

Abstract
Hsp90 populates distinct open and closed conformations mediated by transient N-terminal dimerization. To investigate the mechanistic role of these large conformational changes, we designed Hsp90 with an N-terminal coiled-coil to clamp the termini together and enforce N-domain proximity. Biophysical analyses demonstrate that the coiled-coil effectively maintains N-domain proximity in the absence of ATP, a condition that favors the open state of Hsp90. Enforcing N-domain proximity results in increased ATPase activity, indicating that N-terminal dimerization is a rate-limiting step that is sped-up with the coiled-coil due to increased effective N-domain concentration. The relative difference in ATPase activity between coil-Hsp90 and wt was reduced in the presence of both an ATPase activating (Aha1) and an inhibiting (Sba1) co-chaperone. As both of these co-chaperones bind preferentially to N-terminally dimerized Hsp90, the buffering effect of these co-chaperones demonstrates the biochemical relevance of Hsp90 conformational properties in addition to N-terminal dimerization. Enforcing N-domain proximity is compatible with viability in yeast, underlining the mechanistic relevance of Hsp90 conformational changes that are less dramatic than the transition between fully open and closed.
Keywords: ATPases, Enzyme Mechanisms, Protein Conformation, Protein Folding, Signal Transduction, Heat Shock Protein 90
Introduction
Hsp90 is an essential chaperone in eukaryotes where it is required for the efficient maturation of numerous signal transduction proteins including many kinases and steroid hormone receptors (1–4). Because many of its chaperone substrates or clients are proto oncogenes, Hsp90 has emerged as a promising target for anti-cancer therapeutics (5, 6). The central role of Hsp90 in many important biological pathways along with its role in cancer provides great motivation to understand its mechanism of action.
The maturation of clients by Hsp90 requires ATP hydrolysis by the chaperone. Point mutations in Hsp90 that abrogate either ATP binding or hydrolysis both fail to mature hormone receptors and kinases and do not support viability in yeast (7, 8). All of these Hsp90-dependent processes are also impaired by treatment of cells with small molecule inhibitors that compete for the ATP binding site on Hsp90 (5, 9, 10). Thus, a wealth of experimental evidence demonstrates the requirement of ATP hydrolysis for Hsp90 function.
ATP binding and hydrolysis is widely utilized by proteins to drive conformational changes required for mechanical or regulatory function. The binding of ATP analogues to isolated Hsp90 results in a conformation change that causes a compaction of the protein as ascertained from small-angle x-ray scattering (SAXS)2 and negative stained electron micrographs (11–13). Hsp90 is composed of three stably folded domains refered to as N (N-terminal), M (middle), and C (C-terminal). The ATP binding site is located in the N domain and binding of nucleotide increases the probability of N-N dimerization as assessed by chemical crosslinking and fluorescent resonant energy transfer experiments (14–16). These observations have led to a model where ATP binding and hydrolysis triggers a conformational cycle between closed (N-domain dimerized) and open Hsp90 conformations.
Hsp90 is a slow ATPase (kcat of about 1 min−1 for yeast Hsp90) (7) and the rate-limiting step in this process involves a large protein conformational change (15, 17, 18). Mounting evidence indicates that this rate-limiting conformational change is dimerization of the N-domains induced by ATP binding (12, 19–21). The N-domain dimer is very weak, even in the presence of ATP, and is only readily observed in full-length Hsp90 where the C-domain dimer imposes high effective N-domain concentration. The fragile nature of the N-domain dimer has made it challenging to investigate its role in ATP hydrolysis. Of note, the isolated N-domain of human Hsp90 does not exhibit dimerization-dependent activity even at very high concentrations (22). EM analysis (12) indicates that the strength of N-domain dimerization may be weaker in human Hsp90 than yeast Hsp90 providing a plausible explanation for this result. Weaker N-domain dimerization of human Hsp90 may also explain its ∼10-fold lower ATPase activity than yeast Hsp90 (23).
