

Abstract
Cell-based cancer immunotherapy represents a new and powerful weapon in the arsenal of anticancer treatments. Non-invasive monitoring of the disposition, migration and destination of therapeutic cells will facilitate the development of cell based therapy. The therapeutic cells can be modified intrinsically by a reporter gene or labeled extrinsically by introducing imaging probes into the cells or on the cell surface before transplant. Various advanced non-invasive molecular imaging techniques are playing important roles in optimizing cellular therapy by tracking cells and monitoring the therapeutic effects of transplanted cells in vivo. This review will summarize the application of multiple molecular imaging modalities in cell-based cancer immunotherapy.
1. Introduction
1.1 Cell-based cancer immunotherapy
Cancer is one of the main causes of death worldwide (http://www.cdc.gov), imposing tremendous emotional and financial burdens. Cancer treatment remains an immense challenge despite significant advances in chemotherapy and radiotherapy. Especially, the profound side effects and drug resistance make a potentially lethal combination. Therefore, alternative therapy regimens are under development for better disease control.1 One appealing strategy is cell-based cancer immunotherapy, which is designed to harness the patients’ own immune system to induce a potent anti-tumor response. By doing so, various therapeutic cell types, such as lymphocytes (CD4+ and CD8+ T cells), dendritic cells (DCs) and natural killer cells (NK cells), are locally or systemically administrated to cancer patients after ex vivo manipulation. This strategy results in a highly selective toxic effect directly against cancer cells with significantly less side effects on normal cells.2–6 Over the last decade, advances in the manipulation of immunocompetent cells have spurred the development of exciting new immunotherapeutic strategies. The promising role of cell-based cancer immunotherapy is reflected by the constantly growing number of clinical trials adopting therapeutic cells.7
However, cell-based immunotherapy is still in the early stages of development.6–8 Our current knowledge on the biodistribution and pharmacokinetics of the therapeutic cells relies mainly on the ex vivo examinations of tissue samples obtained by biopsy/autopsy. The mechanisms underlying migration and the role therapeutic cells play in therapy have not been fully elucidated. Thus, it is critical to develop sensitive and reproducible non-invasive methods for real-time assessment of the therapeutic process for the refinement of current cell-based cancer immunotherapy.9
1.2 Molecular imaging
Molecular imaging is defined as the visualization, characterization, and measurement of biologic processes at the cellular/molecular level.10 Driven by the advances in biology, chemistry, nanotechnology and imaging techniques, molecular imaging has grown rapidly in the last 10 years and has now becomes one of the key components of 21st-century disease management.11–14 It enables non-invasive visualization of cellular function and the follow-up of the molecular process in living organisms with the help of advanced probes. The multiple and numerous possibilities of this field are applicable to earlier and more precise diagnosis of diseases as well as to improve treatment by optimizing pre-clinical and clinical tests of new medication.11,15
Molecular imaging is not restricted to any given imaging tool. For example, nuclear imaging techniques, optical imaging, and magnetic resonance imaging (MRI) are all adapted for molecular imaging.11 As summarized in Table 1, each imaging modality has its own advantages and disadvantages in terms of sensitivity, spatial resolution, and tissue penetration depth.13,16,17 To echo each imaging modality, various imaging probes with molecular specificity (targeted/activatable probes) have been designed and synthesized.18–21 The ideal imaging probes should exhibit favorable kinetics, metabolic stability and biocompatibility, while the optimal readout technique should have a minimal background signal and allow for signal amplification to decrease the detection limit. Currently, multifunctional probes are also under development to overcome the limitations of sensitivity and resolution that a single imaging technique may have.22–24
Table 1.
| Modality | Quantification | Depth | Spatial resolution | Relative sensitivity | Assessment of cell viability/function | Cellular tracking in humans |
|---|---|---|---|---|---|---|
| PET | Yes | No limit | 1–2 mm | High | Yes | Yes |
| PETCT | Yes | No limit | 0.5–1 mm | High | Yes | Yes |
| MRI | Yes | No limit | 25–100 μm | Low | No | Yes |
| Optical imaging | No | mm | 1–3 mm | High | Yes | No |
1.3 Molecular imaging in cell-based immunotherapy
Successful application of cell-based approaches in clinical therapies requires techniques to monitor transplanted cell survival and integration non-invasively and dynamically with a high temporal and spatial resolution.25 Consequently, molecular imaging has been extensively adapted to study cell-based cancer immunotherapy. In order to facilitate effective tracking of the movement and function by imaging methods, the therapeutic cells must be pre-labeled with a probe either by intrinsic or extrinsic cell labeling. Intrinsic cell labeling is usually achieved by ex vivo transduction of therapeutic cells with imaging reporter genes.26 This approach allows for reliable, stable, and harmless visualization of cellular trafficking, proliferation, and function at the target site. It could provide a general solution for long-term monitoring of tailored therapeutic cells. However, genetic modification of therapeutic cells without substantially affecting their characteristics could be a challenge. In addition, the sensitivity of this technique is determined by the reporter probe’s pharmacokinetics and by the reporter gene expression level in the transfected cells. For extrinsic cell labeling, imaging probes with radioactive, fluorescent or paramagnetic properties can be incorporated into cells according to standardized protocols.27,28 These methods are primarily a short-term monitoring technique due to probe washout or dilution from cell division. In the following sections, we will summarize the application of molecular imaging in cell-based cancer immunotherapy, categorized by commonly used imaging modalities (Scheme 1).
Scheme 1.

Schematic diagram of molecular imaging of cell-based cancer immunotherapy.
2. Nuclear imaging
Nuclear imaging techniques, including positron emission tomography (PET) and single photon emission computed tomography (SPECT), are the primary choices for molecular imaging because of their inherent high sensitivity.18,29 SPECT uses low energy γ-emitting isotopes (e.g. 123I,111In, and 99mTc) and is relatively inexpensive and widely available. For imaging purposes, a target-to-nontarget ratio of >1.5 is sufficient for SPECT, whereas planar imaging requires much higher ratios. PET imaging is based on annihilation coincidence detection after labeling imaging probes with a positron-emitting radionuclide. Compared with SPECT, PET has the advantages of higher sensitivity and versatility. Moreover, it is also capable of scatter correction. PET imaging leads to high acquisition efficiency and results in high-quality three-dimensional images or maps of functional processes within the body. The major advantage of SPECT imaging is that it can be used for simultaneous imaging of multiple radio-nuclides emitting γ-rays with different energies. Thus, SPECT can potentially allow simultaneous detection of multiple biologic events with multiple isotopes, which is not possible with PET. However, compared with PET, SPECT has lower sensitivity and spatial resolution. Up to now, both imaging methods have been widely used in the field of cellular imaging.
2.1 Intrinsic cell labeling for nuclear imaging
Several reporter genes have been used for radionuclide imaging such as thymidine kinase (tk), an enzyme that biochemically modifies the probe;30,31 dopamine D2 receptor, a cell surface receptor that specifically binds to the probe32 and sodium iodide symporter (NIS), a cell membrane associated transporter that transports the probe across the cell membrane.33,34 The specific interaction between reporter gene encoded products and the administered probes generates a signal accumulation that can be detected by either PET or SPECT. Among them, the herpes simplex virus type 1 thymidine kinase (HSV1-tk) and the mutated gene HSV1-sr39tk are the most commonly used reporter genes for PET imaging.35 These enzymes phosphorylate a wide range of radiolabled substrates including pyrimidine (uracil-based FIAU, FEAU, FFEAU) and purine [acycloguanosine-based penciclovir (PCV), FHBG, FHPG)] nucleoside analogs. After phosphorylation, these compounds are unable to cross the cell membrane and the intracellularly trapped radioactivity is then directly related to the local expression of the reporter genes.36,37
T lymphocytes are important components of the immune response and play important roles in the elimination of cancer cells from the body. After transducing the cells to express HSV1-tk reporter gene, Koehne et al.38,39 demonstrated the feasibility of long-term in vivo tracking of adoptively transferred antigen-specific T cells with PET imaging. After adoptive transfer, HSV-tk+ T cells labeled with 124I-FIAU/131I-FIAU can be noninvasively tracked in tumor-bearing mice by scintigraphy and serial PET images. These T cells selectively accumulate in EBV+ tumors expressing the T cells’ restricting HLA allele. The concentrations of transduced T cells detected in tumors and tissues are closely correlated with the concentrations of label retained at each site. Moreover, 131I labeled T cells can eliminate the targeted tumors selectively due to β emission.
