

Abstract
14-O-cinnamoyl esters of naltrexone (6) were synthesised and evaluated in isolated tissue assays in vitro and in vivo in mouse antinociceptive assays. Their predominant opioid receptor activity was mu receptor (MOR) antagonism but the unsubstituted cinnamoyl derivative (6a) and the p-methylcinnamoyl derivative (6c) had partial MOR agonist activity in vitro and in vivo. When compared to the equivalent 14-cinnamoylaminomorphinones (5) the cinnamoyloxy morphinones (6) as MOR antagonists had shorter duration of action and were less effective as pseudoirreversible antagonists. The antinociceptive activity of the cinnamoyloxycodeinones (7) was not significantly greater than that of the morphinones (6) but they showed no evidence of any pseudoirreversible MOR antagonism. In both respects these profiles differed from those of the equivalent 14-cinnamoylaminocodeinones (4).
Introduction
Naloxone (1b) and naltrexone (1a) are prototype opioid antagonists having some limited selectivity for mu opioid receptors (MOR). They have found clinical utility respectively as a treatment for opiate1 and alcohol dependence2, and to reverse narcotic overdosage.3 14-O-alkyl ethers (2) of naltrexone retain predominant MOR antagonist activity4,5 but the 14-O-3-phenylpropyl ether (2a) has recently been shown to have high efficacy and high potency MOR agonist activity in antinociceptive assays.6 We have made extensive studies of derivatives (3) of 14β-amino-7,8-dihydromorphinone with a major focus on the cinnamoylamino- derivatives (4, 5) of which the MOR-selective irreversible antagonists clocinnamox (C-CAM; 5b) and methcinnamox (M-CAM; 5c) are the most studied examples.7-10 We here report preparation and evaluation of 14-O-cinnamoyl esters of naltrexone (6) and the equivalent codeinones (7) for comparison with the 14-amino derivatives (4, 5) and with the 14-O--phenylpropyl ether of naltrexone (2a).
Synthesis
Acylation of naltrexone 3-O-methyl ether (8a) to give 7a-7c was achieved using the appropriate anhydrides (Scheme 1), themselves prepared from their equivalent acid chlorides by the method of Armesto et al.11 Ligands 6a-6c were similarly prepared from 3-O-(tert-butyldimethylsilyl)naltrexone (8b)12 by acylation and then removal of the protecting group with potassium fluoride to yield the morphinones (Scheme 1).
Scheme 1.

(i) (RCO)2O, toluene, reflux (ii) KF, MeOH, CH2Cl2
Results
The new ligands (6, 7) in opioid receptor binding assays13,14 displayed high affinity for MOR and significantly lower affinity for delta opioid receptors (DOR) and kappa opioid receptors (KOR) (Table 1). Affinity of the morphinones (6) was generally higher than that of the codeinones (7) with DOR affinity showing greater disparity than MOR and KOR affinity. The unsubstituted cinnamoylmorphinone (6a) had the highest affinity for all three receptors, comparable to naltrexone and for MOR, higher than C-CAM (5b).
Table 1.
Binding affinities of new ligands to opioid receptors
| Ki (nM)d | |||||
|---|---|---|---|---|---|
| R | R’ | MOR | DOR | KOR | |
| 6a a | H | H | 0.40±0.05 | 3.4±0.8 | 3.6±2 |
| 6b a | H | Cl | 1.3±0.45 | 15±6 | 8.3±0.2 |
| 6c a | H | CH3 | 3.3±0.1 | 19±0.6 | 8.1±0.7 |
| 7a b | CH3 | H | 2.5±0.7 | 180±58 | 4.3±0.7 |
| 7b b | CH3 | Cl | 3.5±1.0 | 110±20 | 52±12 |
| 7c b | CH3 | CH3 | 4.2±1.5 | 270±80 | 36±8.0 |
| Naltrexonea (1) | 0.40±0.05 | 6.5±1 | 0.6±0.1 | ||
| Naltrexoneb (1) | 0.20±0.0 | 11±3 | 0.4±0.1 | ||
| MC-CAMb (4b) | 4.8±0.6 | 4.8±0.7 | 16±2.5 | ||
| C-CAMb (5b) | 3.0±0.2 | 2.7±0.2 | 1.4±0.5 | ||
| PPN (2a)b,c | 0.34±0.06 | 0.48±0.05 | 0.41±0.09 |
binding to guinea pig brain membranes (method in ref. 13)
binding to cloned human opioid receptors transfected into CHO cells (method in ref. 14)
figures from ref 6.
