

Abstract
TDP-43 is an evolutionarily conserved ubiquitously expressed DNA/RNA-binding protein. Although recent studies have shown its association with a variety of neurodegenerative disorders, the function of TDP-43 remains poorly understood. Here we address TDP-43 function using spermatogenesis as a model system. We previously showed that TDP-43 binds to the testis-specific mouse acrv1 gene promoter in vitro via two GTGTGT-motifs and that mutation of these motifs led to premature transcription in spermatocytes of an otherwise round spermatid-specific promoter. The present study tested the hypothesis that TDP-43 represses acrv1 gene transcription in spermatocytes. Plasmid chromatin immunoprecipitation demonstrated that TDP-43 binds to the acrv1 promoter through GTGTGT motifs in vivo. Reporter gene assays showed that TDP-43 represses acrv1 core promoter-driven transcription via the N-terminal RRM1 domain in a histone deacetylase-independent manner. Consistent with repressor role, ChIP on physiologically isolated germ cells confirmed that TDP-43 occupies the endogenous acrv1 promoter in spermatocytes. Surprisingly, however, TDP-43 remains at the promoter in round spermatids, which express acrv1 mRNA. We show that RNA binding-defective TDP-43, but not splice variant isoforms, relieve repressor function. Transitioning from repressive to active histone marks has little effect on TDP-43 occupancy. Finally, we found that RNA polymerase II is recruited but paused at the acrv1 promoter in spermatocytes. Because mutation of TDP-43 sites caused premature transcription in spermatocytes in vivo, TDP-43 may be involved in pausing RNAPII at the acrv1 promoter in spermatocytes. Overall, our study shows that TDP-43 is a transcriptional repressor and that it regulates spatiotemporal expression of the acrv1 gene during spermatogenesis.
Keywords: Amyotrophic Lateral Sclerosis (Lou Gehrig Disease), Chromatin Immunoprecipitation (ChIP), Chromatin Histone Modification, Gene Regulation, Neurodegeneration, Protein-DNA Interaction, RNA Polymerase II, Spermatogenesis, Transcription Repressor, TDP-43
Introduction
TAR DNA-binding protein of 43 kDa (TDP-43)3 is an evolutionarily conserved, ubiquitously expressed DNA/RNA binding nuclear protein. It was originally identified from a HeLa cell cDNA library as a transcription factor binding to the HIV transactivation response region (1). In vitro transcription as well as transient transfection assays showed that TDP-43 repressed HIV transactivation response mediated transcription (1). Since that initial report, the role of TDP-43 in transcription has not been studied. Subsequent studies have focused on the roles of TDP-43 in mRNA splicing and stability (2). Interest in TDP-43, however, increased exponentially after the discovery in 2006 that aberrantly truncated, phosphorylated, and mislocalized TDP-43 was present in the intracellular ubiquitinated inclusions in the brains of patients with frontotemporal lobar degeneration with ubiquitin-positive inclusions, amyotrophic lateral sclerosis, and Alzheimer disease (3). Although a large number of reports have since confirmed the link between TDP-43 and human neurodegenerative disorders, it is not yet clear how TDP-43 contributes to disease. This is due to the fact that very little is known about the normal nuclear function of TDP-43 (4). Understanding TDP-43 nuclear function is important to determine the contribution of loss-of-function to TDP proteinopathies. The evolutionarily conserved TDP-43 must play a fundamental role in biological processes because knock-out of TDP-43 leads to embryonic lethality in mice (5–7).
TDP-43 contains two RNA recognition motifs in the N-terminal half with which it recognizes UG/TG repeats in RNA/DNA and a C-terminal glycine-rich domain, considered important for protein-protein interactions (8). TDP-43 resembles the heterogeneous nuclear ribonucleoprotein family of RNA-binding proteins in terms of primary structure. Consistent with this, TDP-43 has been shown to bind RNA and regulate mRNA splicing in vitro and in cell culture assays (9). Work from our laboratory on testis-specific gene transcription, however, has shown that TDP-43 plays an additional role as a transcription factor (10, 11).
Our studies utilize the mouse acrv1 gene, which codes for the sperm acrosomal protein SP-10, as a model gene to understand the mechanisms of testis-specific gene transcription. The acrv1 mRNA is transcribed exclusively in the post meiotic round spermatids during spermatogenesis (12). We cloned TDP-43 from a mouse testis cDNA library as a transcription factor binding to the acrv1 promoter (10). The acrv1 promoter contains two GTGTGT motifs, canonical TDP-43 binding sites, at −172 and −160 positions on the antisense strand. EMSAs showed that recombinant TDP-43 binds to this region in a GTGTGT-dependent manner. Furthermore, our previous work using transgenic mice as a reporter system showed that mutation of the GTGTGT motifs in the −186/+28 acrv1 promoter leads to premature expression of a reporter gene in the meiotic spermatocytes, whereas the wild-type −186/+28 acrv1 promoter delivers correct post meiotic round spermatid-specific reporter gene expression (10). TDP-43 is expressed in spermatocytes as well as round spermatids. Based on the above data we hypothesized that TDP-43 represses the acrv1 gene transcription in spermatocytes. The present work addressed the following questions. 1) Does TDP-43 function as a transcriptional repressor, and if so, what are the domains necessary for repression? 2) Does TDP-43 bind to its putative target gene (acrv1) promoter in vivo in a physiological context? 3) How might TDP-43 transcriptional function be modulated? 4) What is the status of histone marks and RNAPII associated with TDP-43 promoter occupancy in vivo? Results presented in this study establish that TDP-43 is a transcriptional repressor and that the mouse acrv1 gene is a bona fide target gene for TDP-43 mediated repression in vivo.
EXPERIMENTAL PROCEDURES
Cell Lines and Culture Conditions
Mouse GC-2 spermatogenic cells (ATCC catalogue number CRL-2196) and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum, 1% l-glutamate, and 1% nonessential amino acids. COS-7 cells were maintained in DMEM with 10% fetal calf serum.
Antibodies
Mouse IgG Whole Molecule (Thermo Fisher Scientific; 31202), anti-guinea pig TDP-43 (in house); anti-rabbit TDP-43 (Abcam (Cambridge, MA); #50502), anti-RNAP II (Covance; Clone 8WG16; MPY-127R), anti-RNAPII phosphoserine 2 (Covance; Clone H5; MMS-129R), anti-RNAPII phosphoserine 5 (Covance; Clone H14; MMS-134R), anti-NELF-E monoclonal antibody raised against human full-length NELF-E (a kind gift from Dr. Yuki Yamaguchi, Yokohama, Japan), anti-pan aceylated-H3 (Ac-H3; Upstate Biotechnology; 06-599), anti-histone H3 lysine 4 trimethylation (H3K4me3; Upstate; 07-473), anti-H3 lysine 9 dimethylation (H3K9me2; Upstate; 07-441), anti-H3 lysine acetylation (H3K9ac; Abcam; ab4441), anti-FLAG (Sigma; F3165), anti-Gal4 DNA binding domain (DBD) (sc-510), anti-α-tubulin (Sigma; T9026), Cy3-conjugated anti-mouse IgG (Jackson ImmunoResearch Laboratories; 115-165-003), Cy3-conjugated anti-guinea pig IgG (Jackson ImmunoResearch Laboratories; 106-165-003), 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes (Eugene, OR); D-1306), and normal goat serum (Jackson ImmunoResearch Laboratories; 005-000-121).
Isolation of Mouse Spermatocytes and Round Spermatids
Pure populations (> 95% purity) of spermatocytes and round spermatids were isolated by Sta-Put gradient as described (10, 11). The outer membrane of each testis was decapsulated using forceps, and the seminiferous epithelia (tubules) were collected in a 10-cm dish and washed in 10 ml of DMEM. Tubules were dissociated in 10 mg of collagenase and 20 μg of DNase in 8.5 ml of DMEM for 10 min in a 37 °C incubator with gentle disruption. Tubules were washed twice with cold DMEM. The germ cells were released by enzymatic treatment with 7 mg of collagenase, 15 mg of hyaluronidase, 10 mg of trypsin, and 20 μg of DNase in 8.5 mg of DMEM for 10 min in the 37 °C incubator as before. Tubules were cut to 5-mm lengths with scissors to further enhance the digestion during enzymatic treatment. The entire volume was transferred to a 50-ml conical tube, reconstituted in 45 ml of DMEM, and allowed to sediment for 10 min on ice to separate the heavier tubule pieces away from the germ cells. The supernatants containing the germ cells were transferred to a fresh conical tube and centrifuged at 900 × g for 10 min at 4 °C. The cells were washed twice with PBS and loaded onto a 2–4% BSA Sta-Put gradient to separate the larger spermatocytes and smaller spermatids by gravity sedimentation for 3 h at 4 °C. Fractions (300 drops per fraction) of the heavier spermatocytes first followed by lighter round spermatids were collected over a 1-h period. Every fifth fraction of an average total of 70 fractions was observed under a light microscope to identify spermatocyte and round-spermatid fractions based on their morphology. Fractions of spermatocytes and round spermatids were centrifuged at 900 x g for 20 min at 4 °C and pooled separately. On average, the testes of 11 Swiss Webster mice (10–12 weeks old) yielded 22 × 106 spermatocytes and 104 × 106 round spermatids. The spermatocytes and round spermatids obtained were fixed in 1% formaldehyde and divided into 10 × 106 spermatocytes and 40 × 106 round spermatid aliquots for chromatin immunoprecipitations (ChIPs).