Quantitative PET provides a potential opportunity to determine the number of therapeutic cells at the target site. In 2004, Su et al.40 determined the correlation between PET signal and cell number and characterized the cellular limit of detection for PET. By PET imaging with 18F-FHBG, they observed a cell number-dependent signal using native mouse T cells transduced with the HSV1-sr39tk reporter gene. Further study demonstrated that early accumulation and apparent proliferation of memory T cells at the antigen-positive tumor could be visualized and quantified by PET imaging.41 Recently, Shu et al.42 also demonstrated that PET can monitor the distribution of self-antigen-specific T cells engineered to express HSV1-sr39tk. PET imaging using 18F-FHBG enabled the detection of transferred T cells in secondary lymphoid organs of recipient mice over a 3-week period. The lower limit of detection was approximately 7 × 105 T cells in the spleen and 1 × 104 T cells in lymph nodes (LNs). However, quantification of transplanted T cells in the tumor was hampered by the sr39tk-independent trapping of 18F-FHBG within the tumor architecture.
All these studies supported the feasibility of using PET to visualize the expansion, homing and persistence of transferred T cells. Indeed, in a recent clinical trial, Yaghoubi et al.43 reported successful utilization of genetically modified CD8 + T-cells carrying IL-13 zetakine and HSV1-tk genes in a patient with glioblastoma multiforme. The distribution of transferred cytolytic T-cells in the tumor as well as other parts of the body can be detected by PET scanning using 18F-FHBG. However, the immune reaction of the viral protein elicited in humans may limit the translation of HSV-tk reporter genes into a clinical setting.
2.2 Extrinsic cell labeling for nuclear imaging
One of the major advantages of radionuclide methods used in cell labeling is that the small probe mass and labeling strategies do not significantly perturb the biological processes under study.44 For scintigraphy and SPECT imaging, several lipophilic compounds such as 111In-Oxyquinoline (Oxine) and 99mTc-exametazime (Ceretec) are most commonly employed for in vitro leukocyte labeling and have found widespread clinical applications in monitoring cellular immunotherapy.45 For example, scintigraphic imaging of the biodistribution patterns of dendritic cells in mice was demonstrated by Kupiec-Weglinski et al.46 using 111In-labeled splenic DCs. Suda et al.47 reported the non-invasive tracking of 111In-labeled DCs using a gamma camera. In a study published by Fisher et al.,48 patients with metastatic malignant melanoma were treated with 111In-labeled tumor-infiltrating lymphocytes, followed by serial whole-body gamma camera imaging and serial biopsies of tumor and normal tissue. It was demonstrated that 111In-labeled lymphocytes can localize preferentially to tumors, providing information about the possible therapeutic mechanism of tumor-infiltrating lymphocytes. Blocklet et al.49 labeled autologous monocyte-derived DCs with 111In-oxine and injected the therapeutic cells into patients with various cancer types. The imaging results showed that transferred DCs rapidly moved to a regional lymph node in which antigen presentation should occur.
FDG is a model PET radiopharmaceutical and has been lauded as the “molecule of the century” in nuclear medicine.50 As a glucose analog, FDG enters the cell through specific glucose transporters on the cell membrane and is converted to deoxyglucose 6-phosphate, which is trapped in the cell in proportion to glucose metabolism.51 The magnitude of FDG uptake in certain tumors relates quite directly to the number of viable cells. Thus, FDG-PET imaging provides high specificity and sensitivity to several kinds of cancer with many clinical applications of patients with malignant diseases. 18F-FDG has also been investigated as a marker to label mesenchymal stem cells for in vivo PET imaging.52 However, the labeling efficiency of 18F-FDG varies from one cell type to another and is typically lower than what can be achieved with 111In-Oxine.53 Other PET tracers are thus being developed as alternatives to FDG. Olasz et al.54 labeled bone marrow-derived DCs with N-succinimidyl 4-[18F]-fluorobenzoate (FSB) and tracked the cell migration from the footpad to the draining popliteal lymph node over the course of nearly 4 h in mice. Radu et al.55 demonstrated that 1-(2′-deoxy-2′-18F-fluoroarabinofuranosyl) cytosine (18F-FAC) could image activation-specific upregulation of the deoxyribonucleotide salvage pathway in lymphoid cells by PET (Fig. 1). The results demonstrated that 18F-FAC enabled visualization of lymphoid organs, specifically sensitive to localized immune activation, in a mouse model of antitumor immunity. 64Cu-pyruvaldehyde-bis-(N4-methylthiosemicarbazone) (64Cu-PTSM) also has been used to radiolabel cells. It has been reported that 64Cu-PTSM labeling efficiency and cell viability were comparable or superior to 111In-Oxine.53 Adonai et al. labeled C6 rat glioma cells with 64Cu-PTSM and intravenously injected the labeled cells in mice. The images indicated that tail-vein-injected labeled C6 cells traffic to the lungs and liver. In addition, transient splenic accumulation of radioactivity was clearly detectable in a mouse scanned at 3.33 h postinfusion of 64Cu-PTSM-labeled lymphocytes.56
Fig. 1.

Noninvasive PET monitoring of immune cell activation using a novel radiotracer 1-(2′-deoxy-2′-18F-fluoroarabinofuranosyl) cytosine (18F-FAC). Images are 1 mm coronal sections from PET/CT scans using 18F-FAC on day-1 (A) and day 15 (B) after oncoretrovirus injection. At the peak of immune response (day 15), increased 18F-FAC retention is observed in lymph nodes and spleen (C). %ID/g = percentage injected dose per gram; B = bone; BL = bladder; GI = gastrointestine; LN = lymph node; SP = spleen; TU = tumor. Reproduced from ref. 55 with permission.
Direct cell labeling with radionuclides can only determine short-term circulation and homing properties of infused therapeutic cells because the imaging signal decreases with radioactive decay, instability of labeling and biological clearance.56,57 With current labeling techniques, relatively low level of radioactivity per cell can be attained and it is difficult to evaluate the proliferation of labeled cells in the body. In addition, PET and SPECT have limited spatial resolution, so they are unable to precisely localize the site of increased tracer uptake.
3. MRI
MRI has been developed into one of the most powerful imaging tools in radiology and biomedical sciences.58–60 The fundamental principle underlying MRI is that unpaired nuclear spins (i.e. hydrogen nuclei of water and nuclei with similar chemical shifts) align themselves when placed into a magnetic field.61 MRI detects the interaction of protons (or certain other nuclei) with each other and with the surrounding molecules in a tissue of interest.62 Different tissues have different relaxation times which can result in endogenous MR contrast. Among the different imaging approaches, MRI is particularly attractive due to its superior spatial resolution and the harmlessness of the magnetic field used. Its exceptional spatial resolution is at least one order of magnitude higher than nuclear and optical imaging, allowing morphological images without in-depth limitations even on clinical machines. Several anatomical, physiological and metabolic parameters can be measured almost simultaneously with multiple MRI methods. To date, MRI has also been widely exploited for molecular/cellular imaging. The non-invasive acquisition of both temporal changes of transplanted cell location and high-resolution anatomy is of great interest in the field of cell-based cancer immunotherapy.
3.1 Intrinsic cell labeling for MRI
The primary requirement of an MR reporter gene is to produce a signal or to provide contrast that can be distinguished from the surrounding tissues by MRI. Several MRI based reporter genes have been investigated.63–67 For example, Louie et al.63 used a gadolinium-based probe to label Xenopus embryos and monitor beta-galactosidase activity. The probe contains a galactose group that hinders the inner sphere relaxation enhancement effect of the paramagnetic chelated gadolinium. In the presence of lacZ-transfected cells expressing beta-galactosidase, the galactose is cleaved off, leading to an increase in water molecules diffusing towards the inner gadolinium atom. This increase leads to an inner sphere relaxation enhancement and amplified contrast in T1 weighted MR images. Nonmetallic and biodegradable artificial MRI reporter genes encoding a lysine-rich protein have also been reported.65,68 Based on the rapid transfer of an amide to a water proton in the lysine-rich protein, this probe can produce chemical-exchange saturation transfer contrast in solution and thus reduce MRI signal intensity. Combining overexpression of transferrin receptor with transferrin-attached iron oxide nanoparticles led to detectable intracellular iron accumulation and corresponding decrease in T2 signals in MRI.64 This method results in improved sensitivity. Liu et al.66 also established transgenic mouse ES (mES) cell lines that carry a human ferritin heavy chain as a reporter gene and succeeded in monitoring the transferred cells in vivo using MRI.