Values are the average from two experiments each carried out in duplicate. Tritiated ligands were [3H]DAMGO (MOR), [3H]Cl-DPDPE (DOR) and [3H]U69593 (KOR)
The morphinones (6) were evaluated for opioid receptor functional activity in the mouse vas deferens (MVD) and guinea pig ileum (GPI) isolated tissue assays.13 In MVD the morphinones displayed little agonist activity, but were very potent opioid receptor antagonists of the standard selective opioid receptor agonists DAMGO (MOR), DPDPE (DOR) and U69593 (KOR), having subnanomolar Ke values for all three opioid receptors (Table 2). Although MOR antagonist potency was higher than DOR and KOR potency, there was no appreciable selectivity for MOR over DOR and KOR. In GPI morphinones 6 partially inhibited the electrically-stimulated contractions of the tissue. This opioid receptor partial agonist effect was not reversed by the selective antagonists CTAP (MOR) or norBNI (KOR) indicating very slow receptor offset such as was observed with the similarly lipophilic opioid ligands buprenorphine (10)15 and C-CAM (5b).16
Table 2.
Antagonist activity of new ligands in the mouse vas deferens
| R | R’ | MOR | Ke (nM) DOR |
KOR | |
|---|---|---|---|---|---|
| 6a | H | H | 0.020±0.007 | 0.25±0.06 | 0.19±0.06 |
| 6b | H | Cl | 0.060±0.03 | 0.060±0.01 | 1.3±0.2 |
| 6c | H | CH3 | 0.12±0.02 | 0.66±0.2 | 0.54±0.07 |
| Naltrexone (1) | 0.44±0.09 | 7.2±0.3 | 8.0±0.6 |
Ke (nM) versus the standard selective agonists DAMGO (MOR), DPDPE (DOR) and U69593 (KOR). Values are the average of at least four experiments.
Opioid receptor functional activity of the codeinones (7) was investigated by their effects on stimulating [35S]GTPγS binding in recombinant human opioid receptor transfected into CHO cells (Tables 3 and 4).14 The unsubstituted cinnamoyl ester (7a) showed partial agonist activity of modest potency for all three opioid receptors (Table 4), whereas the p-chloro analogue (7b) had insignificant agonist activity for any opioid receptor but was an antagonist of the standard agonists DAMGO (MOR), DPDPE (DOR) and U69593 (KOR) (Table 3) which were also used as the standards against which the agonist stimulation of 7a was measured. 7c like 7b had only antagonist activity for MOR (Table 3) but was a KOR partial agonist (Table 4).
Table 3.
Inhibition of agonist stimulated [35S]GTPγS binding in recombinant human opioid receptors
Table 4.
Opioid agonist stimulation of [35S]GTPγS binding in recombinant human opioid receptors.
| EC50 (nM); % stimulation | |||||
|---|---|---|---|---|---|
| R | R’ | MOR | DOR | KOR | |
| 7a | CH3 | H | 34.4±5.0; 35 | 430±110; 64 | 26±4.9; 76 |
| 7c | CH3 | CH3 | ANT | NT | 157±3.4; 37 |
| MC-CAM (4b)a | 17.8±11; 8 | NT | ANT | ||
| Morphine | 15.6±0.5; 93 | 316±4.9; 103 | 484±213; 62 | ||
Percent maximal stimulation with respect to the standard agonists DAMGO (mu), U69593 (kappa) and DPDPE (delta). ANT indicates antagonist activity (Ke values reported in Table 3). Values are the mean of 5 or 6 experiments. N.D. indicates not determined. Data supplied by NIDA Addiction Treatment Discovery Program.
Data from Spagnolo et al, 200819
In vivo activity of the 14-O-acyl derivatives was investigated in mouse antinociceptive tests using assays with thermal (tail withdrawal from 50°C warm water (TW)) or chemical (acetic acid induced writhing (AW)) stimulation.8 None of the naltrexone derivatives (6,7) showed any significant opioid receptor agonist activity in TW but all were effective antagonists of morphine in this assay. A high dose (32 mg/kg given 30 minutes before morphine) of each of the morphinones (6) flattened the morphine dose-response curve up to 320-1000 mg/kg of the agonist (illustrated for 6b in Fig. 1). Pre-treatment (24 h) with the antagonist 6b resulted in a 10-fold parallel rightward shift of the morphine dose-response curve, but the shift produced by 6a and 6c was negligible (Fig. 1). The codeinones (7b, 7c) were antagonists of morphine in TW but at 32 mg/kg only shifted the morphine dose-effect curve in the standard assay 3-4-fold to the right with no evidence of flattening and there was no antagonist effect with 24 h pre-treatment (data not shown).
Figure 1.