Cloning of TDP-43 Splice Variants from Testis Germ Cells
Spermatocytes and spermatids were separated by Sta Put method and flash-frozen in liquid nitrogen. 3 × 106 cells of each cell type were used to isolate RNA using the RNeasy Mini kit (Qiagen; 74104) adding the optional DNase step (Qiagen; 79254). Cells were disrupted using a homogenizer. cDNA was generated with 2 μg of RNA using the AffinityScript Multi Temperature cDNA Synthesis kit (Stratagene; 200436) at a temperature of 55 °C, with the TDP specific primer, TDP nested Rev1, CAGGTGATGAATCCATTTGACTTGA. This primer sits at bp 3138 of NM_145556.
For cloning out potential splice variants, we used the information available on GenBankTM for currently identified splice variants. The splice variants currently in the data base are of three different groups; that is, the annotated full-length protein and some C-terminal deletion (encompassing the Gly domain) variants that end with one of two different novel exons. Using a primer set that starts at the common ATG and ends at the most 3′ exon (the second novel terminal exon) will yield all splice variants currently in the data base (supplemental Fig. S3A). These primers were: TDP-43 forward (ATGTCTGAATATATTCGG) and TDP-43 reverse (v2, TCAAAGACGCAGCCTGT). The latter primer sits at bp 2268 of NM_145556.
The products of the above PCR were cloned using the TOPO TA cloning kit (Invitrogen; K461020). The products that deviated in size from the full-length protein were sequenced. Two major species were identified. One was spermatocyte-specific, and the other was spermatid-specific. These two splice variants differed by only 9 base pairs. There were several additional products that contained TDP sequence; however, these either did not code for a protein or had retained introns.
Extraction of Histones for Western Blot Analysis
Western blotting analysis of specific histone marks was carried out after histone extraction as described by Abcam. In brief, 107 GC-2 cells were harvested and washed in ice-cold PBS supplemented with 5 mm sodium butyrate. Cells were resuspended in Triton extraction buffer (0.5% Triton X-100, 2 mm phenylmethylsulfonyl fluoride (PMSF), 0.02% NaN3 in PBS) for 10 min at 4 °C with gentle end-to-end shaking. Nuclei were pelleted at 6500 × g for 10 min at 4 °C. Nuclei were recovered, washed in Triton extraction buffer, and pelleted again. Acid extraction of histones were carried out in 0.2 n HCl overnight at 4 °C. Samples were centrifuged as before, and supernatants containing histone proteins were recovered.
Immunofluorescence Microscopy
GC-2 cells (0.2 × 106/well) were grown on glass coverslips in 6-well chambers. Cells were transfected with Gal4-TDP-43 (wild-type) and all of the Gal4-TDP43 mutant constructs used in this study (1 μg/well) using Mirus TransIT®-LT1 to determine subcellular localization of the fusion proteins. Transfection of Gal4 DBD alone served as a control. All cells were fixed 48 h post-transfection in 4% buffered paraformaldehyde (Alfa Aaser; 43368) in PBS for 10 min at room temperature. Cells were permeabilized and blocked in 0.2% Triton X-100 and 10% normal goat serum in PBS for 10 min. Primary antibody incubations were carried out with either anti-GAL4 DBD (1:200) or anti-TDP-43 (in-house; 1:400) antibodies at room temperature for 35 min in 10% normal goat serum in PBS-Tween. Secondary antibody incubations were carried out at room temperature for 20 min. Anti-mouse (1:200) CY3-conjugated secondary antibodies diluted in 10% normal goat serum in PBS-Tween was used for visualization of anti-Gal4 DBD. Nuclei were stained with DAPI. Cells were visualized using an Olympus BX50 microscope. Nuclear localization was observed for all of the Gal4 TDP-43 fusion constructs used in this study (supplemental Figs. S1, A and B, and S4).
Gal4 Assay Constructs
Full-length mouse TDP-43 (mTDP-43; amino acids 1–414) was cloned in pFLAG-CMV and pFA-CMV vectors (Stratagene). The pFA-CMV clone was used as a template to generate two N-terminal truncation constructs (104–414, 191–414), two C-terminal truncations (1–200, 1–262), and 4 domain constructs as follows: RNA recognition motif 1 (RRM1) (104–200), RRM2 (191–262), RRM1 + 2 (104–262), Gly (274–414). Human TDP-43 (hTDP-43), ΔRRM1 (hTDP-43 ΔRRM1), and amino acid 147/149 mutant (hTDP-43 F147L/F149L) in pFLAG-CMV-2 vectors were kind gifts from Dr. Emanuele Buratti (13). These 3 hTDP-43 clones were cloned into pFA-CMV vectors (Stratagene). DBD-p53 activation domain was a kind gift from Dr. Rong Li (UT Health Science Center, Dept. of Molecular Medicine, San Antonio, TX).
Luciferase Reporter Gene Constructs
The −91/+28 ACRV1 reporter containing 5 Gal binding sites has been previously described (11). Briefly, 5X Gal element was PCR-amplified from the pFR-Luc plasmid (Stratagene) and ligated into the BGIII site of pGL3 −91/+28 Luc. The c-fos reporter containing four Gal binding sites was a kind gift from Dr. Rong Li and has been described elsewhere (14, 15).
Transient Transfections and Luciferase Assays
Transient transfections were performed in GC-2 and HeLa cells and COS-7 cells using Mirus TransIT®-LT1 (Mirus Corp.). 2 × 105 cells were plated overnight in 6-well tissue culture plates (BD Biosciences; 353046). Cells were 40–50% confluent at the time of transfections. 0.5 μg of reporter and 1 μg of effector (DBD-TDP-43 or empty vector DBD alone) were transfected per well GC-2 cell transfections. 0.2 μg of reporter was used per well of HeLa cell transfections. Renilla Luciferase was co-transfected at a 1:10 ratio. Cells were harvested 48 h post-transfection. Luciferase activities were measured by the Dual Luciferase reporter assay system (Promega) according to the instructions provided with the kit. For experiments using HDAC inhibitors, cells were treated 24 h post-transfection, and drug treatments lasted 24 h.
The reporter luciferase values were first divided by the Renilla luciferase values to normalize for transfection efficiency. These ratios were then expressed as a -fold change of control DBD vector-alone set as 1. Therefore, transcriptional repression with DBD-TDP-43 was defined as a value significantly lower than 1 as determined by one-way ANOVA followed by Bonferroni post-hoc test.
To verify whether the DBD fusion proteins were expressed correctly, the insoluble cellular material from the reporter assay experiments treated with the passive lysis buffer supplied with the Dual Luciferase assays system (Promega) was used. These samples were solubilized in 1× Laemmli buffer, separated by SDS-PAGE, and blotted with anti-DBD antibody. Western blots indicated that all of the DBD-TDP-43 constructs used in reporter assays in this study expressed fusion proteins of the expected molecular weight (supplemental Figs. S1B and S3C and Fig. 5C).
FIGURE 5.

Deletion of RRM1 or mutation of Phe-147 and Phe-149 within RRM1 relieves TDP-43 repressor function. The −91/+28 acrv1 promoter luciferase reporter shown in Fig. 1A was used in this study. A, schematics of hTDP-43, hTDP-43 ΔRRM1, and hTDP-43 F147L/F149L clones are shown. NLS, nuclear localization signal; NES, nuclear export signal. B, transcriptional repressor function of DBD-hTDP-43, DBD-hTDP-43 ΔRRM1, and DBD-hTDP-43 F147L/F149L is shown. 1 μg of empty vector (DBD) or DBD-TDP-43 was co-transfected with 0.5 μg of reporter into GC-2 cells. 0.05 μg of Renilla Luciferase was used to normalize for transfection efficiency. Cells were harvested 48 h later, and luciferase activities were measured. The release of repression observed with ΔRRM1 and F147L/F149L mutants is expressed as -fold release over full-length hTDP-43 set as 1. Results shown are the means ± S.E. for duplicate samples from three separate experiments. Asterisk represent significant difference (p < 0.05) compared with DBD-hTDP-43. C, Western blot (WB) analysis showing expected kDa sizes of DBD-hTDP-43 clones using anti-DBD antibody (1:400) is shown. Note: all DBD-hTDP-43 clones show nuclear localization (supplemental Fig. S4).
