

Abstract
OleA catalyzes the condensation of fatty acyl groups in the first step of bacterial long-chain olefin biosynthesis, but the mechanism of the condensation reaction is controversial. In this study, OleA from Xanthomonas campestris was expressed in Escherichia coli and purified to homogeneity. The purified protein was shown to be active with fatty acyl-CoA substrates that ranged from C8 to C16 in length. With limiting myristoyl-CoA (C14), 1 mol of the free coenzyme A was released/mol of myristoyl-CoA consumed. Using [14C]myristoyl-CoA, the other products were identified as myristic acid, 2-myristoylmyristic acid, and 14-heptacosanone. 2-Myristoylmyristic acid was indicated to be the physiologically relevant product of OleA in several ways. First, 2-myristoylmyristic acid was the major condensed product in short incubations, but over time, it decreased with the concomitant increase of 14-heptacosanone. Second, synthetic 2-myristoylmyristic acid showed similar decarboxylation kinetics in the absence of OleA. Third, 2-myristoylmyristic acid was shown to be reactive with purified OleC and OleD to generate the olefin 14-heptacosene, a product seen in previous in vivo studies. The decarboxylation product, 14-heptacosanone, did not react with OleC and OleD to produce any demonstrable product. Substantial hydrolysis of fatty acyl-CoA substrates to the corresponding fatty acids was observed, but it is currently unclear if this occurs in vivo. In total, these data are consistent with OleA catalyzing a non-decarboxylative Claisen condensation reaction in the first step of the olefin biosynthetic pathway previously found to be present in at least 70 different bacterial strains.
Keywords: Bacteria, Enzyme Catalysis, Fatty Acid, Metabolism, Protein Purification, OleA, Olefin Biosynthesis, Thiolase Superfamily, Xanthomonas
Introduction
Commodity hydrocarbons derive from petroleum, but nature provides a rich source of hydrocarbons for which biosynthetic pathways are being elucidated. Isoprenoid biosynthesis has been well studied (1), and an enzymatic decarbonylation of fatty aldehydes to produce alkanes has recently been demonstrated for cyanobacteria (2). It has been known for more than 40 years that some bacteria biosynthesize long (C23–C33) hydrocarbon chains containing a double bond at the median carbon via a mechanism known as a “head-to-head” condensation of fatty acyl groups (3–8). For example, bacteria from the genus Arthrobacter produce largely C15 fatty acids (9) and make predominantly C29 olefins (10). These observations are consistent with studies in 1969 showing the loss of the 14C label at carbon-1 of one of the acyl groups undergoing head-to-head condensation (6). These early in vitro studies were conducted with crude cell protein extracts. It was not until 2010 that the genes involved in the head-to-head biosynthetic pathway were described in the peer-reviewed literature (11, 12), providing new insights into the biosynthetic pathway based on a bioinformatics analysis of the gene and protein families.
OleA is homologous to proteins in the thiolase or condensing enzyme superfamily (11, 13). This is a very large superfamily of over 13,000 known proteins. The known thiolase superfamily proteins typically catalyze condensation reactions between acyl-thioester substrates, either with or without the loss of a carboxyl group. Approximately 70 bacteria are known to contain genes denoted as oleABCD, and those tested produce long-chain olefinic hydrocarbons (13). The precise role of each ole gene product in the biosynthesis remains to be defined. When the oleC gene is deleted or only the oleA gene is present in vivo, a long-chain ketone(s) is observed. These data supported the idea that OleA is involved in the initial stages of the head-to-head hydrocarbon biosynthetic reactions (11, 13, 14).
There are two alternative proposals in the literature regarding the OleA condensation reaction (Fig. 1). Beller et al. (11) (Fig. 1A) have proposed that OleA catalyzes a decarboxylative condensation between a β-ketoacyl-CoA and a fatty acyl-CoA. Sukovich et al. (13) (Fig. 1B) have proposed that OleA catalyzes a non-decarboxylative Claisen condensation between two fatty acyl-CoA substrates. These two types of condensation reactions are difficult to differentiate in vivo, where both fatty acyl-CoAs and β-ketoacyl-CoAs may be present simultaneously, and many enzymes are present. The study by Beller et al. (11) used a purified OleA enzyme, but their demonstration of activity required the addition of a crude soluble protein extract from Escherichia coli. The proposed β-ketoacyl-CoA substrate was suggested to have been generated from the corresponding acyl-CoA by the proteins present in the E. coli soluble fraction. A clear differentiation between OleA reaction A and B could be obtained using a purified OleA preparation in admixture with defined substrates in vitro. The two types of condensation reactions could also be differentiated by determining the reaction product. OleA reaction A produces a 1,3-diketone, whereas OleA reaction B yields a β-ketoacid (Fig. 1).
FIGURE 1.

Fundamentally different condensation mechanisms have been proposed for OleA: decarboxylative condensation between a β-keto ester and an acyl thioester (11) (A) or non-decarboxylative condensation between two acyl thioesters (13) (B).
There are other important questions that can be answered directly using a purified OleA protein and purified single substrates. These include determining the substrate specificity of OleA with respect to chain length, determining the complete reaction stoichiometry, determining what drives the apparent Claisen condensation to completion, and revealing why cloning oleA genes in heterologous hosts produces monoketones. These issues are addressed in the present work.
The OleA protein from Xanthomonas campestris was cloned, overexpressed in E. coli, and purified to homogeneity. The putative product of the reaction was synthesized chemically to allow comparison with the biochemical product. OleA was shown to react with myristoyl2-CoA to produce the corresponding β-ketoacid via a non-decarboxylative Claisen condensation reaction. This intermediate was shown to react, in the presence of OleC and OleD, to yield a long-chain olefin. In the absence of OleC and OleD, the product of the OleA reaction was shown to undergo spontaneous chemical decarboxylation to yield a ketone. This explains previous in vivo observations of ketone formation with the expression of an oleA gene in a heterologous host (11, 13).
EXPERIMENTAL PROCEDURES
Chemical Synthesis and Analysis
β-Ketocarboxylic acid syntheses have been previously reported (15), but the literature does not describe the synthesis of higher benzyl esters of β-ketocarboxylic acids derived from fatty acids. The detailed procedure used for the synthesis of 2-myristoylmyristic acid (2-dodecyl-3-ketohexadecanoic acid) is described in the supplemental material.
