inverse agonists [78]. For example, the Arg389Gly variant of the human β1AR, which lies within the Gs binding region [79], shows greater sensitivity to carvedilol than the Gly389 variant with regard to inhibition of G protein signaling. Nonetheless, despite a number of population-based studies and post-hoc analyses of randomized controlled trials of beta-blockers, no clear consensus has been reached regarding effects of the Arg389Gly variant and other common polymorphisms on the natural history of disease or pharmacogenomic interactions [80-82].
Conformational changes in GPCR and β-arrestin recruitment
Although there is strong evidence showing β-arrestin recruitment after stimulation with “biased ligands”, the precise molecular mechanism that triggers β-arrestin-biased signaling is not well understood. There is increasing evidence that ligand induced receptor conformation plays a pivotal role in signaling. In fact, current concepts support the notion that ligands such as carvedilol or SII can induce unique conformations within βAR that decrease G protein coupling while promoting selective GRK phosphorylation and β-arrestin mediated signaling. Ligand induced changes in receptor conformation have been directly studied using a modified βAR with fluorophores ligated to the c-terminus, and to either a cysteine residue in the sixth transmembrane region [83], or c-terminus and the third intracellular loop [78]. For the β2AR, it was shown that different ligands induce variable changes in receptor conformation involving the C terminus [83]. Compared with ligands that do not mediate β-arrestin mediated signaling, ligands known to activate β-arrestin mediated MAP kinase signaling seem to induce a unique conformation at the proximal C terminal tail [78, 83, 84].
To understand how ligand-induced receptor conformation affect downstream signaling, biased receptors have been generated by mutating key residues involved in G-protein coupling enabling these receptors to activate only non G-protein signaling pathways. Such mutated receptors include the AT1aR mutant ATIaR (DRY/AAY), in which residues of the highly conserved DRY motif have been mutated to AAY (Asp125-Arg126-Tyr127 to Ala125-Ala126-Tyr) [44] and the β2AR mutants TYY which contain mutations of three residues at positions 68 (T), 132 (Y), and 219 (Y) critical for G protein signaling [43]. Cellular studies using both ATIaR (DRY/AAY) and β2AR TYY mutants demonstrate absent G protein signaling while maintaining β-arrestin recruitment and ERK signaling [44]. Thus availability of these mutant receptors provides a valuable tool for better understanding the mechanisms of biased receptor signaling and for the development of the next generation of biased agonists for the β1AR and AT1aR. The physiological importance of β-arrestin-mediated signaling has so far, been confined to studies in cell culture and experimental knock-out animal systems [31], and therefore the therapeutic impact of biased agonists will not be known until they are tested in clinical trials.
Conformational changes in β-arrestin and functional selectivity to activate only certain pathway
In addition to selective changes in receptor conformation induced by ligand, conformational changes in the β-arrestin molecule itself may be responsible for the various physiologic effects observed in response to biased agonism. Using a novel intramolecular β-arrestin biosensor [85], stimulation of the AT1aR with the biased ligand SII induced a conformational change in β-arrestin that was significantly different from that of the endogenous ligand angiotensin II or the traditional ARB valsartan [86]. Interestingly, the conformational change in β-arrestin induced by SII, was similar to that observed when mechanical stretch was applied to AT1aR expressing cells containing the β-arrestin biosensor [67].
To date, the impact of GRK mediated receptor phosphorylation on β-arrestin conformation has not been directly studied. However, it has been suggested that the sites of receptor phosphorylation may have important implications for the ultimate conformation adopted by β-arrestin and for downstream signaling since different GRKs are associated with different cellular responses [48].