ChIP Analysis
ChIPs were performed as described (16). Cells were fixed with 1% formaldehyde in PBS for 20 min at room temperature, and cross-linking was stopped by adding 0.125 m glycine (Fisher; G45–212) for 5 min. Cells were pelleted at 170 × g for 10 min at 4 °C, washed twice with ice-cold PBS, and resuspended in 1 ml sonication buffer (1% Triton X-100, 0.1% deoxycholate, 50 mm Tris, pH 8.1, 150 mm NaCl, 5 mm EDTA) with protease inhibitors (2 μg of leupeptin (Sigma; L2884), 2 μg of aprotinin (Fisher; BP250310), 0.2 mm PMSF (Sigma; P7626)). Chromatin was sheared using a Sonicator W375 (Heat Systems-Ultrasonics, Inc., Farmingdale, NY). Sheared chromatin was precleared with 60 μl of protein A/G beads (Santa Cruz; sc-2003) and 2 μg of herring sperm DNA (Sigma; D3159) for 1 h at 4 °C. 200 μg of soluble chromatin was used for immunoprecipitation with control IgG antibody or specific target protein antibody (described above). 20 μg of chromatin was used to generate Input DNA for real-time quantitative PCR (qPCR) analysis, used as a reference for quantifying target DNA within immunoprecipitated samples. Chromatin was incubated with antibody overnight at 4 °C with rotation, after which 50 μl of protein A/G beads and 2 μg of herring sperm DNA was added for 2 h. The beads were washed sequentially with 1 ml each of sonication buffer containing high salt (500 mm NaCl), LiCl wash buffer (0.25 m LiCl, 0.5% IGEPAL CA-630, 0.5% deoxycholate, 0.01 m Tris, pH 8.1, 1 mm EDTA), and TE buffer (10 mm Tris, pH 7.5, 1 mm EDTA). Chromatin was eluted twice with 250 μl of elution buffer (1% SDS, 0.1 m NaHCO3, 0.01 mg/ml herring sperm DNA). Input and immunoprecipitated samples were then heated to 65 °C in a water bath for 4 h to reverse the formaldehyde cross-links. DNA fragments were ethanol-precipitated overnight at −20 °C and centrifuged at 16, 060 x g for 20 min at 4 °C to pellet the DNA. Pelleted DNA was washed with 70% ethanol, air-dried for 10 min, resuspended in 100 μl of TE buffer with 11 μl of proteinase K buffer (0.1 m Tris pH 7.5, 0.05 m EDTA, 5% SDS) and 1 μg of proteinase K (Bioline; BIO-37037), and heated to 55 °C in a water bath for 1 h. Samples were then diluted 4 times with TE buffer, extracted once with 500 μl phenol chloroform isoamyl alcohol (25:24:1), and ethanol-precipitated overnight at −20 °C. Precipitated DNA was centrifuged at 16,060 × g for 15 min at 4 °C, washed with 70% ethanol, air-dried for 10 min, and finally resuspended in 100 μl of TE buffer for qPCR.
qPCR
3-μl aliquots of each sample was used in triplicate for qPCR analysis of the acrv1 promoter (−267 to +27) or a far downstream region (+5652 to +5930). Thermal cycling was performed using an iCycler (Bio-Rad). SYBR Green I dye (Molecular Probes) was added at a 1:75,000 dilution in each 25-μl PCR reaction. qPCR was performed in triplicate so that an average correlation time (Tc) was determined for each Input sample, each control immunoprecipitation sample, and each antibody immunoprecipitation sample. Enrichments relative to Input DNA were calculated by using the 2ΔTc formula where ΔTc represents the cycle difference between each IP sample and input. Finally, all relative enrichments of DNA in antibody immunoprecipitation samples were expressed as -fold of control IgG samples set as 1. Factor binding to the acrv1 gene was determined to be significantly enriched using one-way ANOVA followed by the Bonferroni post-hoc test to determine difference from IgG background. Amplification of a downstream region of the acrv1 promoter at +5652 bp was used to indicate specificity of factor binding to the proximal acrv1 promoter.
Plasmid-based ChIPs
For plasmid-based ChIPs, transfections were performed in COS-7 cells. 1.5 × 106 cells were plated overnight in 100-mm tissue culture dishes (Corning Inc.; #430167). 3 μg of plasmid (bearing either wild-type or GTGTGT-mutant −186 + 28 acrv1 promoter) and 3 μg of FLAG TDP-43 were transfected as described above. ChIPs were performed as described above. 500 μg of soluble chromatin was immunoprecipitated with control IgG or anti-FLAG antibody. 3-μl aliquots of each sample was used in triplicate for qPCR analysis using plasmid-specific PCR primers (−186 to +27). Factor binding to the acrv1 plasmid promoter was determined to be significantly enriched using one-way ANOVA followed by the Bonferroni post-hoc test to determine difference from IgG background.
Primers were: −267 acrv1 forward (GACCCTCTGCAAAGAAGTGC), −186 acrv1 forward (AGGATCCGAAGCTACCCCTA), +27 acrv1 reverse (GGCACACTCAAGAGCTGAGA); +5652 acrv1 forward (GAACAAAGTGAATGTTGTGCACAATC), and +5930 acrv1 reverse (TCAGTCATTCCAGGAGCTGG).
Statistical Analysis
All data are expressed as the mean ± S.E. Statistical analysis consisted of one-way ANOVAs followed by Bonferroni post tests to determine which means differ (p < 0.05). All data analyses were performed using the Number Cruncher Statistical System program (NCSS, Kaysville, UT).
RESULTS
TDP-43 Is a Transcriptional Repressor
Here we directly tested the potential of TDP-43 to repress the acrv1 gene promoter using mouse spermatocyte cell line GC-2 and Gal 4 recruitment strategy. A luciferase reporter plasmid bearing five tandem Gal 4 binding sites upstream of the acrv1 core promoter (−91/+28) was constructed as a reporter (Fig. 1A). Full-length mouse TDP-43 was expressed as a fusion protein with the Gal4 DBD. Gal4DBD or Gal4DBD-TDP-43 plasmids were co-transfected with the above reporter plasmid into mouse GC-2 cells. The cells were harvested 48 h later, and luciferase activities were measured and normalized for transfection efficiencies. Transcriptional output from the vector expressing Gal4 DBD alone was used as the base line. The Gal4DBD-TDP-43 fusion protein repressed transcription of the reporter gene in a statistically significant manner, whereas the untargeted TDP-43 (FLAG-TDP-43) had no effect (Fig. 1B), indicating that the repressor effect of DBD-TDP-43 is a direct effect on the reporter gene. The positive control DBD-p53AD (activation domain of p53; a bona fide transcriptional activator) showed elevated reporter gene activity as expected. The DBD part contains a canonical nuclear localization signal that directs the location of the fusion proteins. Nuclear localization (supplemental Fig. S1A) as well as migration at the expected molecular size was verified (supplemental Fig. S1B) for all of the DBD-TDP43 fusion proteins used in reporter assays. The above results showed that TDP-43 represses transcription in the context of the acrv1 core promoter in a cell line (GC-2) of the spermatogenic lineage. Because TDP-43 is a ubiquitously expressed protein, we have also tested its function in the context of the generic c-fos minimal promoter in HeLa cells and found that TDP-43 acts as a repressor in that system as well (supplemental Fig. S2). These data suggest that the ubiquitously expressed TDP-43 protein likely functions as a repressor of transcription in multiple tissues.
FIGURE 1.

A–D, GAL4 recruitment strategy shows that full-length mouse TDP-43 represses transcription and that the N-terminal RRM domain is sufficient for transcriptional repression. A, in the schematic of the reporter gene used in this study five Gal4 binding sites were placed upstream of the −91/+28 acrv1 core promoter, which was fused to a luciferase reporter gene of the pGL3 basic vector to test TDP-43 repressor function. The GAL4 binding sites allow promoter recruitment of the GAL4 DBD-fusion proteins. B–D, transcriptional repressor function of mouse full-length TDP-43 (DBD-mTDP-43), various truncated forms (1–200, 1–262, 104–414, 191–414), and domains (RRM1, RRM2, RRM1 + 2, GLY) is shown. Schematics depicting mouse TDP-43 and major domains as DBD fusion proteins are shown to the left, and the reporter gene activities are shown to the right of each panel. One microgram of empty vector (DBD) or DBD-TDP-43 was co-transfected with 0.5 μg of reporter into GC-2 cells. 0.05 μg of Renilla Luciferase was used to normalize for transfection efficiency. Transcriptional output with DBD alone was used as the base line set as 1. FLAG-mTDP-43 is an untargeted TDP-43 version. p53AD corresponds to the activation domain of p53. Note: all DBD-TDP-43 clones showed nuclear localization (supplemental Fig. S1A) and migrated at the expected kDa sizes (supplemental Fig. S1B). Similar reporter assay results were obtained in the context of the c-fos reporter gene in HeLa cells (supplemental Fig. S2). NLS, nuclear localization signal; NES, nuclear export signal. E and F, HDAC inhibitors do not relieve TDP-43 mediated repression. TDP-43 mediated repression was evaluated in the presence of HDAC inhibitors sodium butyrate and Trichostatin A. Control DBD- and DBD-mTDP-43-transfected cells were treated with various concentrations of drug or vehicle (DMSO) for 24 h. Drug treatment effects are expressed as -fold difference of DMSO treatment. The inset shows dose-dependent hyperacetylation of core histones. HDAC inhibitors did not relieve TDP-43-mediated repression. NaB, sodium butyrate; TSA, Trichostatin A; Ac-H3, acetylated histone H3. All results shown (B–F) are the means ± S.E. for duplicate samples from four separate experiments. Asterisks represent significant difference (p < 0.05) compared with DBD. WB, Western blot.