Dose response curves for morphine alone (■) and after pretreatment with a 32 mg/kg dose of the morphinones 6a, 6b and 6c in the mouse warm water tail withdrawal assay. Pretreatment times (ptt) were 24 h (6a, 6b, 6c) and 30 min (6b).
The p-substituted cinnamoyloxymorphinone 6c and the equivalent codeinone 7c unimpressively inhibited the acetic acid induced writhing effect; whereas 6a was substantially more potent and effective (Figure 2). The only opioid antagonists without any in vivo agonist effects were the p-chlorocinnamoyloxy derivatives (6b, 7b). This data confirms that the chemical nociceptor used in the AW assay presents a less intense challenge than the thermal stimulus in TW.
Figure 2.
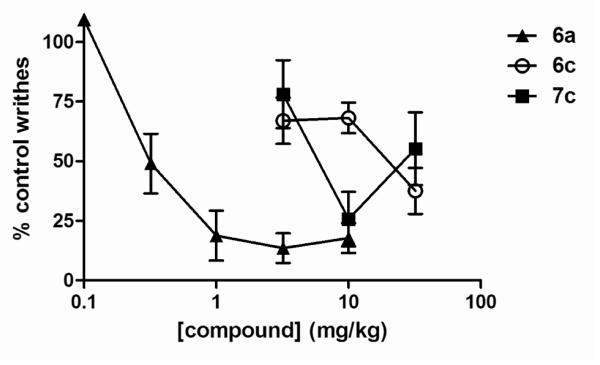
Inhibition of acetic acid induced writhing by 6a, 6c and 7c after s.c. administration.
Agonist selectivity for the individual opioid receptor in AW was determined for 6a by the use of selective antagonists for MOR, DOR and KOR. These were the antagonists β-FNA (MOR), naltrindole (DOR) and norBNI (KOR). β-FNA and norBNI were administered 24 h before 6a to ensure a competitive (and selective) antagonist effect. The agonist effect of 6a in AW was partially antagonized by β-FNA and by naltrindole but not by norBNI (Fig. 3) so that it appears that in AW the agonist effects of 6a are primarily mediated by DOR and MOR. 6b was evaluated as an antagonist versus the agonists morphine (MOR), BW373U86 (DOR) and bremazocine (KOR), only proving effective against morphine at 24h pretreatment (data not shown).
Figure 3.

Agonist selectivity of 6a (10 mg/kg, s.c.) in the writhing assay.
NorBNI (KOR), 32 mg/kg (24 hr pretreatment); β-FNA (MOR), 32 mg/kg (24 hr pretreatment); Naltrindole (DOR), 10 mg/kg (24h pretreatment) t-test P-values
Discussion
Our prime interest in the activity of the naltrexone esters (6) was in comparison to the activity of the equivalent amides C-CAM (5b) and M-CAM (5c). The latter are highly effective and selective MOR antagonists with insignificant agonist effects in vivo.7,8 They are more effective than β-FNA in flattening the dose-response curve of MOR agonists8 but since they do not form covalent bonds in vitro by Michael addition of protein nucleophilic groups they have been termed pseudo-irreversible antagonists.16,17 The very powerful binding to MOR in vivo seems very likely to involve the lipophilic cinnamoylamino group functioning in a manner similar to the t-butyl group in buprenorphine (10).18 The present 14-O-acylmorphinones (6) fell short of C-CAM and M-CAM as pseudo-irreversible antagonists; though in TW they all flattened the morphine dose-response curve 30 minutes after their administration, their MOR antagonist effect was much reduced at 24h, whereas the amides C-CAM (5b) and M-CAM (5c) had very pronounced MOR antagonist effects at 24h and beyond.8 6b, with p-chloro substitution in the cinnamoyl aromatic ring, was the most effective pseudo-irreversible antagonist among the esters; its MOR antagonist profile was comparable to that of β-FNA.8 Since, together with the corresponding codeinone (7b), 6b was the only ester to lack any demonstrable antinociceptive action; the profile of 6b is therefore not dissimilar to that of C-CAM (5b). The unsubstituted morphinone ester 6a in vivo was also basically similar to the equivalent amide 5a.9 This means it showed little agonist activity in TW but substantial activity in AW. Again the most significant difference between 6a and 5a is the duration of morphine antagonist activity in TW. 5a with 24 h pre-treatment produced a 0.5 – 1 log unit shift of the morphine dose-response curve9 whereas the shift from 6a was barely significant.