β- arrestin as a multifunctional regulator of GPCR signaling
One of the ways that β-arrestin conformation contributes to functional selectivity of receptor signaling is through its ability to form multimolecular protein complexes. Recently, Mangmool et al. showed that activation of the β1AR induces a conformational change in β-arrestin that promotes a stable complex between β-arrestin, Ca2+/calmodulin kinase II (CaMKII), and the cAMP-dependent guanine-nucleotide exchange factor (Epac) [13]. The association of β-arrestin with the β1AR stabilizes a β-arrestin-CaMKII-Epac complex and promotes CaMKII signaling [13] (Figure 5). This β-arrestin-CaMKII-EPAC complex is only seen for the β1AR and not the β2AR despite prominent β-arrestin recruitment to both receptors by agonist [13]. The mechanism for this differential effect is that ligand activation of the β2AR does not induce a β-arrestin conformation that allows association with CaMKII and Epac at the cell membrane[13]. Using βAR chimeras (i.e. β2AR with β1AR c-terminal tail), it was shown that structural elements in the c-terminus of the β1AR is responsible for β-arrestin mediated CaMKII signaling [13] (Figure 5).
Figure 5. The carboxyl terminus of β1-AR but not β2-AR is necessary for CaMKII activation and conformational changes of β-arrestin.
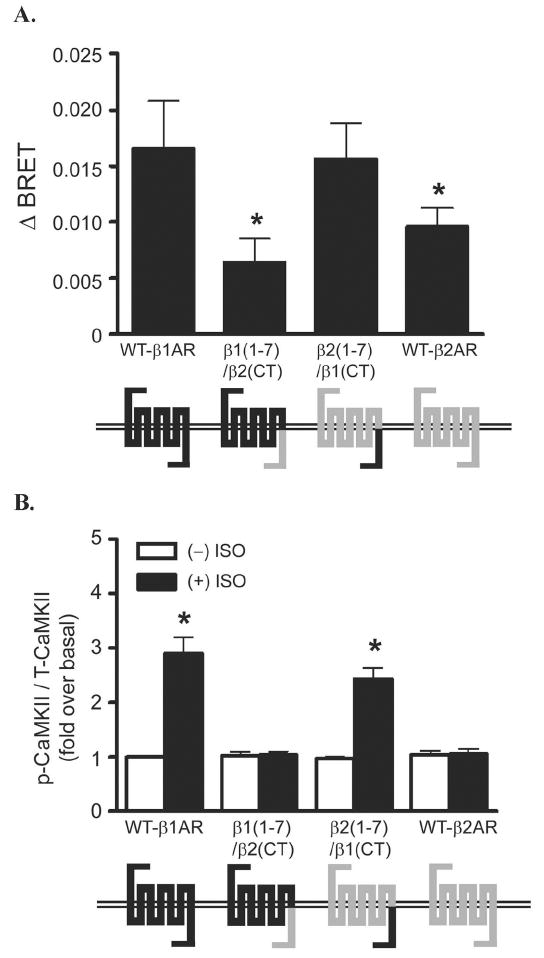
A) Using βAR chimeras (i.e. β2AR with β1AR c-terminal tail), YFP-β arrestin2-Luc biosensor and measuring bioluminescence resonance energy transfer (BRET), Mangmool et al. show that the C-tail of the β1AR is the key regulatory component that promotes unique β- arrestin conformation. B) The c-terminal of the β1AR also plays a key role in promoting β arrestin mediated CAMKII signaling upon ligand stimulation. (Reproduced with permission from [13]).
Additionally, proteomic analyses using a mass-spectrometry based approach further reveals that β-arrestin directly interacts with more than 300 proteins from different families involved in cellular signaling (such as kinases, phosphatases and nucleic acid binding) in the basal state and following AT1aR [12]. This study, and other recent analysis of the β-arrestin phosphoproteome [32, 87], suggests that β-arrestins are critically involved in a complex matrix of protein-protein interactions and cellular signaling events highlighting their pivotal role in cellular physiology and cardiac disease. The unique signaling complexes that may be formed with β-arrestin as a scaffold when more potent biased agonists are developed will further enhance our understanding of the physiological importance if this multifunctional protein and its importance in heart failure.