The ATPase rate of Hsp90 can be dramatically altered by the binding of co-chaperones including Aha1 that increases hydrolysis rate and Sba1 that decreases hydrolysis rate (24, 25). Experimental evidence indicates that both of these co-chaperones preferentially bind to N-domain-dimerized conformations of Hsp90. The co-crystal structure of Sba1 with AMPPNP-bound Hsp90 demonstrates that p23 binds at the dimerization interface of the Hsp90 N-domains (19). The increased affinity of this interaction in the presence of nucleotide (26) is consistent with nucleotide-binding inducing N-domain dimerization that in turn provides an improved binding site for Sba1. The co-chaperone Aha1 also binds to a similar region of the N domain of Hsp90 as determined by chemical shift differences observed in nuclear magnetic resonance experiments (27). Of note, binding of these co-chaperones to similar locations on Hsp90 leads to opposite effects on ATPase rates. These effects are presumably mediated by distinct impacts on the local conformation of the ATP active site (19, 28).
One important prediction of the N-domain dimerization model that has been missing is experimental validation that mutants that promote N-domain association will stimulate ATPase rates in Hsp90 from different organisms. Previous studies have shown that the first eight residues of Hsp90 are involved in regulating N-domain dimerization. Deletion of these eight amino acids in yeast Hsp90 increased the stability of N-domain dimerization and also slightly increased ATPase activity (21). However, N-domain deletions are likely to have multiple biophysical effects in addition to influencing N-domain dimerization (i.e. thermodynamic stability and monomer molecular dynamics). Indeed, larger N-terminal deletions result in either a significant or total loss of ATPase activity. The effect of N-terminal deletions have not been reported for Hsp90 from species other than yeast, so it is not clear if the effect of these deletions are specific to yeast Hsp90 or are a general property of Hsp90 across different species.
In this study, we use a modular approach to enforce N-domain proximity with fully intact Hsp90 that we apply to both yeast and human Hsp90. We attach dimeric coiled-coils directly to the N-domain to enforce N-domain proximity even in the absence of ATP (Fig. 1). Toward this end we chose to utilize a GCN4-based coiled coil that had been optimized for dimer stability (29). To determine if a coiled structure at the N terminus of hsp90 inherently affects the activity of Hsp90, control experiments were also carried out using the monomeric Baldwin coil (30) motif fused to the N terminus of Hsp90.
FIGURE 1.

Conformational models of Hsp90 and the influence of engineered N-terminal coiled-coils. A, wild-type Hsp90 contains a stable C-terminal dimerization domain illustrated as a rectangle, a middle domain shown as an oval, and a transient N-terminal dimerization domain illustrated as a triangle. Transient association of the N-domain leads to open and closed states with large conformational rearrangements. To investigate the role of these large conformational rearrangements, we engineered a stable coiled-coil dimerization domain at the N terminus. B, stable association of the N-terminal coil was designed to enforce N-domain proximity, thus increasing N-domain effective concentration and favoring N-domain self-association.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Hsp82 constructs containing an N-terminal His6 tag were expressed from T7 promoters in BLR(DE3) cells induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside at 30 °C for 5 h. Cell pellets were prepared by centrifugation and resuspended in wash buffer (20 mm KPi pH 6.8, 300 mm KCl, 5 mm imidazole). Cells were then disrupted by sonication on ice and lysates were clarified in a centrifuge at 20,000 × g for 20 min at 4 °C. The supernatant was added to Co2+-nitrilotriacetic acid-agarose beads and placed on a rotator at 4 °C for 15 min. Co2+ resin was collected by centrifugation and extensively rinsed in Wash Buffer. After washing, His6-tagged protein was eluted in 200 mm imidazole, pH 7.5. EDTA was added to 10 mm to chelate any free Co2+ ions and eluates were dialyzed into 20 mm Tris, pH 8.0, 1 mm EDTA. All full-length Hsp90 constructs contained a rhinovirus 3C protease recognition site that enabled removal of the His6 tag. For these constructs, the His6 tag was removed by overnight digestion at 4 °C with a 1:20 molar ratio of protease to Hsp90 in a buffer consisting of 10% glycerol, 50 mm Tris (pH 7.5), 150 mm KCl, 5 mm EDTA. Samples were dialyzed (20 mm Tris, pH 8, 1 mm EDTA) then loaded onto a 15Q-Sepharose ion exchange column (GE Healthcare) and eluted with a linear gradient from 0 to 700 mm KCl. Eluted fractions were collected, pooled, and dialyzed into appropriate buffer conditions. The samples for SAXS analysis were dialyzed into SAXS Buffer (50 mm Tris pH 8.0, 50 mm KCl, 10 mm MgCl2) and further purified with a Superdex200 column (1.0 cm × 30 cm, GE Healthcare) in the same buffer immediately prior to SAXS analysis. Hsp82 N-domain as well as full-length Aha1 and Sba1 with non-cleavable N-terminal His6 tags were expressed and purified as described for Hsp90 constructs except that a 15S-Sepharose column was used for Aha1. Protein concentrations were determined based on absorbance measurements at 280 nm using extinction coefficients based on amino acid composition determined with the program Sednterp (Amgen).