However, all these MRI based reporter genes including transferrin have not been widely used for MRI cell tracking due to several inherent shortcomings. Compared with the radio-nuclide reporter gene method, the sensitivity of MRI based reporter genes is much lower and it is difficult to achieve distinguishable contrast, especially in vivo. For transferrin reporter genes, it takes a relatively long time period to accumulate sufficient amount of iron that lead to detectable contrast and sufficient endogenous iron needs to be present. Also, the sensitivity of detection of MRI is lower at clinical field strengths. Active collaborations between the fields of molecular biology and MRI are needed to further improve the design of MRI reporter genes, such as identification of the optimized mutants with increased transverse relaxivity and larger monitoring window.
3.2 Extrinsic cell labeling for MRI
The most widely used MR contrast agents in cellular MR imaging are superparamagnetic iron oxide nanoparticles (SPIOs). As MRI contrast agents, SPIOs have many advantages:11,59,60,69,70 (1) they can be biologically degraded, metabolized and integrated into serum Fe pool to form hemoglobin or to enter other metabolic processes; (2) they provide the most change in signal per unit of metal, in particular to T2*-weighted images, allowing small numbers of SPIO-loaded cells to be detected in MR images in vivo; (3) they can be magnetically manipulated to change their magnetic properties, which opens new opportunities such as hyperthermia treatment for cancer induced by a high-frequency magnetic field; (4) they have a large surface area for functional modification; (5) they are easily detectable by histology (Perl’s iron staining) or electronic microscopy for cell labeling. For all these reasons, there is an ever increasing interest in using SPIOs to label transplanted cells to monitor their temporal and spatial migration in vivo by MRI.
Generally, the cellular labeling efficiency of SPIOs depends on a number of factors, including cell type, size and surface properties of SPIO.71 SPIOs are usually encapsulated by dextran or poly(ethylene glycol) (PEG) for improved biocompatibility, reduced non-specific protein binding and prolonged circulation in biological systems. To enhance the labeling efficiency of SPIOs, complexing of polycationic transfection agents to SPIOs is often introduced through electrostatic interactions. Transfection agents such as poly-L-lysine,72,73 protamine sulfate74 or cationic liposomes75 form complexes with SPIOs in the labeling solution and greatly improve the endocytosis efficiency of cells. This offers SPIO nanoparticles with more universal applications in imaging and therapy at the cellular level. Another strategy involves modifying the SPIO surface with targeting moieties such as peptides or monoclonal antibodies.20,27,76,77
The use of SPIO-labeled therapeutic cells has illustrated a clear need for image-guided cellular immunotherapy. Kircher et al.78 designed intracellular labels, CLIO-HD nanoparticles, for in vivo MRI tracking of systemically injected cells with near single-cell resolution. They demonstrated for the first time high resolution imaging of T-cell recruitment to intact tumors in vivo. Daldrup-Link et al.79 showed that the human NK cell line NK-92 can be efficiently labeled with clinically applicable iron-oxide contrast agents, and the accumulation of these labeled cells in murine tumors can be monitored in vivo by MR imaging. Similarly, Arbab et al.80 reported the feasibility of imaging the migration and incorporation of SPIO-labeled sensitized splenocytes in an experimental 9L glioma brain tumor model. In another study, de Vries et al.81 labeled DCs with SPIOs or 111In-oxine and monitored the cells in vivo with MRI (Fig. 2). They demonstrated that in vivo MR tracking of magnetically labeled DCs is feasible in humans to detect very low number of cells with detailed anatomical information. Those MR cell tracking techniques may be particularly useful for evaluating novel cell-based therapies in vivo.
Fig. 2.
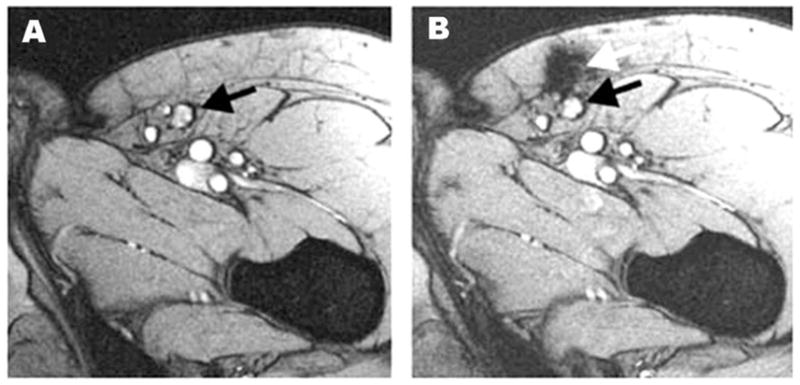
Monitoring of the accuracy of delivery of SPIO-labeled DCs using MRI. Black arrow denotes the inguinal lymph node (A) and white arrow indicates that the location of labeled DCs not in the lymph node but in the perinodular fat (B). Reproduced from ref. 81 with permission.
MRI allows noninvasive and three-dimensional visualization of whole organisms over time and would be ideally suited to quantitatively monitor cell trafficking in vivo. Long et al.82 developed an in vivo labeling method based on cell-to-cell transfer of SPIOs from tumor cells to endogenous antigen-presenting cells. In this study, mice were immunized with a tumor cell-based vaccine that was irradiated and labeled with SPIOs. Antigen-presenting cells that captured SPIOs can be quantitatively imaged over time as they accumulate in lymph nodes. This strategy can be exploited to optimize the current cellular immunotherapy by quantifying antigen-presenting cell numbers through noninvasive MRI.
Although it is promising to monitor cell trafficking with MRI after the cells are labeled with SPIOs, several limitations still exist. For example, once the labeled cells die, SPIOs remain in and around dead cells or are engulfed by cells of the host, which may result in misleading interpretation of MRI-based cell tracking. Furthermore, MRI cell tracking is difficult to perform in certain cases such as traumatic injury in which the endogenous blood derivatives exhibit hypointensity. Gadolinium (Gd)-based contrast agents are considered positive contrast agents for T1-weighted images because they brighten the tissues by shortening the T1 relaxation time of the water protons in the accumulated tissue. They do not have the signal void problems associated with SPIO. Gd-based probes have been used to label and monitor cells in an animal model bearing central nervous system tumors.83 After being labeled with a clinically approved formulation of a Gd-based MR contrast agent, Gd(III)-HP-DO3A (ProHance), injected DCs can be effectively imaged in vivo in established central nervous system tumors. However, compared with SPIOs, Gd based contrast agents are much less sensitive, due to the T1 and T2 signal differences. Although this might be overcome by delivering more Gd to the target cell/tissue, minimization of dose is desirable, especially with growing concern over Gd-based contrast agents related toxicities including nephrogenic systemic sclerosis.84,85
4. Optical imaging
Optical imaging is one of the most widely used molecular imaging techniques in preclinical settings.86 Optical imaging is relatively safe and simple without hazardous radiation. The imaging process is fast and is thus suitable for high throughput screening of a large number of species. Optical imaging has been applied for various applications, such as studying gene-delivery efficiency and gene-expression patterns, measuring protein-protein interactions, determining tumor growth, metastasis and response to therapy, and monitoring the location and the proliferation of therapeutic cells.87 Fluorescence reflectance and bioluminescence imaging are the most frequently employed modalities for cell-based cancer immunotherapy.86 In fluorescence imaging, excitation light illuminates the subject, and a charge-coupled device (CCD) camera collects the emission light at a shifted wavelength.88 The fluorescent probe can be either injected or genetically engineered and no substrate is required for its visualization.89 Bioluminescence imaging (BLI) is based on the expression of a light-emitting enzyme (such as firefly luciferase) in target cells and tissues.90 In the presence of its substrate (such as D-luciferin), an energy-dependent reaction releases photons that can be detected using sensitive detection systems.