Summary
β-arrestin has emerged as a multifunctional molecule that plays a key role in cell signaling via GPCRs and in providing cardioprotection in response to pathogenic stimuli. Since β1AR and AT1aRs are already targets for therapeutics, there is increasing interest in developing the next generation of drugs for heart failure that not only selectively antagonize harmful cardiac signaling but also potentiate the beneficial pathways. In that regard, a greater understanding of the signaling mechanisms by the multifunctional protein β-arrestin should provide the framework for development of powerful new therapies for cardiac diseases, more specifically heart failure.
Acknowledgments
This work was supported by grants from the National Institutes of Health HL-075443 and HL-56687 to H.A.R. and a Duke University Stead Fellowship to N.N.
Footnotes
Disclosures: H.A.R. is a scientific cofounder and consultant for Trevena Inc., a company that is developing G protein coupled receptor–targeted drugs.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hunt SA. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure) J Am Coll Cardiol. 2005 Sep 20;46(6):e1–82. doi: 10.1016/j.jacc.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 2.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002 Jan 10;415(6868):206–12. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 3.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005 Apr 22;308(5721):512–7. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 4.Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ. Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science. 1998 Apr 24;280(5363):574–7. doi: 10.1126/science.280.5363.574. [DOI] [PubMed] [Google Scholar]
- 5.Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation. 1998 Sep 29;98(13):1329–34. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]
- 6.Molkentin JD, Dorn GW., 2nd Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001;63:391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- 7.Rajagopal K, Lefkowitz RJ, Rockman HA. When 7 transmembrane receptors are not G protein-coupled receptors. J Clin Invest. 2005 Nov;115(11):2971–4. doi: 10.1172/JCI26950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990 Jun 22;248(4962):1547–50. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 9.Lefkowitz RJ, Rajagopal K, Whalen EJ. New roles for beta-arrestins in cell signaling: not just for seven-transmembrane receptors. Mol Cell. 2006 Dec 8;24(5):643–52. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–82. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- 11.Wu N, Hanson SM, Francis DJ, Vishnivetskiy SA, Thibonnier M, Klug CS, et al. Arrestin binding to calmodulin: a direct interaction between two ubiquitous signaling proteins. J Mol Biol. 2006 Dec 15;364(5):955–63. doi: 10.1016/j.jmb.2006.09.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, et al. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci U S A. 2007 Jul 17;104(29):12011–6. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mangmool S, Shukla AK, Rockman HA. beta-Arrestin-dependent activation of Ca(2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J Cell Biol. 2010 May 3;189(3):573–87. doi: 10.1083/jcb.200911047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Couchonnal LF, Anderson ME. The role of calmodulin kinase II in myocardial physiology and disease. Physiology (Bethesda) 2008 Jun;23:151–9. doi: 10.1152/physiol.00043.2007. [DOI] [PubMed] [Google Scholar]
- 15.Wilden U, Hall SW, Kuhn H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci U S A. 1986 Mar;83(5):1174–8. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Annu Rev Biochem. 1998;67:653–92. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- 17.Nelson CD, Perry SJ, Regier DS, Prescott SM, Topham MK, Lefkowitz RJ. Targeting of diacylglycerol degradation to M1 muscarinic receptors by beta-arrestins. Science. 2007 Feb 2;315(5812):663–6. doi: 10.1126/science.1134562. [DOI] [PubMed] [Google Scholar]