The biggest difference between the 14-cinnamoyloxy morphinones and equivalent 14-cinnamoylamino morphinones was found in the p-methyl substituted derivatives (6c and 5c). Whereas in the cinnamoylamino series M-CAM (5c) had no agonist activity in TW or AW and was a substantially more effective pseudo-irreversible MOR agonist than the p-chloro analogue C-CAM (5b),8 the p-methylcinnamoyloxy derivative 6c was a less effective MOR antagonist than the p-chloro congener 6b and had measurable agonist activity in AW. However, the SAR established for the 14-cinnamoylamino- series (5) that the 4′-substituted derivatives (5b, 5c) in vivo had lower MOR efficacy than the unsubstituted parent (5a)9 also applied to the present 14-cinnamoyloxy series (6).
The cinnamoyloxy codeinones (7b, 7c) in the antinociceptive assays had no agonist activity in TW and showed parallel rightward shifts of the morphine dose-response curve in this assay indicating a competitive MOR antagonist effect. In AW 7c but not 7b had a weak opioid receptor agonist effect. These profiles are not dissimilar to those of the equivalent morphinones (6b, 6c) in the antinociceptive assays, the main difference being the lack of any flattening of the morphine dose-response curve by the codeinones in the MOR antagonist assay in TW. The similarity of the in vivo agonist effects of the cinnamoyloxycodeinones and morphinones contrasts with the 14-cinnamoylamino series in which the codeinones (4) all had substantially higher MOR efficacy in vivo than the equivalent morphinones (5).9 In the in vitro functional assays (Tables 2, 3), the cinnamoyloxymorphinones (6b, 6c) were very much more potent as MOR antagonists than the equivalent codeinones (7b, 7c). This contrasts with the very small difference in potency between the cinnamoylamino morphinone (C-CAM) and equivalent codeinone (MC-CAM) (Table 3).10
It is of interest to compare the activity of 14-cinnamoylnaltrexone (6a) with the phenylpropyl ether (2a) which is structurally similar in having a 3-carbon chain linking the side chain aromatic ring to the C14-oxygen atom. The ether (2a) in vivo gave a full response in a battery of thermal antinociceptive assays with potency up to 400 times greater than morphine.7 In comparison the cinnamoyl ester has much more modest in vitro and in vivo MOR agonist activity. It must be assumed that the relative conformational restraint of the α,β-unsaturated cinnamoyl ester prevents an optimum interaction with MOR in the preferred agonist conformation.
Conclusions
The 14-O-cinnamoyl esters of naltrexone have predominant opioid receptor antagonist activity both in vitro and in vivo. In this regard they are similar to the equivalent 14-N-cinnamoylamino derivatives, but the latter are more potent antagonists of longer duration. Additionally the naltrexone esters (6) have similar in vivo and in vitro MOR efficacy to the corresponding codeinones (7) whereas the codeinone amides (4) have substantially higher MOR efficacy than the morphinones (5). These differences are less significant than the difference between 14-cinnamoylnaltrexone (6a) and 14-O-phenylpropylnaltrexone (2a). The greater side chain conformational freedom of the latter allows it to display very high potency in vivo MOR agonist activity.
Experimental
Column chromatography was performed under gravity, over silica gel 60 (35-70μm) purchased from Merck. Analytical TLC was performed using aluminium-backed plates coated with Kieselgel 60 F254, from Merck. The chromatograms were visualised using either UV light (UVGL-58, short wavelength), ninhydrin (acidic) or potassium permanganate (basic). Melting points were carried out using a Reichert-Jung Thermo Galen Kopfler block or a Gallenkamp MFB-595 melting point apparatus and are uncorrected. High and low resolution electron impact (EI) mass spectra were recorded using EI ionisation at 70eV, on a VG AutoSpec instrument, equipped with a Fisons autosampler. 1H NMR and 13C NMR spectra were recorded using a JEOL 270 (operating at 270 MHz for 1H and 67.8 MHz for 13C) spectrometer. Chemical shifts (δ) are measured in ppm. Spectra were referenced internally using TMS as the standard. Only diagnostic peaks have been quoted for proton NMR. Microanalysis was performed with a Perkin-Elmer 240C analyser. Chemicals and solvents were purchased from Aldrich chemical company. Compounds were submitted for testing as their oxalate salts, formed by adding one equivalent of oxalic acid to an ethanolic solution of the ligand.