N-terminal Truncations Relieve TDP-43-mediated Repression
TDP-43 contains two RNA recognition motifs (RRM) in the N-terminal and a glycine-rich domain in the C-terminal halves (schematic in Fig. 1B). To identify which part of TDP-43 is responsible for transcriptional repression, we generated N- and C-terminal truncations of TDP-43 and expressed them as DBD fusion proteins. The DBD-TDP-43 fusion protein expression plasmids were cotransfected with 5XGal4 luciferase reporter as before. Deletion of the C-terminal portion did not alter the repressor function of TDP-43, as the 1–200 and 1–262 regions repressed transcription in a statistically significant manner (Fig. 1C). In contrast, removal of the N-terminal 191 amino acids completely abolished transcriptional repression, whereas deletion of only the first 104 amino acids maintained repression (Fig. 1C). These data showed that the 104–191 region corresponding to RRM1 is critical for the repressor function of TDP-43.
RRM1 Alone Is Sufficient for Repression
To test whether RRM1 alone is sufficient for transcriptional repression, we made DBD-RRM1, DBD-RRM2, DBD-RRM1 + 2, and DBD-GLY domain fusion proteins and performed functional assays as above. The RRM1 domain alone or in combination with RRM2 caused statistically significant repression of transcription, whereas the RRM2 domain alone did not (Fig. 1D). The context of amino acids 1–104 also augmented the repressor function of RRM1 (Fig. 1C, DBD 1–200). An earlier study showed that TDP-43 1–95 region by itself does not repress transcription (1). This suggests that the repressor activity of DBD 1–200 is contributed by the RRM1 region. On the other hand, the C-terminal 274–414 region containing the glycine-rich domain up-regulated reporter activity (Fig. 1D). Similar results were obtained by replacing the acrv1 core promoter with the c-fos minimal promoter, indicating that RRM1 is sufficient to cause transcriptional repression (supplemental Fig. S2).
TDP-43 Repressor Function Is HDAC-independent
It is commonly observed that transcriptional repressors mediate repressive effects on gene transcription by recruiting histone deacetylases. To test whether TDP-43 functions in a similar way, we performed Gal4-TDP-43 reporter gene assays in the presence of HDAC inhibitors sodium butyrate and Trichostatin A. The inset in Fig. 1E showing hyperacetylation of core histones in response to sodium butyrate treatment confirms that the HDAC inhibitors were functional in the assay. However, sodium butyrate treatment did not relieve repression caused by TDP-43 (Fig. 1E), suggesting that TDP-43 represses transcription in a HDAC-independent manner. This was confirmed by Trichostatin A treatment (Fig. 1F). In fact, we observed a trend of dosage-dependent increase in repressor activity of TDP-43 with both treatments. Repression mediated by the neuronal repressor REST was also shown to increase in a similar way in response to treatment with HDAC inhibitors (21, 22).
TDP-43 Binding to acrv1 Gene Promoter Is GTGTGT-dependent
The main premise for considering the mouse acrv1 gene as a TDP-43 target is that transgenic mice bearing the acrv1 promoter with GTGTGT site mutations expressed a reporter gene prematurely in spermatocytes, whereas the wild-type acrv1 promoter maintained repression in spermatocytes (10). In a previous study using EMSA we showed that TDP-43 binding to the acrv1 promoter DNA requires the GTGTGT motifs, but these assays could only be performed with single-stranded DNA. TDP-43 failed to bind to double-stranded DNA in vitro (10). We hypothesized that TDP-43 requires the context of chromatin and/or cellular environment to bind to double-stranded target DNA. Therefore, we addressed the requirement of GTGTGT motifs for TDP-43 binding to the acrv1 promoter in a cell culture model using plasmid ChIP. It has been shown that plasmid DNA transfected into mammalian cells becomes partially chromatinized (17). Earlier groups successfully established transcription factor and histone H1 binding to transiently transfected plasmid DNA using plasmid ChIP (18, 35). Plasmids containing either the wild-type −186/+28 acrv1 promoter or a mutant version in which the two GTGTGT motifs at −172 and −160 have been mutated (used in Acharya et al. (10) to generate transgenic mice) were separately transfected into COS-7 cells (Fig. 2A). A second plasmid expressing the full-length TDP-43 with an N-terminal FLAG epitope tag was cotransfected. After 48-h, the cells were harvested, and ChIP was performed using anti-FLAG antibody. A parallel ChIP using IgG alone served as a negative control and provided the background values. After ChIP, the region corresponding to the −186/+27 portion of the mouse acrv1 promoter present in the transfected plasmids was amplified using real-time quantitative PCR. The primer pair chosen was such that only the mouse acrv1 promoter present in the plasmids would be amplified but not the endogenous acrv1 gene of the monkey kidney cell line (COS-7) used for transfections. PCR signal from FLAG antibody ChIP, which represents TDP-43 occupancy, was plotted as -fold enrichment over that from the IgG alone control. We observed 5.3-fold enrichment of TDP-43 occupancy of the wild-type acrv1 promoter, whereas no promoter occupancy was observed on the promoter bearing GTGTGT mutations (Fig. 2B). The levels of FLAG-TDP-43 expression were identical in both transfections (Fig. 2C). Thus, the above data show that TDP-43 in fact binds to the acrv1 promoter DNA in the context of chromatin and that the GTGTGT-motifs are required for binding. One additional interpretation could be that acrv1 promoter occupancy of TDP-43 may be aided by another factor(s) present in the cellular environment.
FIGURE 2.

Plasmid ChIP shows that TDP-43 binding to the acrv1 promoter is GTGTGT sequence-dependent. 3 μg of plasmid bearing either the wild-type −186/+28 mouse acrv1 promoter or a version bearing GTGTGT mutations were separately transfected into monkey kidney COS-7 cells. 3 μg of FLAG-TDP-43 was co-transfected. After 48 h, cells were harvested, and ChIP was performed with 5 μg of anti-FLAG antibody. A parallel ChIP with 5 μg of control IgG alone served as a negative control and provided base-line values. After ChIP, the −186/+27 region of the mouse acrv1 promoter was amplified using real-time quantitative PCR with plasmid-specific primers. PCR signal representing TDP-43 occupancy is plotted as -fold enrichment over IgG alone control. A, shown is a schematic of acrv1 promoter plasmids bearing wild-type and mutant promoter sequences. Nucleotide sequence of the region containing the TDP-43 recognition sequences (5′-GTGTGT) on the antisense strand is shown. B, results shown are the means ± S.E. for triplicate PCR samples from eight separate ChIP experiments. The asterisk represents significant difference (p < 0.05) compared with IgG as determined by one-way ANOVA followed by the Bonferroni post-hoc test. C, shown is a Western blot (WB) analysis of FLAG-TDP-43 expression in wild-type and mutant acrv1 plasmid transfected COS-7 cells.
TDP-43 Occupancy of acrv1 Gene Promoter in Vivo in a Physiological Context
Next, to test whether the acrv1 gene is a TDP-43 target in vivo, we examined the occupancy of TDP-43 at the acrv1 gene promoter in a physiological context. The acrv1 mRNA is testis-specific and is expressed only in round spermatids (12). We previously showed that the acrv1 gene is silenced in somatic tissues by the tethering of the proximal promoter to the nuclear matrix (11). Although this tethering is released in the male germ cells during spermatogenesis, thus, permitting access to transcriptional machinery, the acrv1 gene remains repressed in spermatocytes before transcription in round spermatids. Thus, the status of the acrv1 gene in liver, spermatocytes, and round spermatids will be transcriptionally silent, transcription-ready but repressed, and transcriptionally active, respectively. To investigate TDP-43 occupancy of the acrv1 promoter in the above physiological states we performed ChIP using mouse liver cells, spermatocytes, and round spermatids. ChIP was performed using anti-TDP-43 polyclonal antibodies, which were previously characterized using immunoblotting and immunohistochemistry (10). The acrv1 proximal promoter (−267/+27) region, which includes the two TDP-43 binding sites at −172 and −160, was amplified by real-time PCR. Amplification of a downstream region of the acrv1 gene (+5652/+5930), which lacks the GTGTGT sites, served as a negative control. The data are plotted as -fold increase over the IgG alone control. For all three cell types used here, TDP-43 occupancy at the downstream region was not above the background values obtained with IgG alone, which indicated the specificity of anti-TDP-43 antibody in the ChIP procedure.
ChIP data showed 4.3-fold enrichment of TDP-43 at the acrv1 promoter in liver cells (Fig. 3A). This is consistent with the idea that TDP-43 may be partially responsible for the silencing of the acrv1 gene in somatic tissues (11). Our previous studies, however, predicted a more prominent role for TDP-43 in maintaining repression of acrv1 transcription in spermatocytes (10). Consistent with this, ChIP data showed the highest degree of TDP-43 occupancy of the acrv1 gene promoter in spermatocytes (8.8-fold enrichment) (Fig. 3B). This promoter occupancy data combined with the property of TDP-43 to repress transcription shown in Fig. 1 support the view that TDP-43 represses acrv1 gene transcription in spermatocytes in vivo. This then led to a prediction that TDP-43 will be dislodged from the promoter in round spermatids to permit transcription of the acrv1 gene. Contrary to the prediction, ChIP showed persistence of TDP-43 at the acrv1 proximal promoter in round spermatids (Fig. 3C). The downstream region of acrv1 (right halves of Fig. 3, A–C) or unrelated gene promoters did not show enrichment for TDP-43 binding (data not shown), thus, confirming the specificity of ChIP using TDP-43 polyclonal antibodies. Because the acrv1 mRNA is actively transcribed in round spermatids (12), one interpretation of the continued occupancy is that TDP-43 loses its repressor property in round spermatids. Thus, promoter occupancy study in a physiological context indicated that TDP-43 function is modulated at the promoter.