- 18.Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, et al. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science. 2002 Oct 25;298(5594):834–6. doi: 10.1126/science.1074683. [DOI] [PubMed] [Google Scholar]
- 19.Sibley DR, Strasser RH, Benovic JL, Daniel K, Lefkowitz RJ. Phosphorylation/dephosphorylation of the beta-adrenergic receptor regulates its functional coupling to adenylate cyclase and subcellular distribution. Proc Natl Acad Sci U S A. 1986 Dec;83(24):9408–12. doi: 10.1073/pnas.83.24.9408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daaka Y, Luttrell LM, Ahn S, Della Rocca GJ, Ferguson SS, Caron MG, et al. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998 Jan 9;273(2):685–8. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- 21.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999 Jan 29;283(5402):655–61. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 22.Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996 Oct 3;383(6599):447–50. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 23.Gaidarov I, Krupnick JG, Falck JR, Benovic JL, Keen JH. Arrestin function in G protein-coupled receptor endocytosis requires phosphoinositide binding. EMBO J. 1999 Feb 15;18(4):871–81. doi: 10.1093/emboj/18.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naga Prasad SV, Barak LS, Rapacciuolo A, Caron MG, Rockman HA. Agonist-dependent recruitment of phosphoinositide 3-kinase to the membrane by beta-adrenergic receptor kinase 1. A role in receptor sequestration. J Biol Chem. 2001 Jun 1;276(22):18953–9. doi: 10.1074/jbc.M102376200. [DOI] [PubMed] [Google Scholar]
- 25.Naga Prasad SV, Laporte SA, Chamberlain D, Caron MG, Barak L, Rockman HA. Phosphoinositide 3-kinase regulates beta2-adrenergic receptor endocytosis by AP-2 recruitment to the receptor/beta-arrestin complex. J Cell Biol. 2002 Aug 5;158(3):563–75. doi: 10.1083/jcb.200202113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naga Prasad SV, Jayatilleke A, Madamanchi A, Rockman HA. Protein kinase activity of phosphoinositide 3-kinase regulates beta-adrenergic receptor endocytosis. Nat Cell Biol. 2005 Aug;7(8):785–96. doi: 10.1038/ncb1278. [DOI] [PubMed] [Google Scholar]
- 27.Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000 Jun 2;275(22):17201–10. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Barak LS, Anborgh PH, Laporte SA, Caron MG, Ferguson SS. Cellular trafficking of G protein-coupled receptor/beta-arrestin endocytic complexes. J Biol Chem. 1999 Apr 16;274(16):10999–1006. doi: 10.1074/jbc.274.16.10999. [DOI] [PubMed] [Google Scholar]
- 29.Tan CM, Brady AE, Nickols HH, Wang Q, Limbird LE. Membrane trafficking of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2004;44:559–609. doi: 10.1146/annurev.pharmtox.44.101802.121558. [DOI] [PubMed] [Google Scholar]
- 30.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 31.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010 May;9(5):373–86. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao K, Sun J, Kim J, Rajagopal S, Zhai B, Villen J, et al. Global phosphorylation analysis of {beta}-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR) Proc Natl Acad Sci U S A. 2010 Aug 4; doi: 10.1073/pnas.1008461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007 May 14;26(22):3113–21. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- 34.Pierce KL, Maudsley S, Daaka Y, Luttrell LM, Lefkowitz RJ. Role of endocytosis in the activation of the extracellular signal-regulated kinase cascade by sequestering and nonsequestering G protein-coupled receptors. Proc Natl Acad Sci U S A. 2000 Feb 15;97(4):1489–94. doi: 10.1073/pnas.97.4.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rozengurt E. Mitogenic signaling pathways induced by G protein-coupled receptors. J Cell Physiol. 2007 Dec;213(3):589–602. doi: 10.1002/jcp.21246. [DOI] [PubMed] [Google Scholar]