3-O-(tert-Butyldimethylsilyl)-14β-cinnamoyloxy-N-cyclopropylmethyl-7,8-dihydronormorphinone (9a)
A solution of 8b (593 mg:1.3 mmol) and cinnamoyl anhydride (830 mg: 3.0 mmol) in dry toluene (12 mL) was heated to reflux for 3 h. After cooling, the reaction mixture was washed with sodium bicarbonate solution (2 × 5 mL) and water (5 mL), dried over magnesium sulphate and the solvent removed in vacuo. The residue was purified by silica gel chromatography (CH2Cl2:MeOH, 49:1) to give (9a) (269 mg: 44%); EIMS m/z 585 (M+); HRMS (EI) m/z 585.2925 (M+) C35H43NO5Si requires 585.2910; 1H NMR 0.07 (2H, m), 0.19 (3H, s), 0.28 (3H, s), 0.44 (2H, m), 0.74 (1H, m), 1.00 (9H, s), 4.69 (1H, s), 6.57 (1H, d), 6.59 (1H, d), 6.66 (1H, d), 7.70 (1H, d); 13C NMR δ −4.64, −4.48, 3.75, 4.00, 9.49, 18.29, 23.24, 25.75, 26.99, 30.45, 35.75, 43.97, 51.08, 55.46, 59.31, 82.77, 89.95, 119.31, 119.37, 122.55, 126.39, 128.20, 128.71, 130.39, 134.39, 138.01, 144.01, 146.80, 165.85, 207.22.
14β-Cinnamoyloxy-N-cyclopropylmethyl-7,8-dihydronormorphinone (6a)
A solution of (9a) 140 mg: 0.24 mmol) and potassium fluoride (35 mg: 0.60 mmol) in MeOH (11 mL) and CH2Cl2 (1 mL) was stirred for 1 h at ambient temperature. Solvent evaporation gave a residue that was purified by silica gel column chromatography (CH2Cl2:MeOH, 49:1) to give 6a as a white foam (57%); EIMS m/z 505 (M+); 1H NMR δ 0.05 (2H, m), 0.43 (2H, m), 0.76 (1H, m), 4.83 (1H, s), 6.58 (1H, d), 6.62 (1H, d), 6.80 (1H, d), 7.38 (3H, m), 7.56 (2H, m), 7.72 (1H, d); 13C NMR δ 3.71, 4.00, 9.46, 23.18, 27.11, 30.19, 35.78, 44.00, 51.43, 55.56, 59.31, 82.77, 90.26, 118.29, 119.28, 120.07, 125.18, 128.39, 128.93, 129.31, 130.39, 134.39, 138.90, 143.53, 144.74, 165.88, 209.22; Anal. (C29H29NO5.(CO2H)2.2H2O) CHN.
3-O-(tert-Butyldimethylsilyl)-14β-(4-chlorocinnamoyloxy)-N-cyclopropylmethyl-7,8-dihydronormorphinone (9b)
8b (429 mg: 0.94 mmol) and 4-chlorocinnamoyl anhydride (616 mg: 1.77 mmol) were treated as for (9a) to yield (9b) as a clear oil (228 mg: 21%); EIMS m/z 619 (M+); HRMS (EI) m/z 619.2537 (M+), C35H42NO5Si requires 619.2521; 1H NMR δ 0.04 (2H, m), 0.19 (3H, s), 0.27 (3H, s), 0.44 (2H, m), 0.72 (1H, m), 1.02 (9H, s), 4.67 (1H, s), 6.55 (1H, d), 6.56 (1H, d), 6.66 ((1H, d), 7.28 (2H,d), 7.52 (2H, d), 7.65 (1H, d); 13C NMR d −4.70, −4.54, 3.68, 3.94, 9.46, 18.25, 23.21, 25.68, 26.92, 30.45, 35.68, 43.94, 51.02, 55.43, 59.27, 82.96, 89.53, 119.34, 119.85, 122.55, 126.32, 128.61, 129.18, 129.31, 132.86, 136.23, 137.98, 143.12, 146.72, 165.53, 207.06.
14β-(4-Chlorocinnamoyloxy)-N-cyclopropylmethyl-7,8-dihydronormorphinone (6b)
(9b) was treated with KF as described for 6a to yield 6b as a white foam (77%); EIMS m/z 505 (M+); 1H NMR δ 0.04 (2H, m), 0.45 (2H, m), 0.74 (1H, m), 4.80 (1H, s), 6.57 ((1H, d), 6.62 (1H, d), 6.76 (1H, d), 7.36 (2H, d), 7.50 (2H, d), 7.64 (1H, d); 13C NMR δ 3.68, 3.97, 9.43, 23.11, 27.08, 30.19, 35.75, 43.97, 51.40, 55.46, 59.27, 82.93, 90.19, 118.26, 119.82, 120.01, 125.15, 128.16, 129.18, 129.31, 132.86, 136.23, 138.83, 143.21, 143.50, 165.56, 209.09; Anal. (C29H28NO5.(CO2H)2.2.5H2O) CHN.