4.1 Intrinsic cell labeling for optical imaging
Endogenous reporter proteins for optical imaging such as GFP are widely used in molecular imaging.91 It is particularly useful because of its stability and the fact that its chromophore is formed by autocatalytic cyclization that does not require a cofactor. Furthermore, the fusion of fluorescence proteins to other proteins does not significantly alter its fluorescence properties or the intracellular location of the fusion protein. However, endogenous autofluorescence of tissues frequently results in a substantial background emission that limits the sensitivity and specificity of fluorescence imaging.91,92 Various luciferase genes are the most commonly used bioluminescence reporter systems.93 The luciferase reporter system in combination with their corresponding luminescent substrates are more sensitive than fluorescence reporter imaging due to the absence of auto-bioluminescence and low background activity.94
Intrinsic labeling of immunocompetent cells by optical imaging reporter genes and noninvasive imaging of mice has been extensively reported.94–97 Edinger et al.96 labeled effector NK cells and monitored their homing to sites of tumor growth followed by tumor eradication. They firstly demonstrated all of the steps required for effective adoptive immunotherapy followed by intravenous injection of cells to visualize and study migration and infiltration into tumor tissues by optical imaging. Schimmelpfennig et al91 reported serial trafficking patterns and survival of adoptively transferred allogeneic donor DCs in vivo with optical imaging. Based on the luciferase and GFP reporter gene, they demonstrated that DCs maintain their capability of homing to lymphoid organs and are detectable for more than one month in allogeneic transplant recipients. Recently, using bioluminescence imaging, Helms et al.98 demonstrated that short-term cultured cytokine-induced killer (CIK) cells exhibit full cytotoxicity in vitro, but display different tumor homing properties than fully expanded CIK cells in vivo. These data demonstrate that noninvasive and highly sensitive optical imaging is ideally suited to evaluate complex biologic processes in vivo such as for cell-based cancer immunotherapy.
4.2 Extrinsic cell labeling for optical imaging
Fluorescence imaging probes can be broadly classified as conventional fluorophores (organic dyes, fluorescent proteins, etc.), quantum dots (QDs), and hybrid architectures combining one or more of these emitters in an inert matrix.99–104 Among them, organic fluorophores are widely used for a variety of straightforward shorter term cell labeling applications due to their commercial availability, low cost and easy handling.99,105–107 For example, IRDye800CW has been used to study the trafficking of T cells in a small animal model.105 However, these fluorphores are often subjected to photobleaching and/or quenching and may be sensitive to changes in pH and chemical degradation.
QDs are among the most widely studied nanoparticles for optical imaging. QDs are inorganic fluorescent semiconductor nanoparticles with superior properties for optical imaging compared with those of conventional organic fluorophores,28,108 such as higher quantum yields, better resistance to photobleaching and chemical degradation, wider absorption spectra spanning the ultraviolet to near-infrared region, longer fluorescence lifetime and narrower emission spectra. Recently, numerous cell-based applications have been discovered for QDs. Lim et al.109 labeled human NK cells with antibody-coated QDs for fluorescence imaging. The labeled NK cells showed a therapeutic effect similar to that of unlabeled NK cells. Intratumorally injected labeled NK cells could be visualized using near-infrared optical imaging. In another study, Noh et al.110 demonstrated the use of QDs for noninvasive in vivo tracking of DC migration into lymph nodes (Fig. 3). Furthermore, QDs can be developed as a versatile platform for immunoimaging of DCs and as an efficient nanoparticle-based antigen delivery system for priming an immune response.111
Fig. 3.

Noninvasive imaging of DCs migration into lymph nodes using NI fluorescence imaging. After DCs were labeled with NIR-emitting QDs, they were injected into the hind-leg footpad, and the migrations of injected DCs into lymph node were monitored by in vivo NIR imaging system. Reproduced from ref. 110 with permission.
Advances in optical imaging technology and molecular probes have had a significant impact upon cell-based cancer immunotherapy and will continue to provide insight into therapeutic cell distribution, engraftment and survival. However, optical imaging also has its own limitations, among which depth resolution and signal quantification are the major issues that are impeding optical imaging data interpretation and application. Intracellular exogenous probes may be diluted over time due to cell proliferation, thus causing a decrease in fluorescent signal and limiting long-term tracking. In addition, dead cells and/or released probes may be secondarily phagocytosed by macrophages. This process limits the ability to distinguish viable and non-viable cells as well as host and donor cells while potentially generating a significant background signal.
5. Multimodality imaging
Each imaging modality has its own pros and cons. MRI can provide three-dimensional tomography but is limited by low target sensitivity, whereas PET and optical imaging has good sensitivity but suffer from low spatial resolution or tissue penetration. To harness the strengths of different imaging methods, multimodality imaging has become attractive for both small animal and human studies.112,113 Multimodality imaging enables the combination of anatomical, functional and molecular information by combining images from different modalities taken at the same point. It has emerged as a strategy that combines the strengths of different modalities and yields a hybrid imaging platform with characteristics superior to those of any of its constituents considered alone.114,115 Generally, multimodality imaging is chosen to furnish synergistic, complementary, or clinically useful information beyond that provided by any individual method. For example, co-registration with MRI images provides the anatomic landscape for localizing the molecular information generated by PET.
Currently, there are remarkable efforts being made to progress the development of multimodality noninvasive imaging reporter genes for cell tracking. Triple fusion reporter vectors harboring a bioluminescence luciferase reporter gene, a reporter gene encoding fluorescence protein, and a PET reporter gene HSV1-sr39tk has been constructed and found to preserve the high activity for each protein component.116 With this triple-fusion reporter gene, Kim et al.117 reported multimodality imaging of activated T-lymphocyte migration into an immunogenic sarcoma site by using PET and optical imaging. Also, the advantages of the HSV1-tk-GFP fusion gene have been demonstrated for dual-modality imaging of T-cell activity in vivo.118 T cell activation in vivo could be monitored and assessed using an activation-sensitive HSV1-tk-GFP-encoding genetic reporter system under the control of a nuclear factor of activated T cells, which may be useful in the assessment of cell-based adoptive therapies, vaccination strategies and immunosuppressive drugs.
Mesenchymal stem cells (MSCs) have been shown to target cancer tissue in vivo and thus have the potential to serve as vehicles for the delivery of anticancer therapies.25,119 It is crucial to understand the dynamics of homing and engraftment of infused stem cells with noninvasive imaging in real time for the progression of stem cell therapeutics.120,121 Love et al.122 used lentiviral vector encoding a triple fusion reporter gene to transducer human MSCs. These transduced MSCs were visualized with optical and PET imaging in small-animal models, allowing multimodal imaging of cell therapy for in vivo cellular imaging applications. A similar approach has been used for the monitoring of MSC targeting of microscopic tumors and tumor stroma development.123 As imaged by 18F-FHBG PET, MSCs expressing HSV1-tk and GFP reporter genes could target microscopic tumors, subsequently proliferate and differentiate, and contribute to the formation of a significant portion of tumor stroma.
Multimodality imaging of therapeutic cells also can be achieved after the cells are directly labeled with multifunctional imaging probes.23,124–129 For example, Lim et al.130 synthesized a novel mulitfunctional probe for both 19F-based MRI and near-infrared optical imaging using perfluorooctyl bromide (PFOB) and IRDye800, respectively (Fig. 4). The 1H-based MR provides a whole-body image while the 19F-based MR shows only signals generated from the injected DCs labeled with IRDye800-coated PFOB nanoemulsions. In addition, a strong NIR fluorescence signal was detected at each injection site.
Fig. 4.

Multifunctional perfluorocarbon nanoemulsions (upper panel) was designed and used for labeling and imaging of DCs in vivo with NIR optical imaging and MRI (lower panel). Reproduced from ref. 130 with permission.
6. Conclusion and perspectives
Molecular imaging of cell-based cancer immunotherapy is an active area of investigation in both preclinical animal models and clinical trials. With the use of different imaging modalities and probes, molecular imaging has spurred the development of exciting new immunotherapeutic strategies. Ultimately, molecular imaging will play a critically important role in measuring and elucidating the trafficking pathways of therapeutic cells in vivo, along with their migratory properties, activation status and therapeutic efficacy.