FIGURE 3.

TDP-43 is enriched at the endogenous acrv1 promoter in testicular germ cells. ChIP was performed on mouse liver cells, spermatocytes, and round spermatids using 5 μg of anti-TDP-43 polyclonal antibodies. A parallel ChIP of each cell type with 5 μg of IgG antibody alone served as a negative control and provided base-line values. The −267 to +27 region of the acrv1 proximal promoter, which includes the two TDP-43 binding sites at −172 and −160, was amplified by real-time PCR. Amplification of a downstream region of the acrv1 gene (+5652/+5930), which lacks GTGTGT sites, served as a negative control. PCR signal representing TDP-43 binding to the acrv1 gene is plotted as -fold increase over the IgG alone control. Results shown are the means ± S.E. for triplicate PCR samples from five separate ChIP experiments. The asterisks represent significant difference (p < 0.05) compared with IgG.
Splice Variants of TDP-43 Do Not Relieve TDP-43-mediated Repression
How might TDP-43 repression of the acrv1 gene be released in round spermatids? The continued presence of TDP-43 at the acrv1 promoter in round spermatids (Fig. 3C) wherein the acrv1 mRNA is transcribed suggests that there must be conditions under which TDP-43 does not act as a transcriptional repressor. We investigated the possibility that an alternatively spliced form of TDP-43 without repressor domain may relieve repression.
The NCBI data base listed six splice variants of the tardbp gene (which codes for TDP-43). The wild type as well as all the splice variants share the same 3′-UTR region. To determine whether there are TDP-43 splice variants in the male germ cells, we isolated mouse spermatocytes and round spermatids from adult mice and performed TDP-43 cDNA synthesis as described under “Experimental Procedures.” In addition to the full-length TDP-43 cDNA and splice variants common to both cell types, we have cloned one variant each unique to spermatocytes and round spermatids. The spermatocyte and spermatid splice variants are 299 and 296 amino acids in length, respectively (Fig. 4A and supplemental Fig. S3A). They both contain the N-terminal 1–277 region as in the wild-type TDP-43 but are missing the glycine-rich 278–414 part. Instead, the variants contain 18 additional amino acids (VHLISNVYGRSTSLKVVL) derived from a cryptic exon within the 3′-UTR, not present in the wild-type TDP-43. In addition to this, the spermatocyte variant contains three more amino acids (GNP) at 278–280 position that are missing in the round spermatids splice variant. To determine the function of these variants, we generated Gal4 DBD fusion constructs and performed reporter assays. Nuclear localization of the DBD splice variants (supplemental Fig. S3B) and migration at the expected molecular weight (supplemental Fig. S3C) was verified. In reporter assays, both spermatocyte and round spermatid splice variants repressed transcription similar to the wild-type TDP-43 (Fig. 4B). Replacing the acrv1 core promoter with the c-fos minimal promoter produced similar results (supplemental Fig. S3D). Thus, the splice variants cloned from spermatocytes and round spermatids may not be involved in relieving the repressor function of TDP-43 at the acrv1 promoter. Although the presence of the N-terminal half consisting of RRM 1 and RRM2 within the splice variants predicted a repressor function, the experiment proved that the unique 18-amino acid domain at the C terminus did not alter repressor function.
FIGURE 4.

Splice variants of TDP-43 do not relieve repression. A, shown are schematics of TDP-43 splice variants cloned from mouse spermatocytes and round spermatids depicting that they lack the glycine-rich region but contain an additional 18 amino acids at the C-terminal end, not present in wild-type TDP-43. The spermatocyte splice variant contains three amino acids more than the round spermatid variant at position 278–280. B, shown is an evaluation of TDP-43 splice variants repressor function. 1 μg of empty vector (DBD) or DBD-TDP-43 was co-transfected with 0.5 μg of reporter into GC-2 cells. 0.05 μg of Renilla Luciferase was used to normalize for transfection efficiency. Transcriptional output with DBD alone was used as the base line set as 1. Note: DBD-TDP-43 variants show nuclear localization (supplemental Fig. S3B) and migrated at the expected molecular weight (supplemental Fig. S3C). The variants displayed similar repressor function in the context of the c-fos reporter gene in HeLa cells (supplemental Fig. S3D). Results shown are the means ± S.E. for duplicate samples from four separate experiments. NLS, nuclear localization signal; NES, nuclear export signal. The asterisks represent significant difference (p < 0.05) compared with DBD. Cyte, spermatocyte; Tid, round spermatid.
RNA Binding-defective TDP-43 Relieves Repressor Function
Because TDP-43 is an RRM containing RNA binding protein, we tested whether RNA binding plays a role in mediating TDP-43 repressor function by using RNA binding-defective TDP-43 in reporter assays. It has been shown that TDP-43 requires Phe-147 and Phe-149 within RRM1 to bind RNA (24). We obtained human TDP-43 bearing F147L and F149L mutations (defective in RNA binding), fused it to DBD, and tested for repressor function using our reporter gene assay in GC-2 cells. DBD fused full-length human TDP-43 and TDP-43 lacking the entire RRM1 portion (amino acids 106–175) were used as controls. Schematics of the above three human TDP-43 clones, kind gifts of F. Baralle, Italy, are shown in Fig. 5A. All clones show proper expression at the expected molecular size (Fig. 5C) and nuclear localization (supplemental Fig. S4). In reporter assays, the full-length human TDP-43 repressed transcription in mouse GC-2 cells, which was expected based on the evolutionary conservation of TDP-43 between the human and mouse. This value was set at 1 in Fig. 5B. The ΔRRM1 showed a significant relief of repressor function (Fig. 5B), thus, confirming that RRM1 is in fact critical for TDP-43 transcriptional repression. Interestingly, TDP-43 with F147L, F149L mutations also relieved repression to a similar extent as ΔRRM1 (Fig. 5B). Thus, the point mutations that disable RNA binding compromised TDP-43 repressor function, suggesting that an RNA intermediate may play a role in transcriptional repressor function of TDP-43. It has recently been reported that TDP-43-mediated neuron loss in vivo requires RNA binding (23). Transgenic expression of human TDP-43 with F147L, F149L mutations reproduced the neurodegenerative pathology. Thus, our data using mutant (F147L, F149L) hTDP-43 (Fig. 5) provide a functional basis for understanding the pathogenesis caused by loss of RNA binding of TDP-43. Future work will investigate the nature of the RNA intermediate(s) and the mechanistic link with transcriptional repression by TDP-43.
Histone Modifications Associated with TDP-43 Binding at the acrv1 Gene Promoter in Vivo
The above promoter occupancy results prompted examination of the histone code associated with TDP-43 binding at the acrv1 promoter in a physiological context. We performed ChIP on mouse liver cells, spermatocytes, and round spermatids using antibodies specific for histone H3 trimethylated K4 (H3K4Me3), histone H3 acetylated K9 (H3K9Ac), and histone H3 dimethylated K9 (H3K9Me2) markers or control IgG. Per histone code hypothesis, the H3K4Me3 and H3K9Ac are considered as active and H3K9Me2 as repressive histone marks (19, 20). After immunoprecipitation of chromatin, the proximal promoter −267/+27 or a downstream region (+5652/5930) of the acrv1 gene were amplified by real-time PCR and -fold change over IgG antibody control plotted. The +5652 downstream region was not enriched above background in ChIP with any of the above antibodies (data not shown). In mouse liver, H3K9Me2 mark dominated at the proximal promoter with nearly 23-fold enrichment over background indicating that the acrv1 promoter region is wrapped in a repressive higher order chromatin structure (Fig. 6A). This is consistent with the fact that the acrv1 gene is transcriptionally silent in liver. In spermatocytes and round spermatids, the H3K9Me2 mark was much reduced compared with liver, suggesting that the higher order chromatin structure has given way to a transcription factor-accessible form of chromatin configuration (Fig. 6, B and C). The active histone marks H3K4Me3 and H3K9Ac appeared at the acrv1 promoter in spermatocytes (Fig. 6B) but became highly enriched in round spermatids (Fig. 6C) consistent with the notion that the acrv1 gene becomes readied for transcription in spermatocytes before being actually transcribed in round spermatids. Thus, the 23-fold enrichment of the “inactive” histone mark H3K9Me2 in liver and the 64- and 29-fold enrichment of the “active” histone marks H3K4Me3 and H3K9Ac in round spermatids at the mouse acrv1 gene promoter are in agreement with the histone code hypothesis (19). At the same time, the appearance of active marks in spermatocytes where the acrv1 gene is repressed and the incomplete removal of inactive marks in round spermatids where acrv1 gene is transcribed reflect the flexibility of the histone code in a physiological context. From the point of view of TDP-43 occupancy (Fig. 3), it is interesting to note that the state of histone tail modifications at the acrv1 promoter has little effect on TDP-43 occupancy because TDP-43 remains at the promoter even in the presence of active histone marks (H3K4Me and H3K9Ac).
FIGURE 6.