3-O-(tert-Butyldimethylsilyl)-14β-(4-methylcinnamoyloxy)-N-cyclopropylmethyl-7,8-dihydronormorphinone (9c)
8b (584 mg: 1.28 mmol) and 4-methylcinnamoyl anhydride (690 mg: 2.25 mmol) were treated as for 9a to yield 9c as a clear oil (424 mg: 55%); EIMS m/z 599 (M+); HRMS (EI) m/z 599.3088, C36H45NO5Si requires 599.3067; 1H NMR δ 0.05 (2H, m), 0.21 (3H, s), 0.29 (3H, s), 0.44 (2H, m), 0.75 (1H, m), 0.99 (9H, s), 2.38 (3H, s), 4.67 (1H, s), 6.53 (1H, d), 6.56 (1H, d), 6.65 (1H, d), 7.19 (2H,d), 7.48 (2H, d), 7.68 (1H, d); 13C NMR δ −4.70, −4.51, 3.71, 4.48, 9.46, 18.25, 21.46, 23.24, 25.21, 30.70, 32.73, 35.68, 43.94, 51.05, 55.53, 59.30, 82.57, 89.56, 118.16, 119.31, 122.51, 126.36, 127.69, 128.61, 129.78, 137.98, 140.36, 143.34, 144.56, 146.77, 165.98, 207.09.
14β-(4-Methylcinnamoyloxy)-N-cyclopropylmethyl-7,8-dihydronormorphinone (6c)
9c was treated with KF as for 9a to yield 6c as an oil (99%); EIMS m/z 485 (M+); 1H NMR δ 0.04 (2H, m), 0.44 (2H, m), 0.72 (1H, m), 4.77 (1H, s), 6.53 ((1H, d), 6.60 (1H, d), 6.75 (1H, d), 7.15 (2H, d), 7.48 (2H, d), 7.70 (1H, d); 13C NMR δ 3.62, 3.90, 9.49, 21.46, 24.70, 26.32, 30.99, 32.79, 43.97, 51.46, 55.59, 59.34, 82.61, 90.29, 118.23, 120.01, 125.24, 127.94, 128.23, 129.66, 131.66, 132.04, 138.90, 140.80, 143.53, 144.71, 166.61, 209.06; Anal. (C30H31NO5.(CO2H)2.2H2O) CHN.
N-Cyclopropylmethyl-14β-cinnamoyloxy-7,8-dihydrocodeinone (7a)
A solution of 8a (500 mg, 1.41 mmol) in anhydrous toluene (80 ml) was treated with cinnamoyl anhydride (512 mg, 1.84 mmol) and the resulting mixture heated to reflux and stirred overnight. On cooling, the solution was washed with Na2CO3 solution (2 × 20 ml) and water (20 ml), dried over MgSO4 and evaporated to dryness. Silica gel chromatography (CH2Cl2:MeOH:NH3, 198:1:1) gave 7a as a white solid (303 mg: 44%); ESMS m/z 486 (MH+); HRMS (ES) m/z 486.2259 (MH+), C30H32NO5 requires 486.2275; 1H NMR δ −0.01-0.08 (2H, m), 0.39-0.48 (2H, m), 0.69-0.77 (1H, m), 1.57-1.61 (1H, m), 1.69 (1H, dt), 2.13-2.20 (1H, m), 2.25-2.38 (3H, m), 2.53 (1H, dd), 2.60-2.75 (3H, m), 2.92-2.97 (1H, m), 3.11 (1H, d), 3.89 (3H, s), 4.58 (1H, d), 4.75 (1H, s), 6.57 (1H, d), 6.64 (1H, d), 6.71 (1H, d), 7.37-7.43 (3H, m), 7.56-7.60 (2H, m), 7.70 (1H, d); 13C NMR δ 3.68, 3.90, 9.45, 23.06, 27.19, 30.26, 35.78, 43.93, 51.27, 55.43, 56.73, 59.29, 82.69, 90.13, 114.79, 119.22, 119.51, 125.88, 128.14, 128.64, 128.90, 130.35, 134.33, 142.92, 144.62, 144.88, 165.74, 207.54; m.p. (oxalate) 124-126°C; Anal. (C30H31NO5.(CO2H)2.0.5H2O) CHN.