Analytical SEC
Samples were dialyzed into SEC Buffer (20 mm Tris pH 8, 200 mm KCl, 1 mm EDTA) and concentrated to 40–50 μm. Each sample was injected at 0.5 ml/minute onto a Superdex 200 column (1.0 cm x 30 cm, G.E. Healthcare) pre-equilibrated with SEC Buffer. The column was calibrated using a 1–670 kDa size exclusion marker (Bio-Rad). Elution profiles were monitored by absorbance at 280 nm.
ATPase Activity
ATPase rates were measured using enzymatic coupling to NADH oxidation that was monitored at 340 nm (31). In this assay, Hsp90 hydrolyzes ATP to form ADP, pyruvate kinase then converts ADP and phosphenolpyruvate to pyruvate. Lactose dehydrogenase then converts pyruvate and NADH to lactate and NAD, which results in a drop in absorbance at 340 nm. Reactions were carried out at 37 °C in ATPase buffer (20 mm HEPES, pH 7.5, 5 mm MgCl2, 100 mm KCl) with 1 mm ATP (except for ATP titrations where this concentration was varied), 0.17 mm NADH, 0.67 mm phosphenolpyruvate, 0.01 mg/ml pyruvate kinase, and 0.02 mg/ml lactose dehydrogenase. The 1 mm concentration of ATP was used for comparison to previous publications and to mimic intracellular conditions. Measurements were taken every 15 s (to avoid photobleaching of NADH) for 10 min using a Bio50 spectrophotometer equipped with a Peltier temperature control unit (Cary) in a 1 cm path length cuvette. The change in absorbance versus time was fit to a linear model for the initial rate and converted to molar rates assuming a 1 to 1 correspondence between molecules of ATP hydrolyzed and molecules of NADH oxidized using 6220 m−1 cm−1 as the change in extinction coefficient for NADH oxidation and normalized for Hsp90 concentration. The ATPase activity of isolated yeast Hsp90 was measured at a protein concentration of 5 μm and human Hsp90 at 30–60 μm. Titration experiments with Sba1 were performed in ATPase buffer at 5 μm yeast Hsp90. Titration experiments with Aha1 were performed in Aha1 ATPase buffer (20 mm Tris, pH 7.5, 1.2 mm MgCl2, 4 mm KCl) at 2 μm Hsp90. Because the total concentration of both Sba1 and Aha1 in these experiments does not accurately reflect the free concentration, we used the following quadratic-derived Equation 1 to determine the apparent Kd for co-chaperone binding.
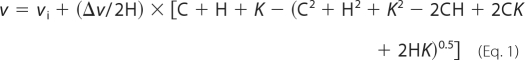 |
where v is the observed ATPase rate, vi is the initial ATPase rate in the absence of co-chaperone, and Δv is the change in ATPase rate upon co-chaperone binding, H is the total concentration of Hsp90, C is the total concentration of co-chaperone, and K is Kd app. All ATPase fits were implemented in the program Kaleidograph (Synergy Software Inc.). Errors were estimated for all measurements. For measurements made at single ATP concentrations, error bars represent standard deviations from a minimum of three independent measurements. In the case of titrations, errors in kcat and Km were based on fits with at least three independent data points for each fitted parameter. Variability in ATPase activity from different preparations was small (less than 20% difference in kcat). The low monovalent salt buffer utilized in Aha1 titrations based on previous methods (25) does result in a reduction in ATPase activity of wild-type Hsp90 (NMC) in the absence of Aha1. However, in the presence of stimulating concentrations of Aha1, the ATPase activity of both wt (NMC) and coilNMC is essentially unaffected by KCl concentration in the range of 0 to 150 mm (data not shown).