In brief, the forced Claisen condensation method described by Briese and McElvain (16, 17) was adapted to the coupling of benzyl myristate, using a half-equivalent of sodium benzyl alcoholate in benzyl alcohol as the basic promoter, removing benzyl alcohol by heating under vacuum to force the condensation nearly to completion. Benzyl 2-myristoylmyristate (benzyl 2-dodecyl-3-ketohexadecanoate) so prepared (70% yield and ∼90% purity after bulb-to-bulb distillation) crystallized, mp 34.5–35.8 °C. The purity of benzyl 2-myristoylmyristate was estimated from NMR data in CDCl3 solution, which show no conclusive evidence for tautomeric enolic forms being present.
Carefully monitored hydrogenolysis of benzyl 2-myristoylmyristate (Pd/C catalyst, methyl t-butyl ether solvent), isolated by filtration and cooling of the filtrate to −80 °C, produced a 5:2 (mol/mol) mixture of 2-myristoylmyristic acid and its decarboxylated product 14-heptacosanone as determined by GC-MS analysis after methylation of the acid (CH2N2). On cold storage (−80 °C), the solution was enriched to 8:1 2-myristoylmyristic acid and the derived ketone, apparently by preferential precipitation of the ketone.
Cloning and Expression of OleA
Synthetic oleA genes were designed based on oleA genes from Congregibacter litoralis KT71 (ZP_01103251.1), Xanthomonas campestris spv. campestris str. ATCC 33913 (NP_635607.1), Xylella fastidiosa 9a5c (NP_299252.1), Plesiocystis pacifica SIR-1 (ZP_01906524.1), and γ-proteobacterium NOR5-3 (ZP_05127044.1) (see supplemental Fig. S1) and purchased from DNA 2.0 (Menlo Park, CA). The genes were cut with NdeI and BamHI restriction enzymes and cloned into pET28b+ (Novagen, Madison, WI). All five genes were separately transformed into E. coli One Shot BL21 (DE3) (Invitrogen). All five recombinant strains were screened for soluble protein expression in 50-ml cultures induced for 4 h at 37 °C. Two of the five constructs expressed soluble protein in E. coli; only X. campestris was found to be active in vitro, and that was selected for further study.
E. coli for OleA purification was cultivated under two different conditions. Small scale cultivations were conducted in 2-liter flasks containing 500 ml of LB with 50 μg/ml kanamycin and induced at an A600 of 0.7–0.85 with 0.1 mm isopropyl-β-d-thiogalactopyranoside (IPTG).3 After 4 h, cells were harvested by centrifugation for 25 min at 3000 × g. Large scale cell cultivation was conducted in the Biotechnology Resource Center, University of Minnesota. A 440-liter culture was prepared in a 550-liter DCI bioreactor (DCI-Biolafitte, St. Cloud, MN) using a Rhapsody digital controller system and induced with 0.5 mm IPTG. Cells were harvested, lyophilized, and then stored at −80 °C.
Purification of OleA
Cells were resuspended in 20 mm sodium phosphate buffer, 500 mm NaCl, pH 7.4, with EDTA-free protease inhibitor tablets (Roche Applied Science). Cells were disrupted by three passes through a chilled French pressure cell at 1200 p.s.i. and centrifuged at 27,000 × g for 90 min to obtain the soluble protein fraction. The soluble fraction was centrifuged at 27,000 × g for 30 min to clear, prior to loading onto an Amersham Biosciences LCC 501 FPLC equipped with a 5-ml Ni(II)-loaded HisTrap HP column (Amersham Biosciences) equilibrated with 20 mm sodium phosphate, 500 mm NaCl, pH 7.4, buffer. The OleA protein was eluted at 135 mm imidazole. Ten g, wet weight, of cell paste yielded 60 mg of purified OleA. Fractions were analyzed by SDS-PAGE and Simply Blue Safestain (Invitrogen). Pooled fractions were concentrated, and imidazole was removed, with three passes through a 50-ml pressure concentrator (Amicon) using a 10,000 molecular weight cut-off membrane (Millipore). Alternatively, after concentration of fractions, the OleA protein was dialyzed three times at 4 °C to remove the imidazole. Protein concentrated up to 30 mg/ml remained soluble and active.
Identification and Expression of Active OleD
Using the NCBI Blast algorithm (18), oleD genes were identified. Sequences from Chloroflexus auranticus (Caur_3530), γ-proteobacterium NOR5-3 (ZP_05127041.1), Xylella fastidiosa Temecula1 (NP_779252.1), and X. campestris spv. campestris str. ATCC 33913 (NP_635614.1) were optimized for expression in E. coli (see supplemental Fig. S2) and cloned into pJexpress expression vectors with a T7 promoter by DNA 2.0. Vectors were transformed into E. coli One Shot BL21 (DE3) (Invitrogen). Proteins were screened for activity and expression using 50-ml LB cultures with 50 μg/ml kanamycin. Cells were induced with 0.1 mm IPTG at an A600 of 0.55–0.75. Soluble cell extracts were combined with OleA, OleC, and cofactors to test for the production of alkenes using the GC-MS enzyme assay described below. The OleD protein originating from X. campestris was the only protein found to support alkene biosynthesis. Cultures were scaled up using 2-liter flasks containing 500 ml of LB with 50 μg/ml kanamycin. Cultures were grown at 37 °C with agitation at 225 rpm. The culture was induced at an A600 of 0.7–0.8 with 0.1–0.4 mm IPTG and grown at 30 °C shaking at 225 rpm for 20 h. Cells were harvested by centrifugation for 25 min at 3000 × g.
Purification of OleC and OleD and Assay of OleD
The cloning, expression, and purification of OleC was described previously (19). For purification of OleD, cell pellets were resuspended in 20 mm sodium phosphate, 500 mm NaCl, pH 7.4, with EDTA-free protease inhibitor tablets (Roche Applied Science) and passed through a chilled French pressure cell three times at 1200 p.s.i. The cell lysate was centrifuged at 27,000 × g for 90 min, and the soluble fraction was centrifuged for an additional 30 min. The soluble fraction was passed though a 0.20-μm syringe filter prior to chromatography. A 5-ml Ni(II)-loaded HisTrap HP column (Amersham Biosciences) equilibrated with 20 mm sodium phosphate, 500 mm NaCl, pH 7.4, buffer was used for purification. Alternatively, 50 mm MOPS, 1% Tween 20, pH 7.0, was used for purification to improve solubility. Fractions were analyzed for purity by SDS-PAGE and Simply Blue Safestain (Invitrogen). Fractions eluting at 450 and 500 mm imidazole were pooled. The pooled protein was concentrated using a 50-ml pressure concentrator (Amicon) with a 10,000 molecular weight cut-off membrane (Millipore) and dialyzed three times at 4 °C. After dialysis, protein was centrifuged at 14,000 × g for 15 min at 4 °C to remove precipitated protein.