Multimodality imaging is a promising technique to overcome the limits of current molecular diagnostics and to permit accurate diagnosis as well as the development of cell-based cancer therapy. However, future investigation is needed to address the issue of sensitivity and determine the detection thresholds for the different modalities. For example, PET is a highly sensitive imaging modality that requires the introduction of only trace amount of probes, whereas a relatively high amount of probes is needed for MRI. Moreover, the design of multifunctional probes needs to be optimized since functional group modification of the probes may change their chemical properties. Fundamental nanoscale understanding of the molecular interactions among the multifunctional probe components will be critical for the assembly into efficacious nanocomposite particles. The influence of one structural component on the performance of the others must be carefully investigated to ensure synergy in the integrated design.
So far, most probes and cell labeling methods have been studied at either an in vitro environment or in preclinical models. In order to be translated into clinical applications, the comprehensive biosafety studies related to toxicity and immunogenicity still need intensive exploration. For example, the gene products of the imaging reporter such as GFP and HSV1-TK are foreign proteins, which will induce immunomediated rejection of the transplanted cells. Humanized variants of the genes or endogenous reporters may reduce the immunological inactivation of genetically modified cells. For extrinsic labeling, more biocompatible imaging probes than QDs and other inorganic nanoparticles are needed to facilitate the clinical translation. In addition, further investigation of in vivo pharmacokinetics, pharmacodynamics, and metabolism is necessary to optimize these imaging probes.
In summary, efficient communication and cooperation are needed from biologists in identifying and validating the crosstalk between tumors and the immune system, from chemists in synthesizing and characterizing imaging probes, and from engineers and physicists in developing high-sensitivity/resolution imaging devices. With continuous efforts by multi-disciplinary approaches, the use of molecular imaging will shed new light on cell-based cancer immunotherapy.
Acknowledgments
This work was partially supported by projects (No. 09ZA036&KFJJ0905) of Sichuan Province, China, the Intramural Research Program (IRP) of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), and the International Cooperative Program of National Science Foundation of China (NSFC) (81028009). G.N. acknowledges an Imaging Sciences Training Fellowship jointly funded by the Radiology and Imaging Sciences Department of NIH Clinical Center and by the IRP/NIBIB, NIH.
Biographies

Gang Liu
Gang Liu received his PhD in Biomedical and Bioengineering from Sichuan University in 2009. He joined the Laboratory of Molecular Imaging and Nanomedicine (LOMIN) of Dr Xiaoyuan Chen at the National Institutes of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH) as a postdoctoral researcher. His current research interests focus on the development of theranostic nanomedicine carrying both chemotherapeutics, gene therapeutics, and imaging tags for cancer research.

Magdalena Swierczewska
Magdalena Swierczewska is a graduate student in the Biomedical Engineering Department at Stony Brook University working towards her PhD in Dr Chen’s Laboratory of Molecular Imaging and Nanomedicine of the NIBIB/NIH. She received her MS in Biomedical Engineering from Stony Brook University. Using her background in nanotechnology and material science, Maggie is developing novel nanoplatforms for ultrasensitive diagnostics.

Gang Niu
Gang Niu received his PhD in the Radiation Oncology Department from University of Iowa, Iowa City. After graduation in December 2005, Dr Niu joined Dr Xiaoyuan Chen’s group in the Molecular Imaging Program at Stanford (MIPS) and moved with Prof. Chen to NIBIB in 2009. Dr Niu is now the leader of Biological Molecular Imaging Section in the LOMIN at NIBIB and his researches focus on new imaging target identification and imaging probe development.

Xiaoming Zhang
Xiaoming Zhang received his MD in Medical Imaging from Shanghai Medical University in 1995, Shanghai, China. He has been served in the Department of Radiology, Affiliated Hospital of North Sichuan Medical College, China. Now he is the head of Sichuan Key Laboratory of Medical Imaging, China. His researches focus on body imaging and molecular imaging.

Xiaoyuan Chen
Xiaoyuan Chen received his PhD in chemistry from the University of Idaho in 1999. After two quick postdoctoral programs at Syracuse University and Washington University in St. Louis, he joined the University of Southern California as an Assistant Professor of Radiology in 2002. He then moved to Stanford University in 2004 and was promoted to Associate Professor in 2008. In the summer of 2009, he joined the Intramural Research Program of NIBIB as a tenured Senior Investigator and Chief of LOMIN. He is interested in developing molecular imaging toolbox for better understanding of biology, early diagnosis of disease, monitoring therapy response, and guiding drug discovery/development. His lab also puts special emphasis on high-sensitivity nanosensors for biomarker detection and theranostic nanomedicine for imaging, gene and drug delivery, and monitoring of treatment.
Contributor Information
Gang Niu, Email: niug@mail.nih.gov.
Xiaoyuan Chen, Email: shawn.chen@nih.gov.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Ca-Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Kapp M, Rasche L, Einsele H, Grigoleit GU. Curr Opin Hematol. 2009;16:437–443. doi: 10.1097/MOH.0b013e32832f57d4. [DOI] [PubMed] [Google Scholar]
- 3.Sutlu T, Alici E. J Intern Med. 2009;266:154–181. doi: 10.1111/j.1365-2796.2009.02121.x. [DOI] [PubMed] [Google Scholar]
- 4.Eksioglu EA, Eisen S, Reddy V. Front Biosci. 2010;15:321–347. doi: 10.2741/3623. [DOI] [PubMed] [Google Scholar]
- 5.Shablak A, Hawkins RE, Rothwell DG, Elkord E. Clin Cancer Res. 2009;15:6503–6510. doi: 10.1158/1078-0432.CCR-09-1605. [DOI] [PubMed] [Google Scholar]
- 6.Disis ML, Bernhard H, Jaffee EM. Lancet. 2009;373:673–683. doi: 10.1016/S0140-6736(09)60404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prados J, Melguizo C, Boulaiz H, Marchal JA, Aranega A. Cell Mol Biol (Noisy-le-grand) 2005;51:23–36. [PubMed] [Google Scholar]
- 8.Markowicz S. Acta Pol Pharm. 2008;65:625–632. [PubMed] [Google Scholar]