N-Cyclopropylmethyl-14β-4′-chlorocinnamoyloxy-7,8-dihydrocodeinone (7b)
8a (310 mg: 0.87 mmol) in anhydrous toluene (50 mL) was added to 4-chlorocinnamoyl anhydride (400 mg: 1.16 mmol) as described for 7a and give 7b as a white solid (110 mg: 24%); EIMS m/z 519 (M+); HRMS (EI) m/z 519.1822 (M+), C30H30NO5Cl requires 519.1813; 1H NMR δ 0.01-0.12 (2H, m), 0.37-0.52 (2H, m), 0.67-0.82 (1H, m), 2.54 (1H, dd), 3.13 (1H, d), 3.90 (3H, s), 4.58 (1H, d), 4.75 (1H, s), 6.56 (1H, d), 6.66 (1H, d), 6.73 (1H, d), 7.38 (2H, m), 7.52 (2H, m), 7.66 (1H, d); 13C NMR δ 3.62, 3.88, 9.40, 23.04, 27.11, 30.23, 35.67, 43.89, 51.17, 55.39, 56.72, 59.22, 82.83, 90.02, 114.88, 119.49, 119.76, 125.78, 128.55, 129.12, 129.25, 132.79, 136.17, 142.88, 143.10, 144.84, 165.42, 207.33; m.p. (oxalate) 122-124°C; Anal. (C30H30NO5Cl.(CO2H)2.0.5H2O) CHN.
N-Cyclopropylmethyl-14β-4′-methylcinnamoyloxy-7,8-dihydrocodeinone (7c)
Using the same procedure as for 7b but with 4-methylcinnamoyl anhydride gave 7c (30%); EIMS m/z 499 (M+); HRMS (EI) m/z 499.2361 (M+), C31H33NO5 requires 499.2359; 1H NMR δ 0.06-0.13 (2H, m), 0.39-0.53 (2H, m), 0.69-0.82 (2H, m), 2.40 (3H, s), 2.55 (1H, dd), 3.13 (1H, d), 3.92 (3H, s), 4.60 (1H, d), 4.77 (1H, s), 6.55 (1H, d), 6.66 (1H, d), 6.74 (1H, d), 7.23 (2H, m), 7.49 (2H, m), 7.70 (1H, d); 13C NMR δ 3.65, 3.85, 9.42, 21.43, 23.05, 27.16, 30.23, 35.73, 43.90, 51.22, 55.46, 56.73, 59.26, 82.50, 90.09, 114.86, 118.07, 119.47, 125.89, 128.09, 128.66, 129.58, 131.57, 140.74, 142.87, 144.57, 144.87, 165.89, 207.49; m.p. (oxalate) 126-128°C; Anal. (C31H33NO5.(CO2H)2.0.5H2O) CHN.
Supplementary Material
Structures.

Acknowledgements
This work was funded through NIDA Grants DA00254 and DA07315 and the in vitro characterisation of compounds carried out through the NIDA Abuse Treatment Discovery Program (ATDP).
Abbreviations
- TW
tail withdrawal from warm water
- AW
acetic acid induced writhing
Footnotes
Supporting Information Available: Full experimental details and spectroscopic data (IR, 1H NMR, 13C NMR, mass spectra and microanalysis data), including biological assay methods. This method is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Kreek MJ. In: Handbook of Experimental Pharmacology. Schuster CR, Kuhar MJ, editors. Vol. 118. Springer; Berlin: 1996. p. 563. [Google Scholar]
- 2.Pettinati HM, O’Brien CP, Rabinowitz AR, Wortman SP, Oslin DW, Kampman KM, Dackis CA. The status of naltrexone in the treatment of alcohol dependence: specific effects on heavy drinking. Journal of Clinical Psychopharmacology. 2006;26:610–625. doi: 10.1097/01.jcp.0000245566.52401.20. [DOI] [PubMed] [Google Scholar]
- 3.Sporer KA. Acute heroin overdose. Annals of internal medicine. 1999;130:84–90. doi: 10.7326/0003-4819-130-7-199904060-00019. [DOI] [PubMed] [Google Scholar]
- 4.Kobylecki RJ, Carling RW, Lord JAH, Smith CFC, Lane AC. Common anionic receptor site hypothesis: Its relevance to the antagonist action of naloxone. J. Med. Chem. 1982;25:116–120. doi: 10.1021/jm00344a005. [DOI] [PubMed] [Google Scholar]
- 5.Schullner F, Meditz R, Krassnig R, Morandell G, Kalinin VN, Sandler E, Spetea M, White A, Schmidhammer H, Berzetei-Gurske I. Synthesis and biological evaluation of 14-alkoxymorphinans. 19. Effect of 14-O-benzylation on the opioid receptor affinity and antagonist potency of naltrexone. Helv. Chim. Acta. 2003;86:2335–2341. [Google Scholar]
- 6.Greiner E, Spetea M, Krassnig R, Schuller F, Aceto MD, Harris LS, Traynor JR, Woods JH, Coop A, Schmidhammer H. Synthesis and biological evaluation of 14-alkoxymorphinans. 18. N-substituted 14-phenylpropyloxymorphinan-6-ones with unanticipated agonist properties: extending the scope of common structure-activity relationships. J. Med. Chem. 2003;46:1758–1763. doi: 10.1021/jm021118o. [DOI] [PubMed] [Google Scholar]