OleD was previously suggested to be a ketone reductase (13). It was shown here to be active in a 250-μl reaction mixture consisting of 100 mm Tris, pH 7.4, containing OleA, OleD, OleC, 8 mm MgCl2, 80 μm ATP, 260 μm myristoyl-CoA, and 120 μm NADPH. NADPH oxidation was followed spectrophotometrically at 340 nm.
Detecting the Release of CoASH Thiol for Assay of OleA Substrate Range
Release of the free thiol group of CoASH was detected by the addition of 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB) measured spectrophotometrically at 412 nm (ϵ412 = 13,600 m−1 cm−1) (20, 21). Acyl-CoA substrates, purchased from Sigma-Aldrich, were reacted with OleA protein in 100 mm Tris, pH 7.4, and incubated at room temperature for 5 min in either 1 ml or 250 μl. DTNB was incubated with the reaction mixture for 2 min and quantified spectrophotometrically.
Hydrocarbon Detection Enzyme Assay
A glass vial containing a 250-μl total volume of 100 mm Tris, pH 7.4, with 200–600 μg of OleA, 5–25 μg of OleD, 66 μg of OleC, 1.4 mm NADPH, 8 mm MgCl2, 3 mm ATP, and 1.2 mm myristoyl-CoA or an excess of 14-heptacosanone was incubated overnight at 30 °C with gentle shaking. Products were extracted with 250 μl of ethyl acetate using 16-hentriacontanone ketone (Tokyo Kasei Kogyo Co., Ltd.) as an internal standard. After vortexing and 5 min of gentle centrifugation, the top solvent layer was transferred to a glass vial and analyzed using a gas chromatograph equipped with a flame ionization detector HP 7890A (Hewlett Packard, Palo Alto, CA) and mass spectrometer HP 5975C (GC-MS-FID). GC was conducted under the following conditions: helium gas, 1.75 ml/min; HP-1ms column (100% dimethylsiloxane capillary; 30 m × 250 μm × 0.25 μm); temperature ramp, 100–320 °C; 10 °C/min, hold at 320 °C for 5 min, 250 °C injection port, and split at the outlet between MS and FID. The mass spectrometer was run under the following conditions: electron impact at 70 eV and 35 μA. The flame ionization detector was set at 250 °C with hydrogen flow set at 30 ml/min, air set at 400 ml/min, and helium makeup gas set at 25 ml/min. GC-MS was also used as described (10) for detecting ketones derived from spontaneous decarboxylation of the OleA β-keto acid products, using 200–600 μg of OleA and 1 mm acyl-CoA substrates.
Radiolabeled Acyl-CoA Assay
Reactions of 200 μl included 0.2 μCi of [1-14C]myristoyl-CoA (40–60 mCi/mmol; American Radiolabeled Chemicals (St. Louis, MO)), 750 μm myristoyl-CoA, and 1 mg of OleA in 100 mm Tris, pH 7.4. Samples were analyzed using high pressure liquid chromatography (HPLC) on a Shimadzu HPLC system equipped with a UV detector (Shimadzu, Columbia, MD) and a β-ram radioflow detector operated with the Laura 4 data acquisition/evaluation software (IN/US Systems, Tampa, FL). UV detection was set at 259 or 274 nm. Unfiltered samples of 50- or 100-μl volume were injected onto an analytical reverse phase Alltima HPC8 column with 5-μm packing (Alltech 250 × 4.6 mm) and a C8 guard column. The column was equilibrated in 50% 20 mm ammonium acetate, pH 5.4 (A) and 50% 85:15 acetonitrile/methanol (B) and the following method adapted from Ref. 22. Linear gradients were as follows: 50% A, 50% B, 0–10 min; ramp to 70% B, 10–15 min; 70% B, 15–30 min; ramp to 100% B, 30–35 min; 100% B, 35–50 min; return to 50% A, 50% B, 50–55 min; and equilibrate, 55–70 min. The flow rate was 1 ml/min. The scintillant (Monoflow X; National Diagnostics, Atlanta, GA) flow rate was 3 ml/min.
Mass Spectrometry Analysis
Mass spectrometry on enzymatically produced and synthetic β-keto acid was performed using an LCQ-classic (Thermo Fisher Scientific) ion trap mass spectrometer with electrospray ionization (ESI) mode. Samples were introduced by loop injection of 5 μl. Product ion spectra for 2-myristoylmyristic acid, m/z 437 (M − H), was identified in negative ion mode, as well two additional ions, m/z 393 and m/z 473/475. The fatty acid HPLC peak was analyzed by direct infusion into a Quantum Discovery Max (Thermo Finnigan) mass spectrometer operated in negative ion mode. ESI−-MS spectra for myristic acid was m/z 227 (M − H). Electron impact mass spectrometry in conjunction with GC (GC-MS) was performed to obtain the spectra of the β-keto acid derivatized with diazomethane. The solvent was evaporated with N2 and then taken up in methyl-t-butyl-ether to run on GC-MS as described previously (10). The ketone molecular ion, m/z 394, was also identified by this method.
Analytical Gel Filtration
A Superdex 75 10/100 GL (Amersham Biosciences) size exclusion column was used on an AKTA (General Electric) FPLC with elution at 0.5 ml/min. The column was equilibrated with 20 mm sodium phosphate, 500 mm NaCl, pH 7.4. Molecular weight standards (Bio-Rad) were used with a range of 1350–670,000 to create a standard curve. Three additional standards were used in a closer Mr range to where OleA eluted, chymotrypsin (Mr = 25,000), albumin (Mr = 67,000), and conalbumin (Mr = 77,000).