- 9.Pham W, Kobukai S, Hotta C, Gore JC. Expert Opin Biol Ther. 2009;9:539–564. doi: 10.1517/14712590902867739. [DOI] [PubMed] [Google Scholar]
- 10.Mankoff DA. J Nucl Med. 2007;48:18N, 21N. [PubMed] [Google Scholar]
- 11.Weissleder R, Mahmood U. Radiology. 2001;219:316–333. doi: 10.1148/radiology.219.2.r01ma19316. [DOI] [PubMed] [Google Scholar]
- 12.Iagaru A, Chen X, Gambhir SS. Nat Clin Pract Oncol. 2007;4:556–557. doi: 10.1038/ncponc0929. [DOI] [PubMed] [Google Scholar]
- 13.Weissleder R, Pittet MJ. Nature. 2008;452:580–589. doi: 10.1038/nature06917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niu G, Chen X. Drugs R&D. 2008;9:351–368. doi: 10.2165/0126839-200809060-00002. [DOI] [PubMed] [Google Scholar]
- 15.Niu G, Chen X. Eur J Radiol. 2009;70:294–304. doi: 10.1016/j.ejrad.2009.01.052. [DOI] [PubMed] [Google Scholar]
- 16.Ai H. Adv Drug Delivery Rev. 2011 doi: 10.1016/j.addr.2011.03.013. accepted. [DOI] [PubMed] [Google Scholar]
- 17.Srinivas M, Aarntzen EH, Bulte JW, Oyen WJ, Heerschap A, de Vries IJ, Figdor CG. Adv Drug Delivery Rev. 2010;62:1080–1093. doi: 10.1016/j.addr.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 18.Gambhir SS. Nat Rev Cancer. 2002;2:683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 19.Duimstra JA, Femia FJ, Meade TJ. J Am Chem Soc. 2005;127:12847–12855. doi: 10.1021/ja042162r. [DOI] [PubMed] [Google Scholar]
- 20.Lewin M, Carlesso N, Tung CH, Tang XW, Cory D, Scadden DT, Weissleder R. Nat Biotechnol. 2000;18:410–414. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- 21.Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi MG, Frangioni JV. Nat Nanotechnol. 2010;5:42–47. doi: 10.1038/nnano.2009.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai W, Chen X. J Nucl Med. 2008;49(Suppl_2):113S–128S. doi: 10.2967/jnumed.107.045922. [DOI] [PubMed] [Google Scholar]
- 23.Lee S, Chen X. Mol Imaging. 2009;8:87–100. [PubMed] [Google Scholar]
- 24.Louie A. Chem Rev. 2010;110:3146–3195. doi: 10.1021/cr9003538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee Z, Dennis JE, Gerson SL. Exp Biol Med. 2008;233:930–940. doi: 10.3181/0709-MR-234. [DOI] [PubMed] [Google Scholar]
- 26.Kang JH, Chung JK. J Nucl Med. 2008;49(Suppl_2):164S–179S. doi: 10.2967/jnumed.107.045955. [DOI] [PubMed] [Google Scholar]
- 27.Bulte JW, Kraitchman DL. Curr Pharm Biotechnol. 2004;5:567–584. doi: 10.2174/1389201043376526. [DOI] [PubMed] [Google Scholar]
- 28.Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Science. 2005;307:538–544. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobrucki LW, Sinusas AJ. Nat Rev Cardiol. 2010;7:38–47. doi: 10.1038/nrcardio.2009.201. [DOI] [PubMed] [Google Scholar]
- 30.Chin FT, Namavari M, Levi J, Subbarayan M, Ray P, Chen X, Gambhir SS. Mol Imaging Biol. 2008;10:82–91. doi: 10.1007/s11307-007-0122-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steffens S, Frank S, Fischer U, Heuser C, Meyer KL, Dobberstein KU, Rainov NG, Kramm CM. Cancer Gene Ther. 2000;7:806–812. doi: 10.1038/sj.cgt.7700173. [DOI] [PubMed] [Google Scholar]
- 32.Liang Q, Gotts J, Satyamurthy N, Barrio J, Phelps ME, Gambhir SS, Herschman HR. Mol Ther. 2002;6:73–82. doi: 10.1006/mthe.2002.0626. [DOI] [PubMed] [Google Scholar]
- 33.Niu G, Gaut AW, Ponto LL, Hichwa RD, Madsen MT, Graham MM, Domann FE. J Nucl Med. 2004;45:445–449. [PubMed] [Google Scholar]
- 34.Kang JH, Chung JK, Lee YJ, Shin JH, Jeong JM, Lee S, Lee MC. J Nucl Med. 2004;45:1571–1576. [PubMed] [Google Scholar]
- 35.Doubrovin M, Serganova I, Mayer-Kuckuk P, Ponomarev V, Blasberg RG. Bioconjugate Chem. 2004;15:1376–1388. doi: 10.1021/bc0498572. [DOI] [PubMed] [Google Scholar]
- 36.Soghomonyan S, Hajitou A, Rangel R, Trepel M, Pasqualini R, Arap W, Gelovani JG, Alauddin MM. Nat Protoc. 2007;2:416–423. doi: 10.1038/nprot.2007.49. [DOI] [PubMed] [Google Scholar]
- 37.Green LA, Nguyen K, Berenji B, Iyer M, Bauer E, Barrio JR, Namavari M, Satyamurthy N, Gambhir SS. J Nucl Med. 2004;45:1560–1570. [PubMed] [Google Scholar]
- 38.Koehne G, Doubrovin M, Doubrovina E, Zanzonico P, Gallardo HF, Ivanova A, Balatoni J, Teruya-Feldstein J, Heller G, May C, Ponomarev V, Ruan S, Finn R, Blasberg RG, Bornmann W, Riviere I, Sadelain M, O’Reilly RJ, Larson SM, Tjuvajev JG. Nat Biotechnol. 2003;21:405–413. doi: 10.1038/nbt805. [DOI] [PubMed] [Google Scholar]
- 39.Zanzonico P, Koehne G, Gallardo HF, Doubrovin M, Doubrovina, Finn R, Blasberg RG, Riviere I, O’Reilly RJ, Sadelain M, Larson SM. Eur J Nucl Med Mol Imaging. 2006;33:988–997. doi: 10.1007/s00259-005-0057-3. [DOI] [PubMed] [Google Scholar]
- 40.Su H, Forbes A, Gambhir SS, Braun J. Mol Imaging Biol. 2004;6:139–148. doi: 10.1016/j.mibio.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Su H, Chang DS, Gambhir SS, Braun J. J Immunol. 2006;176:4459–4467. doi: 10.4049/jimmunol.176.7.4459. [DOI] [PubMed] [Google Scholar]
- 42.Shu CJ, Radu CG, Shelly SM, Vo DD, Prins R, Ribas A, Phelps ME, Witte ON. Int Immunol. 2009;21:155–165. doi: 10.1093/intimm/dxn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yaghoubi SS, Jensen MC, Satyamurthy N, Budhiraja S, Paik, Czernin J, Gambhir SS. Nat Clin Pract Oncol. 2009;6:53–58. doi: 10.1038/ncponc1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ponomarev V. J Nucl Med. 2009;50:1013–1016. doi: 10.2967/jnumed.109.064055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palestro CJ, Love C, Bhargava KK. Q J Nucl Med Mol Imaging. 2009;53:105–123. [PubMed] [Google Scholar]
- 46.Kupiec-Weglinski JW, Austyn JM, Morris PJ. J Exp Med. 1988;167:632–645. doi: 10.1084/jem.167.2.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suda T, Callahan RJ, Wilkenson RA, van Rooijen N, Schneeberger E. J Leukoc Biol. 1996;60:519–527. doi: 10.1002/jlb.60.4.519. [DOI] [PubMed] [Google Scholar]
- 48.Fisher B, Packard BS, Read EJ, Carrasquillo JA, Carter S, Topalian SL, Yang JC, Yolles P, Larson SM, Rosenberg SA. J Clin Oncol. 1989;7:250–261. doi: 10.1200/JCO.1989.7.2.250. [DOI] [PubMed] [Google Scholar]
- 49.Blocklet D, Toungouz M, Kiss R, Lambermont M, Velu T, Duriau, Goldman M, Goldman S. Eur J Nucl Med Mol Imaging. 2003;30:440–447. doi: 10.1007/s00259-002-1001-4. [DOI] [PubMed] [Google Scholar]
- 50.Vallabhajosula S. Semin Nucl Med. 2007;37:400–419. doi: 10.1053/j.semnuclmed.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 51.Sols A, Crane RK. J Biol Chem. 1954;210:581–595. [PubMed] [Google Scholar]
- 52.Drevs J, Hofmann I, Hugenschmidt H, Wittig C, Madjar H, Muller M, Wood J, Martiny-Baron G, Unger C, Marme D. Cancer Res. 2000;60:4819–4824. [PubMed] [Google Scholar]