- 7.Aceto MD, Bowmen ER, May EL, Woods JH, Smith CB, Medzihradsky F, Jacobson AE. Very long-acting narcotic antagonists: the 14β-p-substituted cinnamoylaminomorphinones and their partial mu agonist codeinone relatives. Arzneim-Forsch. Drug Res. 1989;39:570–575. [PubMed] [Google Scholar]
- 8.Broadbear JH, Sumpter TL, Burke TF, Husbands SM, Lewis JW, Woods JH, Traynor JR. Methcinnamox is a potent, long-lasting and selective antagonist of morphine-mediated antinociception in the mouse: comparison with clocinnamox, β-FNA and β-chlornaltrexamine. J. Pharmacol. Exp. Ther. 2000;294:933–940. [PubMed] [Google Scholar]
- 9.Nieland NPR, Moynihan H, Carrington S, Broadbear J, Woods JH, Traynor JR, Husbands SM, Lewis JW. Structural determinants of opioid activity in derivatives of 14-aminomorphinones: effect of substitution in the aromatic ring of cinnamoylaminomorphinones and codeinones. J. Med. Chem. 2006;49:5333–5338. doi: 10.1021/jm0604777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rennison D, Moynihan H, Traynor JR, Lewis JW, Husbands SM. Structural determinants of opioid activity in derivatives of 14-aminomorphinones: effects of changes to the C14-amino to aryl ring linker chain. J. Med. Chem. 2006;49:6104–6110. doi: 10.1021/jm060595u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Armesto N, Ferrero M, Fernández S, Gotor V. Novel enzymatic synthesis of 4-o-cinnamoyl quinic and shikimic acid derivatives. J. Org. Chem. 2003;68:5784–5787. doi: 10.1021/jo034387a. [DOI] [PubMed] [Google Scholar]
- 12.Nagase H, Abe A, Portoghese PS. The facility of formation of a .Δ6 bond in dihydromorphinone and related opiates. J.Org. Chem. 1989;54:4120–4125. [Google Scholar]
- 13.Toll L, Berzetei-Gurske IP, Polgar WE, Brandt SR, Adapa ID, Rodriguez L, Schwartz RW, Haggart D, O’Brian A, White A, Kennedy JM, Craymer K, Farrington L, Auh JS. Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Research Monograph. 1998;178:440–466. [PubMed] [Google Scholar]
- 14.Zaveri N, Polgar WE, Olsen CM, Kelson AB, Grundt P, Lewis JW, Toll L. Characterization of opiates, neuroleptics, and synthetic analogs at ORL1 and opioid receptors. Eur J Pharmacol. 2001;428:29–36. doi: 10.1016/s0014-2999(01)01282-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hambrook JM, Rance MJ. The interaction of buprenorphine with the opiate receptor. In: Kosterlitz HW, editor. Opiates and Endogenous Opioid Peptides. Amsterdam; Elsevier: pp. 295–301. [Google Scholar]
- 16.Zernig G, Butelman ER, Lewis JW, Walker EA, Woods JH. In vivo determination of mu opioid receptor turnover in rhesus monkeys after irreversible blockade with clocinnamox. J. Pharmacol. Exp. Ther. 1994;269:57–65. [PubMed] [Google Scholar]
- 17.Sebastian A, Bidlack JM, Jiang Q, Deecher D, Teitler M, Glick SD, Archer S. 14β-[(p-Nitrocinnamoyl)amino]morphinones, 14β-[(p-Nitrocinnamoyl)amino]-7,8-dihydromorphinones and their codeinone analogues: synthesis and receptor activity. J. Med. Chem. 1993;36:3154–3160. doi: 10.1021/jm00073a015. [DOI] [PubMed] [Google Scholar]
- 18.Husbands SM, Lewis JW. Opioid ligands having delayed long-term antagonist activity: Potential pharmacotherapies for opioid abuse. Mini-Reviews in Med. Chem. 2003;3:137–144. doi: 10.2174/1389557033405395. [DOI] [PubMed] [Google Scholar]
- 19.Spagnolo B, Calo G, Polgar WE, Jiang F, Olsen CM, Berzatei-Gurske I, Khroyan TV, Husbands SM, Lewis JW, Toll L, Zaveri NT. Activities of mixed NOP and □-opioid receptor ligands. Br, J. Pharmacol. 2008;153:609–619. doi: 10.1038/sj.bjp.0707598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.