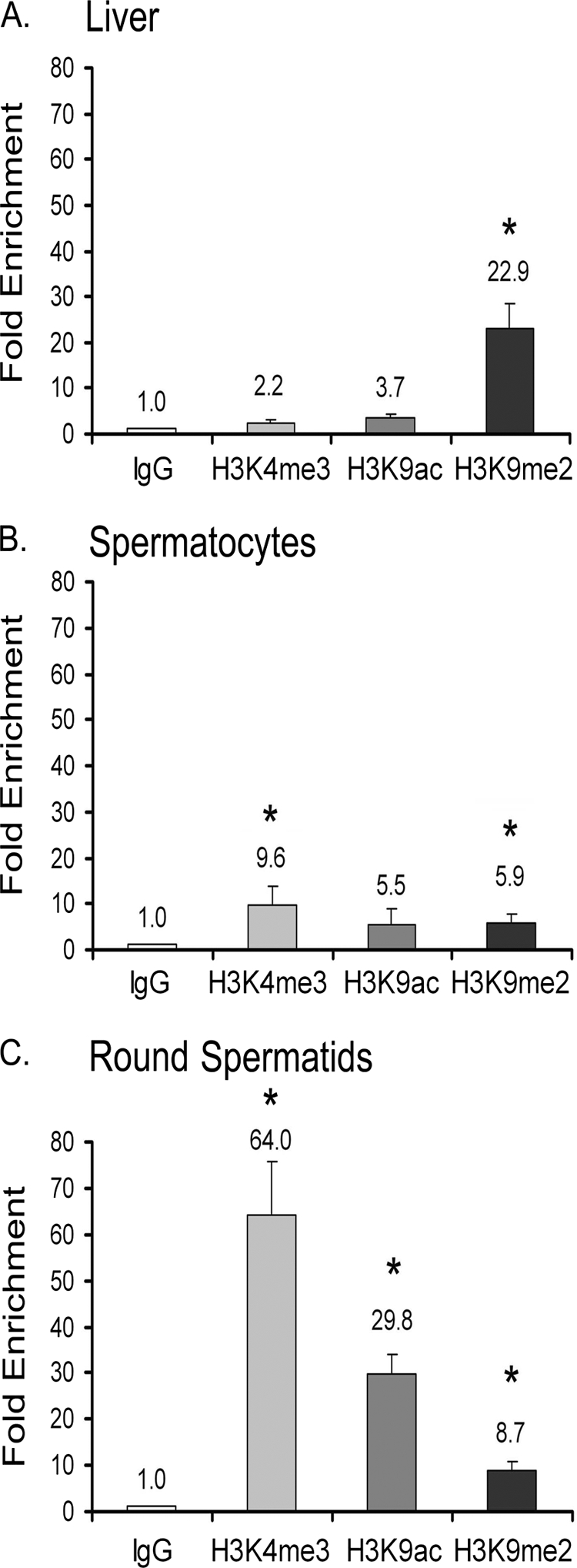
Histone tail modifications associated with TDP-43 promoter occupancy in a physiological context. ChIP was performed on liver cells, spermatocytes, and round spermatids using 3 μg of IgG, H3K4me3, H3K9ac, and H3K9me2 antibodies. The −267 to +27 region of the acrv1 proximal promoter was amplified as before to determine factor binding. Amplification of a downstream region of the acrv1 gene (+5652/+5930) showed no factor binding (data not shown). A PCR signal representing chromatin marks associated with the acrv1 gene is plotted as -fold increase over the IgG alone control. Results shown are the means ± S.E. for triplicate PCR samples from four separate ChIP experiments. The asterisks represent significant difference (p < 0.05) compared with IgG.
RNA Polymerase II Is Paused at the acrv1 Promoter in Spermatocytes
Our previous study showed that TDP-43 binding site-mutant acrv1 promoter expressed a reporter gene prematurely in spermatocytes in vivo. This suggested that the endogenous acrv1 promoter may in fact harbor RNA polymerase II (RNAPII) in spermatocytes even though production of acrv1 mRNA does not begin until round spermatid formation. Next, we investigated RNAPII occupancy of the endogenous acrv1 promoter in a physiological context. ChIP showed that in mouse spermatocytes, which do not yet synthesize acrv1 mRNA, RNAPII is already present at the acrv1 proximal promoter (Fig. 7B). In comparison, liver tissue did not show any significant enrichment over background (Fig. 7A). In round spermatids, which produce acrv1 mRNA, RNAPII enrichment increased 4-fold compared with spermatocytes, consistent with active gene transcription (Fig. 7C). Thus, the presence of RNAPII at the acrv1 promoter in spermatocytes before the actual transcription of acrv1 mRNA generated the hypothesis that RNAPII is paused or stalled at the acrv1 gene promoter in spermatocytes. The RNAPII-pausing phenomenon has received considerable attention in recent years, and molecular markers for “pausing” and “elongation” of transcription have been firmly established. Ser-5 residues of the heptad repeat of the C-terminal domain of RNAPII are phosphorylated during the pause phase, and Ser-2 residues become phosphorylated during the elongation phase (37). In addition, negative elongation factor, NELF, occupies the promoter in the paused state and is dislodged from the promoter during the elongation phase of transcription (37, 38). Examination of these markers at the acrv1 promoter in a physiological context by ChIP showed that the Ser-5 phosphorylation mark is predominant over the Ser-2 mark in spermatocytes (Fig. 7B), whereas the Ser-2 phosphorylation mark increased by 2.3-fold in round spermatids over spermatocytes (Fig. 7C). Furthermore, we found high NELFe occupancy of the acrv1 promoter in spermatocytes (Fig. 7B), which decreased 4-fold in round spermatids (Fig. 7C). Taken together, our data indicate that RNAPII is recruited to the acrv1 gene promoter in spermatocytes but is held in a paused state and that it enters a phase of transcription elongation in round spermatids. TDP-43 occupancy of the acrv1 promoter shown in the present study combined with the previous finding that GTGTGT mutant acrv1 promoter prematurely expressed a reporter gene in spermatocytes in transgenic mice (10) provide a basis for the hypothesis that TDP-43 may play a role in pausing RNAPII at the acrv1 promoter in spermatocytes. Future studies will investigate if a mechanistic link exists between RNAPII pausing and TDP-43.
FIGURE 7.
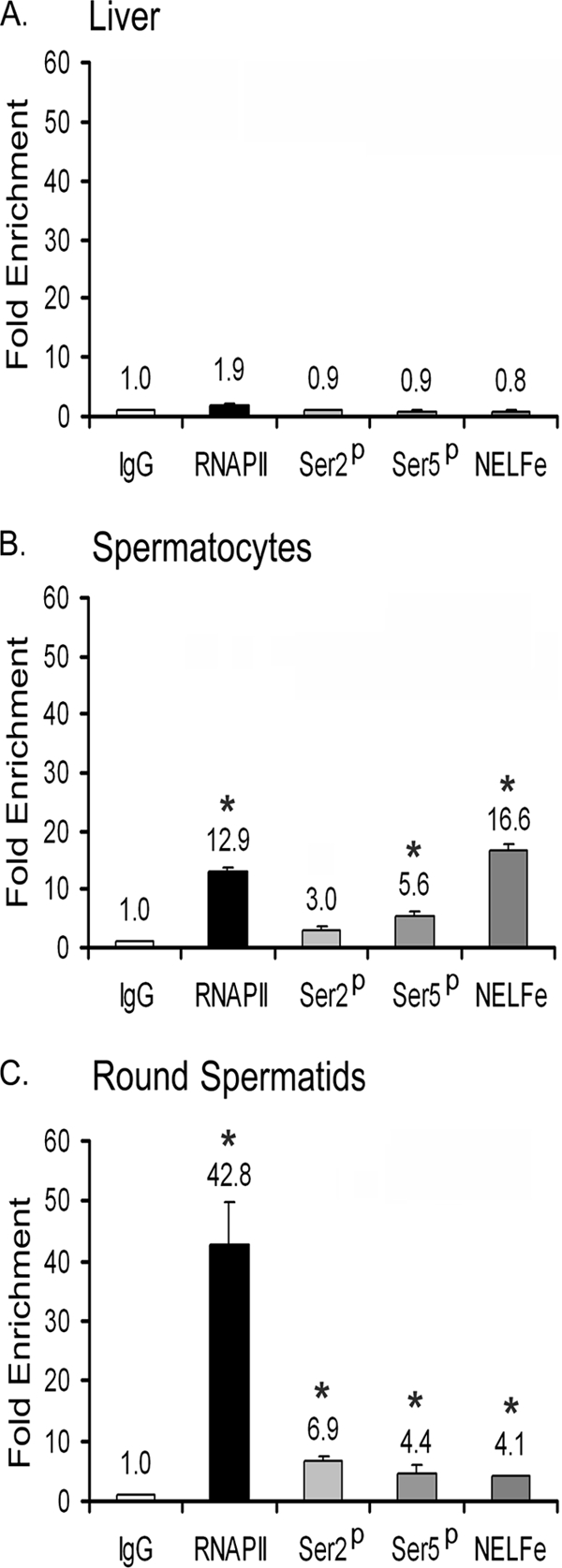
Evidence for RNAPII pausing at the acrv1 promoter in spermatocytes and for its release in round spermatids. ChIP on liver cells, spermatocytes, and round spermatids was performed using 5 μg of IgG, total RNAPII-, RNAPII phosphoserine-2, phosphoserine 5-specific, and NELF-E antibodies. The −267 to +27 region of the acrv1 proximal promoter was amplified as before to determine factor binding. Amplification of a downstream region of the acrv1 gene (+5652/+5930) showed no factor binding (data not shown). PCR signal representing specific protein interactions with the acrv1 gene is plotted as -fold increase over the IgG alone control. Enrichment of phosphorylated Ser-5 of RNAPII and NELF-E at the promoter is indicative of paused RNAPII. Enrichment of phosphorylated Ser-2 mark and decrease of NELF-E occupancy in round spermatid is indicative of transcriptionally elongating RNAPII. Results shown are the means ± S.E. for triplicate PCR samples from 4 separate ChIP experiments. The asterisks represent significant difference (p < 0.05) compared with IgG. Ser2p, phosphoserine 2-specific RNAPII; Ser5p, phosphoserine 5-specific RNAPII.