Native-PAGE
Hsp90 constructs were dialyzed into 10 mm Hepes pH 7.4. Protein samples at 5 μm were prepared in 7.5 mm Hepes, pH 7.4, 10% sucrose, 0.1% bromphenol blue, and loaded onto a polyacrylamide gel (30 mm Hepes, 30 mm imidazole, pH 7, 6% polyacrylamide, 1 mm EDTA). Gels were run at 120 mAmp for 1 h. Protein mobility through the gel was visualized using Coomassie Brilliant Blue.
Analytical Ultracentrifugation
Hsp90 samples were prepared at a subunit concentration of 12 μm for full-length constructs or 40 μm for N-domain constructs in 20 mm KPi pH 6.8, 150 mm KCl, 5 mm MgCl2, 1 mm EDTA. Full-length Hsp82 samples were spun at 8,000 RPM and N-domain constructs at 15,000 RPM at 20 °C in a Beckman XLI ultracentrifuge and a Ti60 rotor. Absorbance at 280 nm was measured every 12 h until equilibrium was achieved as determined by overlapping absorbance traces. A single species fit was used as a model to determine the molecular mass using Equation 2,
where c2(x) is the concentration at a radial distance of x, x0 is a reference radial distance, M is the molecular weight, V is the partial specific volume, ρ is the buffer density, ω is the angular velocity, R is the universal gas constant, and T is the temperature. Data were fit in Kaleidograph (Syenergy Software Inc.) using buffer density, and V values based on the buffer and amino acid composition as determined by the Sednterp program (Amgen).
Small Angle X-ray Scattering
SAXS measurements were collected at BioCAT beamline (18-ID) at the Advanced Photon Source (APS) at Argonne National Laboratory (ANL). Protein samples collected directly from size-exclusion chromatography at 1.5–3.0 mg/ml in 50 mm Tris, 50 mm KCl, 10 mm MgCl2 were shipped on wet ice to APS. Samples were analyzed in the same buffer at 1 mg/ml with or without 5 mm AMPPNP at 25 °C. Samples were exposed while flowing in a capillary tube of 1.5 mm diameter with a detector distance of 2.6 m. For each sample, 15 consecutive images were captured with an exposure time of 2 s. The data were collected on a Mar165 CCD detector. The data were circularly averaged over the detector. Raw scattering images were data reduced using Igor Pro software (Wavemetrics) to obtain I(Q) values. For each sample, the 15 data sets were graphed as I(Q) versus Q. After removing outlying traces (less than 30%), the remaining highly clustered data sets were averaged. Buffer scattering was experimentally obtained using the same procedure and subtracted from the protein samples in order to obtain the scattering due to protein. Radius of gyration (Rg) values were estimated from Guinear plots using BioCAT macros implemented in Igor Pro. The probability density [P(r)] values were determined using the program GNOM (32).
Plasmid-swap Experiments in Yeast
ECU82 cells whose only copy of Hsp90 resides on a URA3-marked plasmid (33) were transformed with TRP1-marked plasmids expressing Hsp90 variants and tested as previously described (34). Briefly, cells were selected for transformation with the TRP-marked plasmid, grown in liquid culture in the presence of uracil and serially diluted onto 5-fluoroorotic acid (FOA) plates to select for loss of the URA3-marked plasmid. All growth experiments were carried out at 25 °C.
v-Src Experiments
To analyze the maturation of a kinase client by Hsp90 we monitored v-src activity essentially as previously described (34). Briefly, a plasmid containing the v-src gene with a v5 epitope tag under a galactose-inducible promoter was introduced into yeast whose sole copy of Hsp90 was a plasmid-borne copy of either wild-type yeast Hsp90 or coilNMC. These cells were grown in selective media with raffinose as the sugar source (where expression of v-Src is off) at 25 °C to early log phase. These cells were pelleted and split into selective media at 25 °C under three different conditions: v-Src expression off (raffinose), v-Src expression on (raffinose plus galactose), and v-Src expression on with Hsp90 inhibition (raffinose plus galactose plus radicicol). In each condition, cells were grown for 5 h before harvesting for analysis. Whole cell lysates were prepared and analyzed for phosphotyrosine and v-Src content by Western blotting. Radicicol treatment included 10 min incubation at 100 μm with concentrated cells and 10 μm throughout the rest of the 5-h growth experiment. All media contained 2% sugar as either a 1:1 ratio of raffinose and galactose or pure raffinose.