RESULTS
Cloning and Expression of oleA Genes and Purification of OleA Protein
The oleA genes from C. litoralis KT71, X. campestris spv. campestris str. ATCC 33913, X. fastidiosa 9a5c, P. pacifica SIR-1, and γ-proteobacterium NOR5–3 were each cloned into E. coli and tested for the production of soluble OleA protein. The recombinant E. coli containing the X. campestris gene showed a high amount of soluble OleA protein, as determined by SDS-PAGE, and was found to be active in purified form, and that strain was therefore selected for further studies. E. coli cells expressing a His-tagged X. campestris OleA protein were grown in a 550-liter bioreactor vessel, harvested, and lysed. Following chromatography on a nickel column, the protein was shown to be homogenous, as indicated by SDS-PAGE (Fig. 2B).
FIGURE 2.
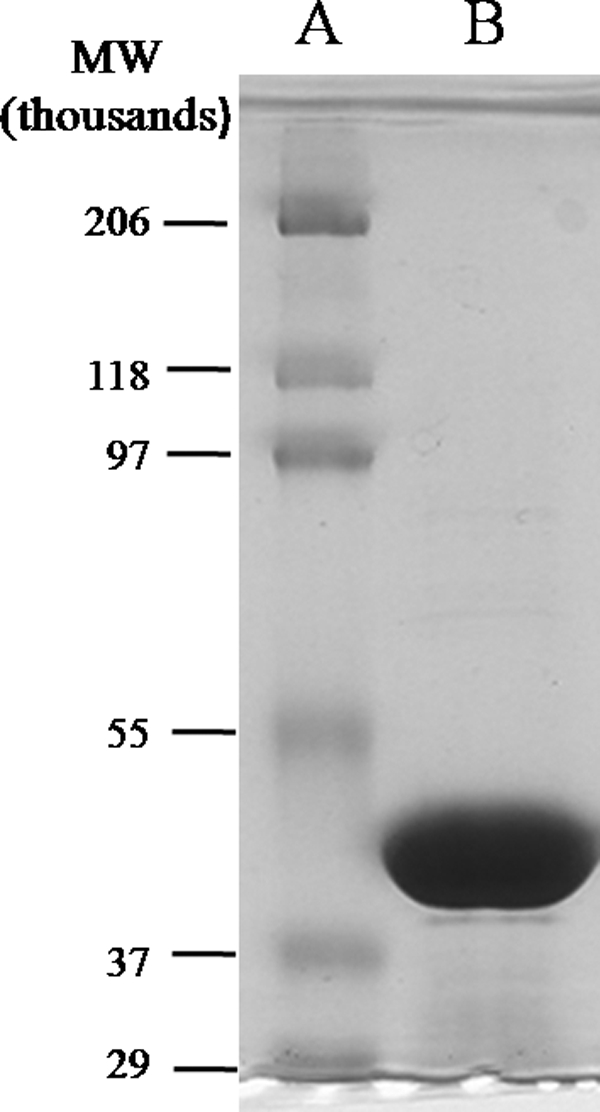
SDS-polyacrylamide gel showing standard molecular weight markers (A) and purified OleA (B).
General Characteristics of OleA
The subunit molecular weight of the native OleA protein is 36,629, but, as engineered here with the His tag, it is 38,792. The protein migrates somewhat higher than this on SDS-PAGE (Fig. 2B). The OleA protein subunit Mr is near the middle of the range found in homologous proteins from the thiolase superfamily (Table 1). The Mycobacterium Pks13 protein is a large multidomain protein with the condensing enzyme domain defined as 44,122 in the annotation on the NCBI server. The native molecular weight of OleA was estimated to be 62,000 by gel filtration chromatography. This is suggestive of a subunit stoichiometry of 2 for the native enzyme. Hydroxymethylglutaryl-CoA reductase, FabH, and the Mycobacterium Pks13 are all dimers, and the Zoogloea thiolase is a tetramer (23–26).
TABLE 1.
Properties of OleA compared with homologous proteins in the thiolase superfamily
| Protein (organism)a | Accession number | Sequence identityb | Calculated Mr | Calculated pI | Cellular function | Claisen mechanism | Sequence signaturec |
|---|---|---|---|---|---|---|---|
| % | |||||||
| OleA (X. campestris) | NP_635607 | 100 | 36,629 | 5.6 | Alkene biosynthesis | Proposed | NACLAFING |
| Non-decarboxylative | |||||||
| OleA (M. luteus) (11) | YP_002957382.1 | 38 | 36,653 | 4.8 | Alkene biosynthesis | Proposed decarboxylative | NACLGFVNG |
| Thiolase (Zoogloea ramigera) (23) | AAA27706.1 | 19 | 40,416 | 5.9 | PHB biosynthesis | Non-decarboxylative | QLCGSGLRA |
| HMG-CoA synthase (Homo sapiens) (26) | 1XPL_A | 16 | 43,204 | 5.0 | Mevalonate pathway | Non-decarboxylative | EACYAATPA |
| FabH (E. coli) (25) | 1EBL_A | 24 | 33,523 | 5.1 | Fatty acid biosynthesis | Decarboxylative | AACAGFTYA |
| Mycobacterium Pks13d (24) | CAA17864 | 19 | 44,122 | 5.2 | Mycolic acid biosynthesis | Decarboxylative | TACSSSLVA |
a Numbers in this column represent references from which the data in the table were obtained.
b Via Needleman-Wunch and BLAST algorithms and comparison with OleA from X. campestris.
c Amino acid sequence surrounding the active site cysteine conserved in thiolase superfamily proteins.
d Alignment to keto-acyl synthase domain only, as defined by NCBI.
The OleA protein shows a low sequence relatedness with homologous proteins in the thiolase superfamily (Table 1). With such divergence, it is not surprising that the cellular functions of the proteins are quite different. However, the general biochemical reaction catalyzed by all of the enzymes shown in Table 1 involves the condensation of acyl substrates. These condensation reactions occur by either a decarboxylative or non-decarboxylative (27) mechanism, and the results described below are consistent with a non-decarboxylative mechanism for OleA. In both cases, thiolase superfamily proteins use a conserved active site cysteine and generate an acyl enzyme intermediate (28). OleA shares this conserved active site cysteine residue (Table 1). In the vicinity of the conserved cysteine, the amino acids in the OleA from X. campestris and Micrococcus luteus (11) are highly conserved (Table 1).