DISCUSSION
The present study systematically addressed the role of TDP-43 in gene transcription and identified a bona fide TDP-43 target gene in vivo. First, reporter gene assays in transiently transfected mouse and human cells showed that 1) TDP-43 is a transcriptional repressor and that the N-terminal RRM1 is critical for repressor function, 2) TDP-43 repression is not mediated by the action of histone deacetylases, and 3) loss of RNA binding may play a role in relieving the repressor function of TDP-43. Second, chromatin immunoprecipitation studies performed in a physiological context established that 1) the testis-specific mouse acrv1 gene, which codes for the sperm acrosomal protein SP-10, is a bona fide TDP-43 target gene in vivo, 2) TDP-43 binds to acrv1 promoter via GTGTGT motifs, 3) promoter occupancy by TDP-43 is not affected by the transitioning of histone code from repressive to active state, and 4) TDP-43 occupancy of acrv1 promoter in spermatocytes coincides with paused RNAPII.
TDP-43 Is a Transcriptional Repressor
TDP-43 repressed transcription in the context of two different types of core promoters (belonging to the testis-specific acrv1 gene and the widely expressed c-fos gene) and in two different cell types (mouse GC-2 and human HeLa) as shown in Fig. 1 and supplemental Fig. S2. This indicates that the ubiquitously expressed TDP-43 is capable of regulating the transcription of a large number of target genes. A previous study showed that overexpression of TDP-43 repressed transcription from the HIV proviral vector (1). Our approach of recruiting TDP-43 to the core promoter via Gal4 DNA binding domain ensured that the observed transcriptional repression is a direct effect of TDP-43 on transcription. Typically, RRM-containing proteins such as TDP-43 are thought of as RNA-binding proteins with roles limited to mRNA stability and splicing. In line with this, TDP-43 has been shown to regulate mRNA splicing of the CFTR gene and stability of the NFL mRNA (26). Our data, however, demonstrate an additional function for TDP-43 as a transcriptional repressor. There have been other examples of RRM-containing proteins acting as transcriptional repressors (27–30). Reinforcing this, a recent study exploring protein-DNA interactions of conventional (transcription factors) and unconventional (RNA-binding proteins, kinases) proteins showed that a large number of RNA binding proteins in fact bind double-stranded DNA in vitro and to gene promoters in vivo (31). Because gene transcription and mRNA splicing are coupled events within the nucleus and given that TDP-43 can bind to both DNA and RNA, it is possible that TDP-43 participates in both functions at some target genes.
The RRM1 Is Critical for Transcriptional Repression
Use of N- and C-terminal truncated versions of TDP-43 in reporter gene assays narrowed down the repressor activity to the N-terminal RRM1. Several lines of evidence proved the critical role of RRM1 in transcriptional repression; Gal4-DBD fused RRM1 domain alone was sufficient to repress transcription (Fig. 1D). Reciprocally, a deletion mutant of TDP-43 lacking the RRM1 domain relieved repression (Fig. 5B). Consistent with the above, naturally occurring splice variants of TDP-43, which contain the RRM domains but lack the C-terminal Gly domain, also repressed transcription (Fig. 4B). Taken together, our data indicate that RRM1 mediates the transcriptional repressor function of TDP-43. We favor the hypothesis that TDP-43 assembles a corepressor complex via RRM1. Use of the RNA binding-defective mutant of TDP-43 (F147L and F149L) relieved repressor function compared with the wild-type TDP-43 (Fig. 5B), suggesting that an RNA intermediate may be involved. Alternatively, binding to RNA via RRM1 may induce allosteric changes to TDP-43, which in turn regulates interactions with transcriptional complexes. Previous reports suggested that TDP-43 may bind to messenger RNA as well as small microRNAs (25, 26, 32). Our future work will focus on identification of the RNA molecules that bind to TDP-43 to regulate its transcriptional function. This study also shows that the C-terminal portion, when freed from the RRMs, can in fact activate transcription (Fig. 1D). This finding may be relevant to TDP-43 pathologies where TDP-43 cleavage products are generated. Proteolytic cleavage sites have been identified in brain tissues of frontotemporal lobar degeneration patients mapping to the 208–246 region (33, 34). The fate of the truncated TDP-43 C-terminal fragments is not fully known except that eventually they form ubiquitinated aggregates. Our work suggests that the C-terminal fragments have the potential to reverse the transcription status of TDP-43 target genes leading to abnormal cell physiology, which may precipitate in pathological conditions.
TDP-43 Binds Promoter DNA via GTGTGT Motifs
In vitro binding studies previously established that TDP-43 binds to single-stranded DNA via (TG)n repeats (10). Several studies reported failure of TDP-43 binding to double-stranded DNA in vitro (2, 9, 10). This raised the question of whether TDP-43 can function as a transcription factor controlling gene expression as most transcription factor binding sites exist in a double-stranded form in vivo. We reasoned that TDP-43 may require a chromatinized DNA template within the cellular context to bind to double-stranded DNA and tested this by performing chromatin immunoprecipitation on transiently transfected DNA. It had been shown that upon transient transfection, plasmid DNA becomes associated with core histones and assumes higher order chromatin structure (17, 18). Plasmid ChIP had been successfully used in the past to demonstrate transcription factor binding (18, 35, 36). The present study showed for the first time that TDP-43 binds to double-stranded DNA target (acrv1 gene promoter) and that it requires the GTGTGT motifs for promoter recognition (Fig. 2B). Thus, chromatin and/or association with another cellular factor(s) may be critical for TDP-43 binding to double-stranded DNA. The plasmid ChIP assay used here can be further exploited to determine the minimal DNA binding region of TDP-43.
The Mouse acrv1 Gene Is a Bona Fide TDP-43 Target Gene
Our previous studies suggested that acrv1 may be a TDP-43 target gene. First, we cloned TDP-43 from a mouse testis library based on its binding to the acrv1 gene promoter sequence and showed that recombinant TDP-43 binds to the acrv1 promoter in vitro. Second, mutation of the GTGTGT motifs of the acrv1 gene promoter, which abrogated TDP-43 binding in vitro, caused loss of spatiotemporal specificity of transcription during spermatogenesis in transgenic mice (10). The above data predicted a role for TDP-43 in regulation of acrv1 gene transcription during spermatogenesis. The present study directly addressed whether TDP-43 in fact binds to the acrv1 gene promoter in vivo. ChIP experiments performed in a physiological context prove that TDP-43 occupies the acrv1 gene promoter in spermatocytes where the acrv1 gene is transcriptionally repressed (Fig. 3). This combined with the functional data that TDP-43 represses transcription of a reporter gene driven by the acrv1 core promoter (Fig. 1B), strongly argue that TDP-43 occupancy of the acrv1 promoter keeps the gene in a repressed state in spermatocytes. ChIP also showed that TDP-43 remains at the acrv1 promoter within the round spermatids, which express the acrv1 mRNA. This suggested the possibility that TDP-43 repressor function must be alleviated in round spermatids to accommodate the transcription of acrv1 mRNA.
Modulation of TDP-43 Repressor Function
Use of HDAC inhibitors Trichostatin A or sodium butyrate did not relieve TDP-43-mediated repression in a cell culture system (Fig. 1, E and F), suggesting that reversing the acetylation status of histones alone does not cause loss of TDP-43-mediated repression. The hypothesis that alternative splice variants may replace the full-length TDP-43 in round spermatids to relieve repression also did not prove correct (Fig. 4). The splice variants cloned from mouse spermatocytes and round spermatids contained the N-terminal RRM region and functioned as potent repressors of transcription (Fig. 4B). Mutation of amino acids critical for binding to RNA (F147L, F149L), however, relieved TDP-43 repressor function (Fig. 5). We speculate that RNA binding may induce an allosteric or conformational change in TDP-43 in a way that prevents interaction with the putative corepressor complex. Under these conditions, the activator potential of the C-terminal Gly domain (Fig. 1D) may allow TDP-43 to function as a transcriptional activator or as a facilitator of transcription. Post-translational modifications of TDP-43 also may facilitate modulation of function.
Finally, ChIP addressing RNAPII occupancy of the TDP-43 target gene acrv1 within the physiological context of spermatogenesis provided a unique insight that TDP-43 function may be linked to RNAPII pausing. The study showed that RNAPII is in a paused state at the acrv1 gene promoter in spermatocytes (Fig. 7). TDP-43 is also present at the acrv1 promoter in spermatocytes. When these data are viewed in the context of evidence that mutation of TDP-43 binding sites of the acrv1 promoter caused transcription to occur prematurely in spermatocytes in vivo, it leads to a prediction that TDP-43 may assist in retaining RNAPII in a paused state in spermatocytes. Modulation of TDP-43 function must then permit RNAPII to enter transcription elongation phase. Future work will investigate the mechanistic link between TDP-43 and RNAPII pausing. Overall, our study has shown that TDP-43 is a transcriptional repressor and that the acrv1 gene is a bona fide target gene of TDP-43 in vivo.