GR Experiments
To analyze the maturation of a nuclear steroid hormone receptor client, we monitored GR transcriptional activation of a β-galactosidase reporter in yeast essentially as previously described (34). Briefly, a plasmid containing constitutively expressed GR with a FLAG tag and β-galactosidase driven by glucocorticoid response elements was introduced into yeast whose sole copy of Hsp90 was a plasmid-borne copy of either wild-type yeast Hsp90 or coilNMC. These cells were grown in selective dextrose media at 25 °C to early log phase. These cells were then pelleted and split into selective dextrose media under three different conditions: uninduced (without hormone agonist), GR-induced (with deoxycorticosterone agonist), and GR-induced with Hsp90 inhibited (deoxycorticosterone plus radicicol). Cells were grown for 1 h under these conditions and harvested for further analysis. Cell lysates were prepared, and β-galactosidase levels monitored based on enzymatic activity and GR expression levels monitored by Western blotting. Deoxycorticosterone was used at 10 μm, and radicicol treatment was as described for the v-Src experiments.
RESULTS
For our strategy to work, it is important that the dimeric coiled-coil that we fused to the N-domain be a sufficiently strong dimer to maintain the N-domains in close proximity. To assess the dimerization capability of our system, we analyzed the ability of the coiled-coil to induce dimerization of isolated N-domain. Equilibrium analytical ultracentrifugation (AUC) clearly demonstrates that appending the coiled-coil to the N-domain effectively induced dimerization (Fig. 2, A and B). Importantly, the AUC profiles fit using single molecular weight models result in small and randomly distributed residuals indicating that the N-domain behaves as a homogenous monomer and coil-N as a homogenous dimer. Appending the monomeric Baldwin coil to the N-domain results in a molecule that is monomeric in solution as determined by AUC (Fig. 2C). Results from analytical size-exclusion chromatography (Fig. 2D) and native PAGE (Fig. 2E) are also consistent with the coiled-coil inducing dimerization of the N-domain. From these results we conclude that the coiled-coil is a sufficiently strong dimer to induce association of the N-domain and that the Baldwin coil does not induce dimerization.
FIGURE 2.

Coiled-coil promotes dimerization of N-domain. Analytical ultracentrifugation of Hsp90 N-domain (A), N-domain appended with a stable dimeric coiled-coil (B), and the N-domain appended with a monomeric “Baldwin” α-helical peptide (C). Single species molecular weight fits are consistent with the coiled-coil inducing dimerization of the N-domain. D, size exclusion chromatography profiles are consistent with the oligomeric states observed in AUC. E, native-PAGE analysis shows that coil-N mobility is retarded relative to N-domain, also consistent with the coiled-coil inducing dimerization of the N-domain.
Encouraged by the ability of the coiled-coil to induce dimerization of the N-domain, we set out to determine if we could use coiled-coil as a molecular clamp to enforce N-domain proximity in full-length Hsp90. We appended the coiled-coil to the N terminus of full-length Hsp90 to generate a construct that we refer to as coilNMC. To directly analyze the conformational properties of coilNMC we used SAXS. We compared a construct with the coiled-coil at the N terminus (coilNMC) to a construct with the coiled-coil at the C-terminal end (NMCcoil) (8). These constructs are virtually identical in their amino acid composition and molecular weight and any observed SAXS differences can therefore be attributed to conformational differences. Because the C-domain of Hsp90 is constitutively dimerized, NMCcoil function is virtually indistinguishable from wild-type Hsp90 (8). As previously observed for wild-type Hsp90 (35), apo NMCcoil populates a conformational ensemble that is extended compared with the AMPPNP-bound form (Fig. 3A). These experiments indicate that AMPPNP binding induces dimerization of the N-domain in NMCcoil that results in compaction of the conformational ensemble. In contrast, the N-terminal coiled-coil imposes N-domain proximity in the absence of nucleotide binding resulting in a SAXS profile that is compacted compared with NMCcoil by both SAXS (Fig. 3A) and native PAGE (Fig. 3B). We find that coilNMC does undergo a slight compaction upon binding to AMPPNP (Fig. 3C). This observation clearly demonstrates that nucleotide binding impacts the conformation of coilNMC and is consistent with the coiled-coil imposing N-domain proximity distinct from the conformation of the nucleotide-bound closed state. The coilNMC construct restrains the N-domains to be in close proximity and is compatible with the nucleotide bound fully closed-state, making it useful in understanding how N-domain proximity impacts Hsp90 function.
FIGURE 3.