- 36.Jarpe MB, Widmann C, Knall C, Schlesinger TK, Gibson S, Yujiri T, et al. Anti-apoptotic versus pro-apoptotic signal transduction: checkpoints and stop signs along the road to death. Oncogene. 1998 Sep 17;17(11 Reviews):1475–82. doi: 10.1038/sj.onc.1202183. [DOI] [PubMed] [Google Scholar]
- 37.Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol. 2003 Jul;5(7):647–54. doi: 10.1038/ncb1005. [DOI] [PubMed] [Google Scholar]
- 38.Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, et al. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003 Oct 2;22(43):6785–93. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 39.Graves LM, Guy HI, Kozlowski P, Huang M, Lazarowski E, Pope RM, et al. Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature. 2000 Jan 20;403(6767):328–32. doi: 10.1038/35002111. [DOI] [PubMed] [Google Scholar]
- 40.Stefanovsky V, Langlois F, Gagnon-Kugler T, Rothblum LI, Moss T. Growth factor signaling regulates elongation of RNA polymerase I transcription in mammals via UBF phosphorylation and r-chromatin remodeling. Mol Cell. 2006 Mar 3;21(5):629–39. doi: 10.1016/j.molcel.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 41.Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. Embo J. 1997 Apr 15;16(8):1909–20. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004 Aug 20;279(34):35518–25. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- 43.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, et al. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006 Jan 13;281(2):1261–73. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 44.Gaborik Z, Jagadeesh G, Zhang M, Spat A, Catt KJ, Hunyady L. The role of a conserved region of the second intracellular loop in AT1 angiotensin receptor activation and signaling. Endocrinology. 2003 Jun;144(6):2220–8. doi: 10.1210/en.2002-0135. [DOI] [PubMed] [Google Scholar]
- 45.Tilley DG, Kim IM, Patel PA, Violin JD, Rockman HA. beta-Arrestin mediates beta1-adrenergic receptor-epidermal growth factor receptor interaction and downstream signaling. J Biol Chem. 2009 Jul 24;284(30):20375–86. doi: 10.1074/jbc.M109.005793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, et al. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003 Feb 21;278(8):6258–67. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 47.Cervantes D, Crosby C, Xiang Y. Arrestin orchestrates crosstalk between G protein-coupled receptors to modulate the spatiotemporal activation of ERK MAPK. Circ Res. Jan 8;106(1):79–88. doi: 10.1161/CIRCRESAHA.109.198580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, et al. Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest. 2007 Sep;117(9):2445–58. doi: 10.1172/JCI31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996 Feb 8;379(6565):557–60. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 50.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999 Dec 23-30;402(6764):884–8. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 51.Shah BH, Catt KJ. Matrix metalloproteinase-dependent EGF receptor activation in hypertension and left ventricular hypertrophy. Trends Endocrinol Metab. 2004 Aug;15(6):241–3. doi: 10.1016/j.tem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 52.Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, et al. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci U S A. 2003 Sep 30;100(20):11406–11. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim J, Ahn S, Rajagopal K, Lefkowitz RJ. Independent beta-arrestin2 and Gq/protein kinase Czeta pathways for ERK stimulated by angiotensin type 1A receptors in vascular smooth muscle cells converge on transactivation of the epidermal growth factor receptor. J Biol Chem. 2009 May 1;284(18):11953–62. doi: 10.1074/jbc.M808176200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goel R, Phillips-Mason PJ, Raben DM, Baldassare JJ. alpha-Thrombin induces rapid and sustained Akt phosphorylation by beta-arrestin1-dependent and -independent mechanisms, and only the sustained Akt phosphorylation is essential for G1 phase progression. J Biol Chem. 2002 May 24;277(21):18640–8. doi: 10.1074/jbc.M108995200. [DOI] [PubMed] [Google Scholar]