- 53.Bhargava KK, Gupta RK, Nichols KJ, Palestro CJ. Nucl Med Biol. 2009;36:545–549. doi: 10.1016/j.nucmedbio.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olasz EB, Lang L, Seidel J, Green MV, Eckelman WC, Katz SI. J Immunol Methods. 2002;260:137–148. doi: 10.1016/s0022-1759(01)00528-2. [DOI] [PubMed] [Google Scholar]
- 55.Radu CG, Shu CJ, Nair-Gill E, Shelly SM, Barrio JR, Satyamurthy N, Phelps ME, Witte ON. Nat Med. 2008;14:783–788. doi: 10.1038/nm1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adonai N, Nguyen KN, Walsh J, Iyer M, Toyokuni T, Phelps ME, McCarthy T, McCarthy DW, Gambhir SS. Proc Natl Acad Sci U S A. 2002;99:3030–3035. doi: 10.1073/pnas.052709599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tseng JR, Kang KW, Dandekar M, Yaghoubi S, Lee JH, Christensen JG, Muir S, Vincent PW, Michaud NR, Gambhir SS. J Nucl Med. 2008;49:129–134. doi: 10.2967/jnumed.106.038836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weissleder R, Cheng HC, Bogdanova A, Bogdanov A., Jr J Magn Reson Imaging. 1997;7:258–263. doi: 10.1002/jmri.1880070140. [DOI] [PubMed] [Google Scholar]
- 59.Xie J, Huang J, Li X, Sun S, Chen X. Curr Med Chem. 2009;16:1278–1294. doi: 10.2174/092986709787846604. [DOI] [PubMed] [Google Scholar]
- 60.Gao J, Gu H, Xu B. Acc Chem Res. 2009;42:1097–1107. doi: 10.1021/ar9000026. [DOI] [PubMed] [Google Scholar]
- 61.Weissleder R. Nat Rev Cancer. 2002;2:11–18. doi: 10.1038/nrc701. [DOI] [PubMed] [Google Scholar]
- 62.Pathak AP, Gimi B, Glunde K, Ackerstaff E, Artemov D, Bhujwalla ZM. Methods Enzymol. 2004;386:3–60. doi: 10.1016/S0076-6879(04)86001-4. [DOI] [PubMed] [Google Scholar]
- 63.Louie AY, Huber MM, Ahrens ET, Rothbacher U, Moats R, Jacobs RE, Fraser SE, Meade TJ. Nat Biotechnol. 2000;18:321–325. doi: 10.1038/73780. [DOI] [PubMed] [Google Scholar]
- 64.Wang K, Shen B, Huang T, Sun X, Li W, Jin G, Li L, Bu L, Li R, Wang D, Chen X. Mol Imaging Biol. 2009;12:520–529. doi: 10.1007/s11307-009-0286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gilad AA, McMahon MT, Walczak P, Winnard PT, Jr, Raman V, van Laarhoven HW, Skoglund CM, Bulte JW, van Zijl PC. Nat Biotechnol. 2007;25:217–219. doi: 10.1038/nbt1277. [DOI] [PubMed] [Google Scholar]
- 66.Liu J, Cheng EC, Long RC, Yang SH, Wang L, Cheng PH, Yang J, Wu D, Mao H, Chan AW. Tissue Eng, Part C. 2009;15:739–747. doi: 10.1089/ten.tec.2008.0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zurkiya O, Chan AW, Hu X. Magn Reson Med. 2008;59:1225–1231. doi: 10.1002/mrm.21606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gilad AA, Ziv K, McMahon MT, van Zijl PC, Neeman M, Bulte JW. J Nucl Med. 2008;49:1905–1908. doi: 10.2967/jnumed.108.053520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weissleder R, Stark DD, Engelstad BL, Bacon BR, Compton CC, White DL, Jacobs P, Lewis J. AJR Am J Roentgenol. 1989;152:167–173. doi: 10.2214/ajr.152.1.167. [DOI] [PubMed] [Google Scholar]
- 70.Wang YX, Hussain SM, Krestin GP. Eur Radiol. 2001;11:2319–2331. doi: 10.1007/s003300100908. [DOI] [PubMed] [Google Scholar]
- 71.Thorek DL, Tsourkas A. Biomaterials. 2008;29:3583–3590. doi: 10.1016/j.biomaterials.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arbab AS, Bashaw LA, Miller BR, Jordan EK, Lewis BK, Kalish H, Frank JA. Radiology. 2003;229:838–846. doi: 10.1148/radiol.2293021215. [DOI] [PubMed] [Google Scholar]
- 73.Liu G, Yang H, Zhang XM, Shao Y, Jiang H. Contrast Media Mol Imaging. 2010;5:53–58. doi: 10.1002/cmmi.362. [DOI] [PubMed] [Google Scholar]
- 74.Arbab AS, Yocum GT, Kalish H, Jordan EK, Anderson SA, Khakoo AY, Read EJ, Frank JA. Blood. 2004;104:1217–1223. doi: 10.1182/blood-2004-02-0655. [DOI] [PubMed] [Google Scholar]
- 75.Arbab AS, Yocum GT, Rad AM, Khakoo AY, Fellowes V, Read EJ, Frank JA. NMR Biomed. 2005;18:553–559. doi: 10.1002/nbm.991. [DOI] [PubMed] [Google Scholar]
- 76.Zhang C, Jugold M, Woenne EC, Lammers T, Morgenstern, Mueller MM, Zentgraf H, Bock M, Eisenhut M, Semmler W, Kiessling F. Cancer Res. 2007;67:1555–1562. doi: 10.1158/0008-5472.CAN-06-1668. [DOI] [PubMed] [Google Scholar]
- 77.Ahrens ET, Feili-Hariri M, Xu H, Genove G, Morel PA. Magn Reson Med. 2003;49:1006–1013. doi: 10.1002/mrm.10465. [DOI] [PubMed] [Google Scholar]
- 78.Kircher MF, Allport JR, Graves EE, Love V, Josephson L, Lichtman AH, Weissleder R. Cancer Res. 2003;63:6838–6846. [PubMed] [Google Scholar]
- 79.Daldrup-Link HE, Meier R, Rudelius M, Piontek G, Piert M, Metz S, Settles M, Uherek C, Wels W, Schlegel J, Rummeny EJ. Eur Radiol. 2005;15:4–13. doi: 10.1007/s00330-004-2526-7. [DOI] [PubMed] [Google Scholar]
- 80.Arbab AS, Rad AM, Iskander AS, Jafari-Khouzani K, Brown SL, Churchman JL, Ding G, Jiang Q, Frank JA, Soltanian-Zadeh H, Peck DJ. Magn Reson Med. 2007;58:519–526. doi: 10.1002/mrm.21343. [DOI] [PubMed] [Google Scholar]
- 81.de Vries IJ, Lesterhuis WJ, Barentsz JO, Verdijk P, van Krieken JH, Boerman OC, Oyen WJ, Bonenkamp JJ, Boezeman JB, Adema GJ, Bulte JW, Scheenen TW, Punt CJ, Heerschap A, Figdor CG. Nat Biotechnol. 2005;23:1407–1413. doi: 10.1038/nbt1154. [DOI] [PubMed] [Google Scholar]
- 82.Long CM, van Laarhoven HW, Bulte JW, Levitsky HI. Cancer Res. 2009;69:3180–3187. doi: 10.1158/0008-5472.CAN-08-3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sengar RS, Spokauskiene L, Steed DP, Griffin P, Arbujas N, Chambers WH, Wiener EC. Magn Reson Med. 2009;62:599–606. doi: 10.1002/mrm.22030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grobner T. Nephrol, Dial, Transplant. 2006;21:1104–1108. doi: 10.1093/ndt/gfk062. [DOI] [PubMed] [Google Scholar]
- 85.Rydahl C, Thomsen HS, Marckmann P. Invest Radiol. 2008;43:141–144. doi: 10.1097/RLI.0b013e31815a3407. [DOI] [PubMed] [Google Scholar]
- 86.Sutton EJ, Henning TD, Pichler BJ, Bremer C, Daldrup-Link HE. Eur Radiol. 2008;18:2021–2032. doi: 10.1007/s00330-008-0984-z. [DOI] [PubMed] [Google Scholar]
- 87.Pierce MC, Javier DJ, Richards-Kortum R. Int J Cancer. 2008;123:1979–1990. doi: 10.1002/ijc.23858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Massoud TF, Gambhir SS. Genes Dev. 2003;17:545–580. doi: 10.1101/gad.1047403. [DOI] [PubMed] [Google Scholar]
- 89.Wunderbaldinger P, Bogdanov A, Weissleder R. Eur J Radiol. 2000;34:156–165. doi: 10.1016/s0720-048x(00)00196-0. [DOI] [PubMed] [Google Scholar]
- 90.Beckham JT, Mackanos MA, Crooke C, Takahashi T, O’Connell-Rodwell, Contag CH, Jansen ED. Photochem Photobiol. 2004;79:76–85. [PubMed] [Google Scholar]
- 91.Chudakov DM, Lukyanov S, Lukyanov KA. Trends Biotechnol. 2005;23:605–613. doi: 10.1016/j.tibtech.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 92.Yang X, Liu H, Li D, Zhou X, Jung WC, Deans AE, Cui Y, Cheng L. Radiology. 2001;219:171–175. doi: 10.1148/radiology.219.1.r01ap23171. [DOI] [PubMed] [Google Scholar]