Relevance of This Work to Neurodegenerative Disease
In the recent past a significant number of studies have shown that TDP-43 is associated with neurodegenerative diseases including frontotemporal lobar degeneration with ubiquitin-positive inclusions, amyotrophic lateral sclerosis, and activation domain (Ref. 4 and references therein). The main clinical features are that TDP-43, which is normally a nuclear protein, becomes mislocalized to the cytoplasm of motor neurons and glial cells where it forms insoluble aggregates. Whether the neurodegenerative disease is caused by the loss of nuclear function of TDP-43 or by the toxic gain of function within the cytoplasm is not clear. Based on our study we predict that TDP-43 plays a broad role as a transcriptional regulator within the neuronal cells, and the loss of this function as well as that of mRNA splicing must severely affect the physiology of the neuronal cells. This may initiate the pathology, and the subsequent aggregation of TDP-43 within the ubiquitinated inclusions may eventually lead to a toxic gain of function.
Supplementary Material
Acknowledgments
We thank Drs. David Auble, Akhilesh Nagaich, and Terry Turner for critical reading of the manuscript. We thank Stacy McDowell and Ravi Durga for technical assistance with preparation of three of the constructs used in this study, Drs. Emanuele Buratti and Francisco Baralle (International Centre for Genetic Engineering and Biotechnology, Trieste, Italy) for providing the human TDP-43 clones, and Dr. Yuki Yamaguchi for the NELF-E antibody.
This work was supported, in whole or in part, by National Institutes of Health Grant R01HD36239 (to P. P. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- TDP-43
- 43-kDa transactivation response (TAR) DNA-binding protein
- hTDP-43
- human TDP-43
- HDAC
- histone deacetylase
- DBD
- DNA binding domain
- qPCR
- quantitative PCR
- RRM1
- RNA recognition motif 1
- H3K4me3
- histone H3 lysine 4 trimethylation
- H3K9Ac
- histone H3 acetylated K9
- H3K9Me2
- histone H3 dimethylated K9
- RNAPII
- RNA polymerase II
- NELF
- negative elongation factor
- ANOVA
- analysis of variance.
REFERENCES
- 1. Ou S. H., Wu F., Harrich D., García-Martínez L. F., Gaynor R. B. (1995) J. Virol. 69, 3584–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Buratti E., Baralle F. E. (2008) Front. Biosci. 13, 867–878 [DOI] [PubMed] [Google Scholar]
- 3. Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., Feiden W., Kretzschmar H. A., Trojanowski J. Q., Lee V. M. (2006) Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 4. Chen-Plotkin A. S., Lee V. M., Trojanowski J. Q. (2010) Nat. Rev. Neurol. 6, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sephton C. F., Good S. K., Atkin S., Dewey C. M., Mayer P., 3rd, Herz J., Yu G. (2010) J. Biol. Chem. 285, 6826–6834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu L. S., Cheng W. C., Hou S. C., Yan Y. T., Jiang S. T., Shen C. K. (2010) Genesis 48, 56–62 [DOI] [PubMed] [Google Scholar]
- 7. Kraemer B. C., Schuck T., Wheeler J. M., Robinson L. C., Trojanowski J. Q., Lee V. M., Schellenberg G. D. (2010) Acta Neuropathol. 119, 409–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buratti E., Brindisi A., Pagani F., Baralle F. E. (2004) Am. J. Hum. Genet. 74, 1322–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buratti E., Baralle F. E. (2001) J. Biol. Chem. 276, 36337–36343 [DOI] [PubMed] [Google Scholar]
- 10. Acharya K. K., Govind C. K., Shore A. N., Stoler M. H., Reddi P. P. (2006) Dev. Biol. 295, 781–790 [DOI] [PubMed] [Google Scholar]
- 11. Abhyankar M. M., Urekar C., Reddi P. P. (2007) J. Biol. Chem. 282, 36143–36154 [DOI] [PubMed] [Google Scholar]
- 12. Reddi P. P., Flickinger C. J., Herr J. C. (1999) Biol. Reprod. 61, 1256–1266 [DOI] [PubMed] [Google Scholar]
- 13. Ayala Y. M., Zago P., D'Ambrogio A., Xu Y. F., Petrucelli L., Buratti E., Baralle F. E. (2008) J. Cell Sci. 121, 3778–3785 [DOI] [PubMed] [Google Scholar]
- 14. Miyake T., Hu Y. F., Yu D. S., Li R. (2000) J. Biol. Chem. 275, 40169–40173 [DOI] [PubMed] [Google Scholar]
- 15. Salghetti S. E., Kim S. Y., Tansey W. P. (1999) EMBO J. 18, 717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lalmansingh A. S., Uht R. M. (2008) Endocrinology 149, 346–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cereghini S., Yaniv M. (1984) EMBO J. 3, 1243–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hebbar P. B., Archer T. K. (2008) J. Biol. Chem. 283, 4595–4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jenuwein T., Allis C. D. (2001) Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 20. Wang Y., Fischle W., Cheung W., Jacobs S., Khorasanizadeh S., Allis C. D. (2004) Novartis Found. Symp. 259, 3–17; discussion 17–21, 163–169 [PubMed] [Google Scholar]
- 21. Ooi L., Belyaev N. D., Miyake K., Wood I. C., Buckley N. J. (2006) J. Biol. Chem. 281, 38974–38980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Otto S. J., McCorkle S. R., Hover J., Conaco C., Han J. J., Impey S., Yochum G. S., Dunn J. J., Goodman R. H., Mandel G. (2007) J. Neurosci. 27, 6729–6739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Voigt A., Herholz D., Fiesel F. C., Kaur K., Müller D., Karsten P., Weber S. S., Kahle P. J., Marquardt T., Schulz J. B. (2010) PLoS ONE 5, e12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Buratti E., Brindisi A., Giombi M., Tisminetzky S., Ayala Y. M., Baralle F. E. (2005) J. Biol. Chem. 280, 37572–37584 [DOI] [PubMed] [Google Scholar]
- 25. Buratti E., Dörk T., Zuccato E., Pagani F., Romano M., Baralle F. E. (2001) EMBO J. 20, 1774–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Volkening K., Leystra-Lantz C., Yang W., Jaffee H., Strong M. J. (2009) Brain Res. 1305, 168–182 [DOI] [PubMed] [Google Scholar]
- 27. Newberry E. P., Latifi T., Towler D. A. (1999) Biochemistry 38, 10678–10690 [DOI] [PubMed] [Google Scholar]
- 28. Hatchell E. C., Colley S. M., Beveridge D. J., Epis M. R., Stuart L. M., Giles K. M., Redfern A. D., Miles L. E., Barker A., MacDonald L. M., Arthur P. G., Lui J. C., Golding J. L., McCulloch R. K., Metcalf C. B., Wilce J. A., Wilce M. C., Lanz R. B., O'Malley B. W., Leedman P. J. (2006) Mol. Cell 22, 657–668 [DOI] [PubMed] [Google Scholar]
- 29. De Leeuw F., Zhang T., Wauquier C., Huez G., Kruys V., Gueydan C. (2007) Exp. Cell Res. 313, 4130–4144 [DOI] [PubMed] [Google Scholar]
- 30. Wang X., Arai S., Song X., Reichart D., Du K., Pascual G., Tempst P., Rosenfeld M. G., Glass C. K., Kurokawa R. (2008) Nature 454, 126–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hu S., Xie Z., Onishi A., Yu X., Jiang L., Lin J., Rho H. S., Woodard C., Wang H., Jeong J. S., Long S., He X., Wade H., Blackshaw S., Qian J., Zhu H. (2009) Cell 139, 610–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Buratti E., De Conti L., Stuani C., Romano M., Baralle M., Baralle F. (2010) FEBS J. 277, 2268–2281 [DOI] [PubMed] [Google Scholar]
- 33. Winton M. J., Igaz L. M., Wong M. M., Kwong L. K., Trojanowski J. Q., Lee V. M. (2008) J. Biol. Chem. 283, 13302–13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y. J., Xu Y. F., Cook C., Gendron T. F., Roettges P., Link C. D., Lin W. L., Tong J., Castanedes-Casey M., Ash P., Gass J., Rangachari V., Buratti E., Baralle F., Golde T. E., Dickson D. W., Petrucelli L. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 7607–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wells J., Farnham P. J. (2002) Methods 26, 48–56 [DOI] [PubMed] [Google Scholar]
- 36. Weinmann A. S., Farnham P. J. (2002) Methods 26, 37–47 [DOI] [PubMed] [Google Scholar]
- 37. Core L. J., Lis J. T. (2008) Science 319, 1791–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Narita T., Yamaguchi Y., Yano K., Sugimoto S., Chanarat S., Wada T., Kim D. K., Hasegawa J., Omori M., Inukai N., Endoh M., Yamada T., Handa H. (2003) Mol. Cell. Biol. 23, 1863–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.