- 55.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007 Jan;292(1):C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 56.Nakashima H, Suzuki H, Ohtsu H, Chao JY, Utsunomiya H, Frank GD, et al. Angiotensin II regulates vascular and endothelial dysfunction: recent topics of Angiotensin II type-1 receptor signaling in the vasculature. Curr Vasc Pharmacol. 2006 Jan;4(1):67–78. doi: 10.2174/157016106775203126. [DOI] [PubMed] [Google Scholar]
- 57.Ahn S, Kim J, Hara MR, Ren XR, Lefkowitz RJ. {beta}-Arrestin-2 Mediates Anti-apoptotic Signaling through Regulation of BAD Phosphorylation. J Biol Chem. 2009 Mar 27;284(13):8855–65. doi: 10.1074/jbc.M808463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mallat Z, Tedgui A. Apoptosis in the vasculature: mechanisms and functional importance. Br J Pharmacol. 2000 Jul;130(5):947–62. doi: 10.1038/sj.bjp.0703407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim J, Zhang L, Peppel K, Wu JH, Zidar DA, Brian L, et al. Beta-arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration. Circ Res. 2008 Jul 3;103(1):70–9. doi: 10.1161/CIRCRESAHA.108.172338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korshunov VA, Berk BC. Smooth muscle apoptosis and vascular remodeling. Curr Opin Hematol. 2008 May;15(3):250–4. doi: 10.1097/MOH.0b013e3282f97d71. [DOI] [PubMed] [Google Scholar]
- 61.Violin JD, Ren XR, Lefkowitz RJ. G-protein-coupled receptor kinase specificity for beta-arrestin recruitment to the beta2-adrenergic receptor revealed by fluorescence resonance energy transfer. J Biol Chem. 2006 Jul 21;281(29):20577–88. doi: 10.1074/jbc.M513605200. [DOI] [PubMed] [Google Scholar]
- 62.Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD, et al. Beta-blockers alprenolol and carvedilol stimulate beta-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci U S A. 2008 Sep 23;105(38):14555–60. doi: 10.1073/pnas.0804745105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, et al. Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci U S A. 2005 Feb 1;102(5):1442–7. doi: 10.1073/pnas.0409532102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brodde OE, Michel MC. Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev. 1999 Dec;51(4):651–90. [PubMed] [Google Scholar]
- 65.Ungerer M, Bohm M, Elce JS, Erdmann E, Lohse MJ. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation. 1993 Feb;87(2):454–63. doi: 10.1161/01.cir.87.2.454. [DOI] [PubMed] [Google Scholar]
- 66.Xiang Y, Kobilka BK. Myocyte adrenoceptor signaling pathways. Science. 2003 Jun 6;300(5625):1530–2. doi: 10.1126/science.1079206. [DOI] [PubMed] [Google Scholar]
- 67.Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal. 2010;3(125):ra46. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhai P, Yamamoto M, Galeotti J, Liu J, Masurekar M, Thaisz J, et al. Cardiac-specific overexpression of AT1 receptor mutant lacking G alpha q/G alpha i coupling causes hypertrophy and bradycardia in transgenic mice. J Clin Invest. 2005 Nov;115(11):3045–56. doi: 10.1172/JCI25330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Galandrin S, Bouvier M. Distinct signaling profiles of beta1 and beta2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Mol Pharmacol. 2006 Nov;70(5):1575–84. doi: 10.1124/mol.106.026716. [DOI] [PubMed] [Google Scholar]
- 70.Kenakin T. Ligand-selective receptor conformations revisited: the promise and the problem. Trends Pharmacol Sci. 2003 Jul;24(7):346–54. doi: 10.1016/S0165-6147(03)00167-6. [DOI] [PubMed] [Google Scholar]
- 71.Kenakin T. Principles: receptor theory in pharmacology. Trends Pharmacol Sci. 2004 Apr;25(4):186–92. doi: 10.1016/j.tips.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 72.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007 Jan;320(1):1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 73.Baker JG, Hall IP, Hill SJ. Agonist and inverse agonist actions of beta-blockers at the human beta 2-adrenoceptor provide evidence for agonist-directed signaling. Mol Pharmacol. 2003 Dec;64(6):1357–69. doi: 10.1124/mol.64.6.1357. [DOI] [PubMed] [Google Scholar]