- 93.Niu G, Chen X. Mol Imaging Biol. 2009;11:61–63. doi: 10.1007/s11307-008-0190-z. [DOI] [PubMed] [Google Scholar]
- 94.Hardy J, Edinger M, Bachmann MH, Negrin RS, Fathman CG, Contag CH. Exp Hematol. 2001;29:1353–1360. doi: 10.1016/s0301-472x(01)00756-1. [DOI] [PubMed] [Google Scholar]
- 95.Costa GL, Sandora MR, Nakajima A, Nguyen EV, Taylor-Edwards C, Slavin AJ, Contag CH, Fathman CG, Benson JM. J Immunol. 2001;167:2379–2387. doi: 10.4049/jimmunol.167.4.2379. [DOI] [PubMed] [Google Scholar]
- 96.Edinger M, Cao YA, Verneris MR, Bachmann MH, Contag CH, Negrin RS. Blood. 2003;101:640–648. doi: 10.1182/blood-2002-06-1751. [DOI] [PubMed] [Google Scholar]
- 97.Schimmelpfennig CH, Schulz S, Arber C, Baker J, Tarner I, McBride J, Contag CH, Negrin RS. Am J Pathol. 2005;167:1321–1331. doi: 10.1016/S0002-9440(10)61219-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Helms MW, Prescher JA, Cao YA, Schaffert S, Contag H. Cancer Immunol Immunother. 2010;59:1325–1334. doi: 10.1007/s00262-010-0860-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sevick-Muraca EM, Houston JP, Gurfinkel M. Curr Opin Chem Biol. 2002;6:642–650. doi: 10.1016/s1367-5931(02)00356-3. [DOI] [PubMed] [Google Scholar]
- 100.Zhao X, Hilliard LR, Mechery SJ, Wang Y, Bagwe RP, Jin S, Tan W. Proc Natl Acad Sci U S A. 2004;101:15027–15032. doi: 10.1073/pnas.0404806101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ow H, Larson DR, Srivastava M, Baird BA, Webb WW, Wiesner U. Nano Lett. 2005;5:113–117. doi: 10.1021/nl0482478. [DOI] [PubMed] [Google Scholar]
- 102.Burns A, Ow H, Wiesner U. Chem Soc Rev. 2006;35:1028–1042. doi: 10.1039/b600562b. [DOI] [PubMed] [Google Scholar]
- 103.Shcherbo D, Merzlyak EM, Chepurnykh TV, Fradkov AF, Ermakova GV, Solovieva EA, Lukyanov KA, Bogdanova EA, Zaraisky AG, Lukyanov S, Chudakov M. Nat Methods. 2007;4:741–746. doi: 10.1038/nmeth1083. [DOI] [PubMed] [Google Scholar]
- 104.Larson D, Ow H, Vishwasrao H, Heikal A, Wiesner U, Webb W. Chem Mater. 2008;20:2677–2684. [Google Scholar]
- 105.Boddington S, Henning TD, Sutton EJ, Daldrup-Link HE. J Vis Exp. 2008;14:686. doi: 10.3791/686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Foster AE, Kwon S, Ke S, Lu A, Eldin K, Sevick-Muraca E, Rooney CM. Appl Opt. 2008;47:5944–5952. doi: 10.1364/ao.47.005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tavri S, Jha P, Meier R, Henning TD, Muller T, Hostetter D, Knopp C, Johansson M, Reinhart V, Boddington S, Sista A, Wels WS, Daldrup-Link HE. Mol Imaging. 2009;8:15–26. [PubMed] [Google Scholar]
- 108.Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Nat Mater. 2005;4:435–446. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- 109.Lim YT, Cho MY, Noh YW, Chung JW, Chung BH. Nanotechnology. 2009;20:475102. doi: 10.1088/0957-4484/20/47/475102. [DOI] [PubMed] [Google Scholar]
- 110.Noh YW, Lim YT, Chung BH. FASEB J. 2008;22:3908–3918. doi: 10.1096/fj.08-112896. [DOI] [PubMed] [Google Scholar]
- 111.Sen D, Deerinck TJ, Ellisman MH, Parker I, Cahalan MD. PLoS One. 2008;3:e3290. doi: 10.1371/journal.pone.0003290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen X. Mini-Rev Med Chem. 2006;6:227–234. doi: 10.2174/138955706775475975. [DOI] [PubMed] [Google Scholar]
- 113.Pichler BJ, Wehrl HF, Judenhofer MS. J Nucl Med. 2008;49(Suppl_2):5S–23S. doi: 10.2967/jnumed.108.045880. [DOI] [PubMed] [Google Scholar]
- 114.Pichler BJ, Judenhofer MS, Pfannenberg C. Handb Exp Pharmacol. 2008:109–132. doi: 10.1007/978-3-540-72718-7_6. [DOI] [PubMed] [Google Scholar]
- 115.Iagaru A, Mittra E, Yaghoubi SS, Dick DW, Quon A, Goris ML, Gambhir SS. J Nucl Med. 2009;50:501–505. doi: 10.2967/jnumed.108.058339. [DOI] [PubMed] [Google Scholar]
- 116.Ray P, De A, Min JJ, Tsien RY, Gambhir SS. Cancer Res. 2004;64:1323–1330. doi: 10.1158/0008-5472.can-03-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kim YJ, Dubey P, Ray P, Gambhir SS, Witte ON. Mol Imaging Biol. 2004;6:331–340. doi: 10.1016/j.mibio.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 118.Ponomarev V, Doubrovin M, Lyddane C, Beresten T, Balatoni J, Bornman W, Finn R, Akhurst T, Larson S, Blasberg R, Sadelain M, Tjuvajev JG. Neoplasia. 2001;3:480–488. doi: 10.1038/sj.neo.7900204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Benedetti S, Pirola B, Pollo B, Magrassi L, Bruzzone MG, Rigamonti D, Galli R, Selleri S, Di Meco F, De Fraja C, Vescovi A, Cattaneo E, Finocchiaro G. Nat Med. 2000;6:447–450. doi: 10.1038/74710. [DOI] [PubMed] [Google Scholar]
- 120.Cao F, Lin S, Xie X, Ray P, Patel M, Zhang X, Drukker M, Dylla SJ, Connolly AJ, Chen X, Weissman IL, Gambhir SS, Wu JC. Circulation. 2006;113:1005–1014. doi: 10.1161/CIRCULATIONAHA.105.588954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang H, Cao F, De A, Cao Y, Contag C, Gambhir SS, Wu JC, Chen X. Stem Cells. 2009;27:1548–1558. doi: 10.1002/stem.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Love Z, Wang F, Dennis J, Awadallah A, Salem N, Lin Y, Weisenberger A, Majewski S, Gerson S, Lee Z. J Nucl Med. 2007;48:2011–2020. doi: 10.2967/jnumed.107.043166. [DOI] [PubMed] [Google Scholar]
- 123.Hung SC, Deng WP, Yang WK, Liu RS, Lee CC, Su TC, Lin RJ, Yang DM, Chang CW, Chen WH, Wei HJ, Gelovani JG. Clin Cancer Res. 2005;11:7749–7756. doi: 10.1158/1078-0432.CCR-05-0876. [DOI] [PubMed] [Google Scholar]
- 124.Chen K, Li ZB, Wang H, Cai W, Chen X. Eur J Nucl Med Mol Imaging. 2008;35:2235–2244. doi: 10.1007/s00259-008-0860-8. [DOI] [PubMed] [Google Scholar]
- 125.Cormode DP, Skajaa T, van Schooneveld MM, Koole R, Jarzyna P, Lobatto ME, Calcagno C, Barazza A, Gordon RE, Zanzonico P, Fisher EA, Fayad ZA, Mulder WJ. Nano Lett. 2008;8:3715–3723. doi: 10.1021/nl801958b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang P, He J, Wang PN, Chen JY. Photomed Laser Surg. 2010;28:201–205. doi: 10.1089/pho.2009.2546. [DOI] [PubMed] [Google Scholar]
- 127.Xie J, Chen K, Huang J, Lee S, Wang J, Gao J, Li X, Chen X. Biomaterials. 2010;31:3016–3022. doi: 10.1016/j.biomaterials.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen K, Xie J, Xu H, Behera D, Michalski MH, Biswal S, Wang A, Chen X. Biomaterials. 2009;30:6912–6919. doi: 10.1016/j.biomaterials.2009.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hsu AR, Cai W, Veeravagu A, Mohamedali KA, Chen K, Kim S, Vogel H, Hou LC, Tse V, Rosenblum MG, Chen X. J Nucl Med. 2007;48:445–454. [PubMed] [Google Scholar]
- 130.Lim YT, Noh YW, Kwon JN, Chung BH. Chem Commun. 2009;(45):6952–6954. doi: 10.1039/b914006a. [DOI] [PubMed] [Google Scholar]