Initial Defining of Substrate Specificity and Reaction Products of OleA
Enzymes catalyzing condensation or hydrolysis reactions with acyl-CoA substrates release coenzyme A that can be assayed colorimetrically using DTNB (20, 29–31). Both proposed mechanisms (Fig. 1, A and B) showed OleA-catalyzed coenzyme A release, and this assay was used to monitor enzyme activity during purification. With purified OleA, DTNB was used to determine the stoichiometry of coenzyme A formation and to begin to discern the substrate specificity of OleA.
First, the coenzyme A product stoichiometry was determined using either myristoyl-CoA or palmitoyl-CoA and allowing the substrate to completely react. With either substrate, the reaction stoichiometry was 1.0 mol of coenzyme A released/mol of acyl-CoA consumed. In this manner, acyl-CoA chains of different lengths were tested with OleA using a time of incubation in which palmitoyl-CoA reacts completely as described under “Experimental Procedures.” Under those conditions (Table 2), palmitoyl-CoA reacted more completely than myristoyl-CoA. Octanoyl-, decanoyl-, lauroyl-, palmitoleoyl-, and stearoyl-CoA were also found to undergo reaction to release coenzyme A (Table 2), but acetyl-CoA did not.
TABLE 2.
Substrate specificity of OleA as determined by CoA release
Values shown are the average of triplicate determinations with S.E.
| Substrate |
CoA producta | Percentage of theoretical yieldb | |
|---|---|---|---|
| Common name | Carbon chain length | ||
| μm | % | ||
| Palmitoyl-CoA | 16 | 65.0 ± 0.9 | 100 |
| Myristoyl-CoA | 14 | 63.2 ± 0.4 | 97 |
| Lauroyl-CoA | 12 | 51.4 ± 1.9 | 79 |
| Palmitoleoyl-CoA | 16 | 36.9 ± 0.9 | 57 |
| Decanoyl-CoA | 10 | 27.2 ± 1.6 | 42 |
| Stearoyl-CoA | 18 | 18.7 ± 1.8 | 29 |
| Octanoyl-CoA | 8 | 8.0 ± 2.2 | 12 |
| Acetyl-CoA | 2 | NDc | ND |
a Free coenzyme A detected as described under “Experimental Procedures.”
b Starting substrate was 65 μm; 65 μm product is 100% of theoretical yield.
c ND, no detectable activity.
Subsequent experiments were conducted to examine if coenzyme A was formed as a consequence of acyl-group condensation, thioester bond hydrolysis, or some mixture of the two reactions. Based on previous observations (11, 13, 14), it was known that long-chain ketones were the observed condensation products. In subsequent experiments in this study, it was shown that β-keto acids are the initial products, and those decarboxylate quantitatively to the corresponding ketone. In this context, reaction mixtures were solvent-extracted and subjected to GC-MS to identify ketones derived from condensation and/or fatty acids derived from acyl chain hydrolysis.
Previous in vivo experiments identified asymmetric ketones, indicating that fatty acyl chains of different chain lengths could be condensed (13). In this context, experiments were conducted with mixtures of fatty acyl-CoA substrates. All pairwise combinations of saturated C10, C12, C14, and C16 and C16 monounsaturated (C16:1) acyl-CoA substrates were incubated, extracted, and analyzed for products by GC-MS and GC-FID. In all, 15 product mixtures were analyzed. The results are shown in Table 3. It was found that acyl-CoA hydrolysis to the corresponding fatty acid was a major reaction in most cases. Only with C12 acyl condensation and C14 plus C16:1 condensation were the major products derived from a condensation of fatty acyl chains. In the case of C14 condensations (myristoyl-CoA), the ketone 14-heptacosanone was produced at only slightly lower levels than the hydrolysis product, myristic acid.
TABLE 3.
Product ratios determined by GC-MS for reactions of OleA with acyl-CoA substrates of different carbon chain lengths as indicated by the left-hand column and the top row
The products, ketones (Cx) and fatty acids (FAx), are indicated in order of decreasing abundance as determined by peak area integration as described under “Experimental Procedures.” The observed partitioning between condensation of similar or different chains or acyl-CoA hydrolysis is illustrated at the bottom.

The identification of ketones as the condensation products by use of GC-MS led to the question of whether the decarboxylation was enzymatic or whether the decarboxylation occurs due to the labile nature of the reaction intermediate preceding the formation of the ketone. Further investigations were conducted using a C1-labeled acyl-CoA substrate to track the carboxyl carbon.
Identification of Initial Condensation Product
OleA reactions with [1-14C]myristoyl-CoA were analyzed using HPLC fitted with a radioflow detector. A major peak eluting at 22.4 min was identified as myristic acid (Fig. 3). The HPLC peak eluting at 44.3 min (compound 2) was analyzed by GC-MS and found to be 14-heptacosanone, but more of the radioactivity co-migrated with a more polar product eluting at 40.0 min (Fig. 3). The major peak eluting at 40.0 min (compound 1) showed very little absorbance at 259 nm, consistent with the absence of a coenzyme A moiety. Over time, the peak at 40 min diminished with a concomitant increase in the peak at 44.3 min (Fig. 3, inset). This observation was consistent with a decarboxylation of the compound at 40.0 min giving rise to increasing concentrations of 14-heptacosanone over the course of 6.5 h. This was also indicated because the compound eluting at 44.3 min contained only half of the 14C as did the compound at 40 min, consistent with a loss of a carbon atom as carbon dioxide. Moreover, β-keto acids are known to be labile, and the decarboxylation of 2-myristoylmyristic acid would be expected to produce 14-heptacosanone.
FIGURE 3.

OleA reaction products with [14C]myristoyl-CoA as the substrate. A, HPLC profile showing radioactive peaks. Inset, plot of the radioactivity detected in product 1 and product 2 over the course of 6 h when a reaction mixture was incubated at room temperature. B, schematic of the reactions leading to the formation of product 1 and product 2.
To investigate this further, the benzyl ester of 2-myristoylmyristic acid was synthesized. It was hydrogenolyzed with palladium and hydrogen to produce 2-myristoylmyristic acid. This latter compound was observed to undergo rapid decarboxylation to produce 14-heptacosanone.