- 74.Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci U S A. 2007 Oct 16;104(42):16657–62. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, et al. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci U S A. 2003 Sep 16;100(19):10782–7. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aplin M, Christensen GL, Schneider M, Heydorn A, Gammeltoft S, Kjolbye AL, et al. The angiotensin type 1 receptor activates extracellular signal-regulated kinases 1 and 2 by G protein-dependent and -independent pathways in cardiac myocytes and langendorff-perfused hearts. Basic Clin Pharmacol Toxicol. 2007 May;100(5):289–95. doi: 10.1111/j.1742-7843.2007.00063.x. [DOI] [PubMed] [Google Scholar]
- 77.Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, et al. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci U S A. 2006 Oct 31;103(44):16284–9. doi: 10.1073/pnas.0607583103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rochais F, Vilardaga JP, Nikolaev VO, Bunemann M, Lohse MJ, Engelhardt S. Real-time optical recording of beta1-adrenergic receptor activation reveals supersensitivity of the Arg389 variant to carvedilol. J Clin Invest. 2007 Jan;117(1):229–35. doi: 10.1172/JCI30012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mialet Perez J, Rathz DA, Petrashevskaya NN, Hahn HS, Wagoner LE, Schwartz A, et al. Beta 1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat Med. 2003 Oct;9(10):1300–5. doi: 10.1038/nm930. [DOI] [PubMed] [Google Scholar]
- 80.Lanfear DE, Jones PG, Marsh S, Cresci S, McLeod HL, Spertus JA. Beta2-adrenergic receptor genotype and survival among patients receiving beta-blocker therapy after an acute coronary syndrome. JAMA. 2005 Sep 28;294(12):1526–33. doi: 10.1001/jama.294.12.1526. [DOI] [PubMed] [Google Scholar]
- 81.Sehnert AJ, Daniels SE, Elashoff M, Wingrove JA, Burrow CR, Horneu B, et al. Lack of association between adrenergic receptor genotypes and survival in heart failure patients treated with carvedilol or metoprolol. J Am Coll Cardiol. 2008 Aug 19;52(8):644–51. doi: 10.1016/j.jacc.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 82.Shin J, Lobmeyer MT, Gong Y, Zineh I, Langaee TY, Yarandi H, et al. Relation of beta(2)-adrenoceptor haplotype to risk of death and heart transplantation in patients with heart failure. Am J Cardiol. 2007 Jan 15;99(2):250–5. doi: 10.1016/j.amjcard.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 83.Granier S, Kim S, Shafer AM, Ratnala VR, Fung JJ, Zare RN, et al. Structure and conformational changes in the C-terminal domain of the beta2-adrenoceptor: insights from fluorescence resonance energy transfer studies. J Biol Chem. 2007 May 4;282(18):13895–905. doi: 10.1074/jbc.M611904200. [DOI] [PubMed] [Google Scholar]
- 84.Ghanouni P, Gryczynski Z, Steenhuis JJ, Lee TW, Farrens DL, Lakowicz JR, et al. Functionally different agonists induce distinct conformations in the G protein coupling domain of the beta 2 adrenergic receptor. J Biol Chem. 2001 Jul 6;276(27):24433–6. doi: 10.1074/jbc.C100162200. [DOI] [PubMed] [Google Scholar]
- 85.Charest PG, Terrillon S, Bouvier M. Monitoring agonist-promoted conformational changes of beta-arrestin in living cells by intramolecular BRET. EMBO Rep. 2005 Apr;6(4):334–40. doi: 10.1038/sj.embor.7400373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shukla AK, Violin JD, Whalen EJ, Gesty-Palmer D, Shenoy SK, Lefkowitz RJ. Distinct conformational changes in beta-arrestin report biased agonism at seven-transmembrane receptors. Proc Natl Acad Sci U S A. 2008 Jul 22;105(29):9988–93. doi: 10.1073/pnas.0804246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Christensen GL, Kelstrup CD, Lyngso C, Sarwar U, Bogebo R, Sheikh SP, et al. Quantitative phosphoproteomics dissection of seven-transmembrane receptor signaling using full and biased agonists. Mol Cell Proteomics. 2010 Jul;9(7):1540–53. doi: 10.1074/mcp.M900550-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]