Treatment of synthetic 2-myristoylmyristic acid with diazomethane yielded the methyl ester. The synthetic methyl ester was compared with the enzyme-produced compound collected at 40.0 min that had been immediately reacted with diazomethane. Both methylated compounds showed a GC retention time of 20.6 min and essentially identical mass spectra (Fig. 4). The parent ion at m/z 452 is present in both, but it is a minor ion. In this context, electrospray ionization mass spectrometry was conducted on the free acid product 1 from the OleA reaction with myristoyl-CoA (Table 4, top) and the synthetic standard 2-myristoylmyristic acid (Table 4, bottom). In this case, a major negative ion was observed (m/z 437) with a mass of 1 Da less than the molecular mass of 2-myristoylmyristic acid in both the biological product and the standard. A second major fragment of m/z 393 found in both is consistent with the loss of carbon dioxide in the mass spectrometer. Another ion fragment was detected at m/z 473/475, suggested to be [M − H + HCl].
FIGURE 4.

Mass spectra for products from the reaction of diazomethane with product 1 (40 min retention time) from the reaction of OleA with 2-myristoylmyristic acid (A) and synthetic 2-myristylmyristic acid (B).
TABLE 4.
ESI mass spectrometry of product 1 from reaction of OleA with myristoyl-CoA as shown in Fig. 3 and synthetic 2-myristoylmyristic acid
| Sample | Molecular mass | ESI, negative ion mode m/z (intensity) |
|---|---|---|
| Da | ||
| Product 1 | 393 (52), 437 (29), 473 (19) | |
| 2-Myristoylmyristic acid | 438 | 393 (45), 437 (36), 473 (19) |
Role of OleA in Olefin Biosynthesis
OleA has been proposed to function with other Ole proteins to produce olefins (11, 13, 14). Other Ole proteins were purified as described under “Experimental Procedures” and tested in admixture with OleA and with myristoyl-CoA as the substrate. Gas chromatography-mass spectrometry was used because it can detect both the OleA product following its decarboxylation to 14-heptacosanone and the expected olefin 14-heptacosene if the entire biosynthetic pathway were functional. Fig. 5 shows that OleA and OleC in admixture produced only 14-heptacosanone (elution time 21.8 min), the product observed with OleA alone. However, when OleA was incubated with myristoyl-CoA, OleC, and OleD, the olefin 14-heptacosene (20.4 min) was observed in addition to the peak corresponding to 14-heptacosanone. The identities of the products were confirmed by mass spectrometry.
FIGURE 5.
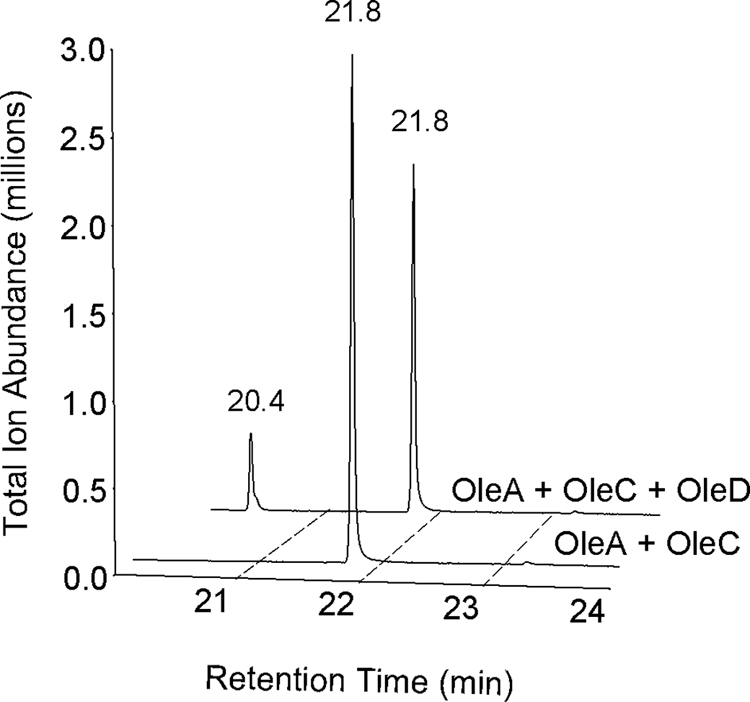
Gas chromatogram with MS detector showing products observed upon co-incubations with OleA and OleC (foreground trace) and OleA, OleC, and OleD (background trace).
To directly demonstrate that 2-myristoylmyristic acid was the intermediate giving rise to the olefin, we incubated synthetic 2-myristoylmyristic acid with OleC and OleD. The experiment yielded 14-heptacosanone and the expected olefinic product from head-to-head condensation, 14-heptacosene (Fig. 6, peak A, 20.4 min). The identity of the compound was confirmed by the mass spectrum shown above the GC chromatogram in Fig. 6A, with the major ion, m/z 378, representing the molecular ion.
FIGURE 6.

Gas chromatogram and accompanying mass spectra of peaks eluting between 20.3 and 20.4 min from extract of reaction mixture containing OleC and OleD incubated with 2-myristoylmyristic acid (A), extract of reaction mixture containing OleC and OleD incubated with 14-heptacosanone (B), and extract of reaction mixture containing OleC incubated with 14-heptacosanone (C).
Next, it was tested if the ketone 14-heptacosanone could also give rise to 14-heptacosene or any other discernible product. In this experiment (Fig. 6B), OleC and OleD were incubated with 14-heptacosanone under the same conditions as described above, and the olefinic product 14-heptacosene was not detected. A minor peak was observed at 20.35 min that had a mass spectrum different than that of 14-heptacosene (m/z 356). The small 20.35 min peak was also present in incomplete reaction mixtures that lacked OleD (Fig. 6C), further demonstrating that it is a contaminant and not relevant to olefin biosynthesis. It was found to be a minor impurity in the synthetic ketone preparation.
Overall, these data suggest that X. campestris OleA produced β-ketoacid intermediates from acyl-CoAs (C8–C18) and that the ketone is a non-physiological product arising from spontaneous decarboxylation. Note that ketones have been observed in vivo in recombinant bacteria containing heterologous oleA genes (11, 13). Additionally, Albro et al. (8) incubated ketones with crude protein fractions and failed to observe olefins. However, when a full suite of ole genes are present in native hosts, olefinic products, and not ketones, are typically observed (12). Thus, previous in vivo results are fully consistent with the in vitro data obtained in the present study.
DISCUSSION
In this study, the OleA protein from X. campestris was purified to homogeneity and shown to condense fatty acyl-CoA substrates to produce a condensed β-ketoacid with the release of 2 mol of CoA. The β-ketoacid, synthesized chemically or enzymatically, was shown to undergo further metabolism to yield a long-chain olefin in the presence of OleC and OleD. These studies confirmed that OleA catalyzes the first reaction in alkene biosynthesis with acyl-CoA substrates and carries out a non-decarboxylative Claisen condensation reaction.
An OleA protein was previously purified from M. luteus, and it was proposed to catalyze a different reaction (11) than the one demonstrated here with the OleA protein from X. campestris. The Xanthomonas and Micrococcus OleA proteins showed 38% sequence identity (Table 1) in a pairwise alignment of their amino acid sequences (18), so they could conceivably catalyze different reactions. The oleA genes from both organisms cluster with oleBCD genes. In the Micrococcus genome, the oleB and oleC genes are fused and likely produce a multidomain protein. However, the OleA, OleB, OleC, and OleD domains are present in both organisms. It was shown in the present study that OleC and OleD proteins act on the β-ketoacid product generated by X. campestris OleA to produce a long-chain olefin. When the Micrococcus oleA gene was cloned and expressed in E. coli, long-chain ketones were observed (11). In the present study, the recombinant E. coli strain expressing the X. campestris OleA protein alone was also observed to produce long-chain ketones that were not observed in the wild-type E. coli (data not shown). The in vitro data in this study showed that the ketones readily arise from the decarboxylation of a corresponding β-ketoacid intermediate. These observations are all consistent with a non-decarboxylative Claisen condensation as shown in Fig. 1B and difficult to reconcile with the proposed decarboxylative reaction shown in Fig. 1A.
The reaction catalyzed by OleA is somewhat reminiscent of the Zoogloea thiolase reaction that catalyzes the first step in the biosynthesis of polyhydroxybutyrate (23). In the latter reaction, however, the condensed product is a β-ketoacetyl-CoA, acetoacetyl-CoA, and with OleA, the product is a β-keto acid. Several lines of evidence strongly suggested that OleA does not produce a β-ketoacetyl-CoA that is hydrolyzed to the acid by another enzyme. First, the oleA gene was cloned as a single open reading frame (ORF) from synthetic DNA and expressed in E. coli, a bacterium that does not natively synthesize hydrocarbons. Enzymes capable of hydrolyzing 2-myristoylmyristoyl-CoA are not likely to be present in E. coli. Second, OleA was highly purified as shown by SDS-PAGE (Fig. 2), so even the unlikely E. coli hydrolytic enzyme would have been removed. Last, our HPLC conditions would have detected 2-myristoylmyristoyl-CoA, and this was never detected.
Based on the data obtained and the known role of the conserved cysteine found in other members of the thiolase superfamily, a working reaction mechanism can be presented for the OleA-catalyzed reaction (Fig. 7). We propose that initially an active site cysteine in the resting enzyme (Fig. 7A) is acylated, and coenzyme A is liberated (Fig. 7B). Subsequently, the tethered substrate is probably activated by an active site base to yield a carbanion on the tethered substrate (Fig. 7C). The carbanion then can react at the active site with the carbonyl carbon of a non-covalently bound acyl-CoA (Fig. 7D). That reaction forms a carbon-to-carbon bond with the condensed product still tethered to the enzyme cysteine and producing the second molecule of coenzyme A formed in the reaction cycle (Fig. 7E). The covalently bound condensation product can then undergo hydrolysis to yield the final β-ketoacid product and regenerate the free cysteine residue of the resting enzyme state (Fig. 7A). Although several features of this proposed mechanism are not yet demonstrated directly, there are multiple data that support this proposal. First, this mechanism explains the observed stoichiometry in which 2 mol of coenzyme A are observed/mol of condensed product. Second, the observed high rate of hydrolysis of acyl-CoAs to produce fatty acids is not unexpected if the enzyme has a mechanism to hydrolyze thioester-linked intermediates during its normal reaction cycle. Thus, there could be a kinetic competition between hydrolysis of the initially bound acyl group (Fig. 7B) and the tethered condensation product (Fig. 7E). Depending upon the binding affinity for the different length acyl-CoA used in the experiment described in Table 3, hydrolysis of intermediate 7B or 7E would occur preferentially. Last, proteins in the thiolase superfamily typically use an active site cysteine to acquire an acyl chain to initiate catalysis (27, 28), and the region around the cysteine residue shown in Table 1 is the most highly conserved region of OleA with other members of the superfamily.
FIGURE 7.

Proposed reaction cycle for OleA. The top of the cycle (A) shows the resting enzyme that reacts with an acyl-CoA to start the reaction cycle; B, enzyme with the covalently attached acyl chain; C, enzyme having abstracted a proton from the acyl chain to activate it; D, activated acyl chain reacting with the second acyl chain bound to the enzyme; E, condensed fatty acyl chains still bonded to the enzyme system just prior to hydrolytic release. CoASC(O)CH2R1 and CoASC(O)CH2R2, first and second reacting acyl-coenzyme A, respectively. B: attached by a line to Enz represents an enzyme base. The products of the reaction, two molecules of coenzyme A (CoASH) and a β-keto acid, are highlighted by boxes.
There are significant questions that remain to be addressed regarding this proposed mechanism (Fig. 7). First, the identity of the proposed cysteine nucleophile has not been directly demonstrated here. Second, the suggested generation of a carbanion (Fig. 7B) requires a general base that remains to be identified. Additionally, this mechanism would be supported by the identification of the binding sites for the acyl chains and that the chains are covalently and non-covalently bound, respectively.
This study identified the product of the OleA-catalyzed reaction to be a β-keto acid. The production of olefins required the presence of OleC and OleD in addition to OleA. These data indicated that OleC and OleD catalyze further reactions with the β-ketoacid intermediate generated by OleA. This was supported by experiments in which 2-myristoylmyristic acid was transformed to an olefin by OleC and OleD. The corresponding ketone was not transformed to an olefin, consistent with the idea that the ketone is not a physiologically relevant intermediate. There is also the issue that C-2 in 2-myristoylmyristic acid is a chiral center. The synthetic 2-myristoylmyristic acid is racemic, and it is plausible that only one enantiomer will react with OleD. The chirality of the reaction is currently under investigation.
Supplementary Material
Acknowledgments
We thank David Sukovich for providing a selection of oleA genes used in these studies. We acknowledge Fred Schendel and Mary Pruss for fermentation and cell harvesting. We especially thank Drs. Sharon Murphy and Linda von Weymarn for assistance with the HPLC radioflow equipment. We thank Brandon Goblirsch and Carrie Wilmot for insightful discussions regarding the OleA mechanism described here. We thank Dr. Burckhard Seelig and laboratory members J. Haugen and L. Hagman for the use of and for instruction in the use of the FPLC and size exclusion column. Mass spectrometry (ESI only) was conducted in the Center for Mass Spectrometry and Proteomics and Mass Spectrometry Services in the Masonic Cancer Center with the assistance of Tom Krick, Brock Matter, and Peter Villalta.
This work was supported by a University of Minnesota doctoral dissertation fellowship (to J. A. F.) and by Department of Energy ARPA-E Award DE-AR0000007 and the Initiative for Renewable Energy and the Environment (to L. P. W.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Method S1 and Figs. S1 and S2.
Myristoyl is equivalent to tetradecanoyl.
- IPTG
- isopropyl-β-d-thiogalactopyranoside
- DTNB
- 5,5′-dithio-bis-(2-nitrobenzoic acid)
- FID
- flame ionization detection
- ESI
- electrospray ionization.
REFERENCES
- 1. Muntendam R., Melillo E., Ryden A., Kayser O. (2009) Appl. Microbiol. Biotechnol. 84, 1003–1019 [DOI] [PubMed] [Google Scholar]
- 2. Schirmer A., Rude M. A., Li X., Popova E., del Cardayre S. B. (2010) Science 329, 559–562 [DOI] [PubMed] [Google Scholar]
- 3. Tornabene T. G., Oró J. (1967) J. Bacteriol. 94, 349–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Albro P. W., Dittmer J. C. (1969) Biochemistry 8, 394–404 [DOI] [PubMed] [Google Scholar]
- 5. Albro P. W., Dittmer J. C. (1969) Biochemistry 8, 953–959 [DOI] [PubMed] [Google Scholar]
- 6. Albro P. W., Dittmer J. C. (1969) Biochemistry 8, 1913–1918 [DOI] [PubMed] [Google Scholar]
- 7. Albro P. W., Dittmer J. C. (1969) Biochemistry 8, 3317–3324 [DOI] [PubMed] [Google Scholar]
- 8. Albro P. W., Meehan T. D., Dittmer J. C. (1970) Biochemistry 9, 1893–1898 [DOI] [PubMed] [Google Scholar]
- 9. Unell M., Kabelitz N., Jansson J. K., Heipieper H. J. (2007) FEMS Microbiol. Lett. 266, 138–143 [DOI] [PubMed] [Google Scholar]
- 10. Frias J. A., Richman J. E., Wackett L. P. (2009) Appl. Environ. Microbiol. 75, 1774–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beller H. R., Goh E. B., Keasling J. D. (2010) Appl. Environ. Microbiol. 76, 1212–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sukovich D. J., Seffernick J. L., Richman J. E., Hunt K. A., Gralnick J. A., Wackett L. P. (2010) Appl. Environ. Microbiol. 76, 3842–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sukovich D. J., Seffernick J. L., Richman J. E., Gralnick J. A., Wackett L. P. (2010) Appl. Environ. Microbiol. 76, 3850–3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Friedman L., Rude M. (September 18, 2008) International Patent WO2008/113041
- 15. Detalle J. F., Riahi A., Steinmetz V., Hénin F., Muzart J. (2004) J. Org. Chem. 69, 6528–6532 [DOI] [PubMed] [Google Scholar]
- 16. Briese R. R., McElvain S. M. (1933) J. Am. Chem. Soc. 55, 1697–1700 [Google Scholar]
- 17. Adams R., Bachmann W. E., Fieser L. F., Johnson J. R., Snyder H. R. (1942) Organic Reactions, Vol. 1, 9th Ed., John Wiley & Sons, Inc., New York [Google Scholar]
- 18. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Frias J. A., Goblirsch B. R., Wackett L. P., Wilmot C. M. (2010) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 1108–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alexson S. E., Nedergaard J. (1988) J. Biol. Chem. 263, 13564–13571 [PubMed] [Google Scholar]
- 21. Ellman G. L. (1958) Arch. Biochem. Biophys. 74, 443–450 [DOI] [PubMed] [Google Scholar]
- 22. Trivedi O. A., Arora P., Sridharan V., Tickoo R., Mohanty D., Gokhale R. S. (2004) Nature 428, 441–445 [DOI] [PubMed] [Google Scholar]
- 23. Davis J. T., Moore R. N., Imperiali B., Pratt A. J., Kobayashi K., Masamune S., Sinskey A. J., Walsh C. T., Fukui T., Tomita K. (1987) J. Biol. Chem. 262, 82–89 [PubMed] [Google Scholar]
- 24. Gavalda S., Léger M., van der Rest B., Stella A., Bardou F., Montrozier H., Chalut C., Burlet-Schiltz O., Marrakchi H., Daffé M., Quémard A. (2009) J. Biol. Chem. 284, 19255–19264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davies C., Heath R. J., White S. W., Rock C. O. (2000) Structure 8, 185–195 [DOI] [PubMed] [Google Scholar]
- 26. Clinkenbeard K. D., Sugiyama T., Reed W. D., Lane M. D. (1975) J. Biol. Chem. 250, 3124–3135 [PubMed] [Google Scholar]
- 27. Heath R. J., Rock C. O. (2002) Nat. Prod. Rep. 19, 581–596 [DOI] [PubMed] [Google Scholar]
- 28. Haapalainen A. M., Meriläinen G., Wierenga R. K. (2006) Trends Biochem. Sci. 31, 64–71 [DOI] [PubMed] [Google Scholar]
- 29. Skaff D. A., Miziorko H. M. (2010) Anal. Biochem. 396, 288–296 [DOI] [PubMed] [Google Scholar]
- 30. Sleeman M. C., Schofield C. J. (2004) J. Biol. Chem. 279, 6730–6736 [DOI] [PubMed] [Google Scholar]
- 31. Yashiro K., Kameyama Y., Mizuno-Kamiya M., Shin S. O., Fujita A. (1995) Biochim. Biophys. Acta 1258, 288–296 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.