N-terminal coiled-coil promotes compact conformation of Hsp90 in the absence of nucleotide. A, interatomic distance distributions observed by SAXS of coilNMC without nucleotide resembles that of NMCcoil with AMPPNP. B, Coomassie-stained native-PAGE analysis of coilNMC and NMCcoil in the absence of nucleotide. The faster relative mobility of coilNMC is consistent with the formation of a compact conformation. C, interatomic distance distributions of coilNMC is compacted slightly upon binding to AMPPNP. The distribution of NMC with AMPPNP is shown for comparison. D, during equilibrium analytical ultracentrifugation coilNMC distribution is consistent with a single molecular mass species with a molecular mass (178 kDa) similar to a theoretical dimer (172 kDa).
Enforcing N-domain proximity with the coiled-coil has the potential to impart strain on a structure that would otherwise energetically prefer a more open conformation. This strain could transiently weaken the coiled-coil interface and lead to higher order coilNMC oligomers that would complicate our analyses. To investigate this possibility, we examined the oligomeric state of coilNMC by AUC (Fig. 3D). In equilibrium AUC, coilNMC distributes as a dimer with no observable higher order oligomers, indicating that strain does not cause the coiled-coil to come apart enough to cause higher order oligomers. This observation is consistent with the structural flexibility of Hsp90 that includes a large flexible linker between the N and M domains (19).
The N-domain dimerization model posits that N-domain association is rate-limiting for ATP hydrolysis. Based on this model, the enforced proximity of the N-domains in coilNMC should hasten N-domain association and result in increased ATPase rates compared with NMC. To test this prediction we performed Michaelis-Menten analyses of ATPase rates as a function of ATP concentration (Fig. 4). Consistent with the N-domain dimerization model we observe that coilNMC has a maximal ATPase rate elevated by about 3-fold compared with NMC. In addition to enforcing N-domain proximity, the coiled-coil adds helical bulk to the N terminus of Hsp90. To distinguish the impact of adding helical bulk on ATPase rates, we analyzed BaldwinNMC that has a monomeric coil at the N terminus. Importantly, BaldwinNMC has a maximal ATPase rate that is similar to NMC. From these results we conclude that imparting N-domain proximity in coilNMC is responsible for the observed increase in ATPase rate.
FIGURE 4.

Enforcing N-domain proximity of Hsp90 increases both the apparent affinity for ATP and the hydrolysis rate. A, Michaelis-Menten analysis of the rate of ATP hydrolysis by wild-type yeast Hsp90 (NMC), Hsp90 with a dimeric coiled-coil appended to the N terminus (coilNMC) and Hsp90 with a monomeric α-helix appended to the N terminus (Bald-NMC). B, addition of a dimeric coiled-coil to human Hsp90 also results in increased ATPase activity relative to human Hsp90 lacking the coiled-coil.
The N-domain dimerization model also predicts that ATP binding and N-domain dimerization are coupled, therefore coil-NMC should favor ATP binding. We observed that coil-NMC has a Km for ATP of 90 nm as compared with wild type and Baldwin-NMC which are approximately the same (290 nm and 360 nm, respectively). Our observation that enforcing N-domain proximity decreases the Km for ATP is consistent with coupling between N-domain dimerization and ATP binding. However, because ATP hydrolysis by Hsp90 involves multiple distinct kinetic steps (15), the Km is a function of both the binding affinity (Kd) and the kinetics of the steps involved in hydrolysis. Further analyses would be required to distinguish the impact of the coiled-coil on ATP affinity.
To examine if the N-domain dimerization model is general, we used the coiled-coil approach to analyze human Hsp90. Human Hsp90 is about an order of magnitude slower ATPase than yeast Hsp90 and this has been proposed to be caused by weaker N-domain dimerization. Our system allowed us to test this model by examining the rate of ATP hydrolysis by human Hsp90 with and without a coiled-coil. With the addition of the coiled-coil motif at the N terminus of human Hsp90, the ATPase rate at 1 mm ATP increased 18-fold (from 0.04 ± 0.01 to 0.7 ± 0.1 min−1). Thus, imposing N-domain proximity in human Hsp90 has a larger fold impact than in yeast Hsp90. These results are consistent with structural studies that found that yeast Hsp90 has a higher propensity to dimerize at the N-domain than human Hsp90 (12). The predisposition of yeast Hsp90 to dimerize at the N terminus limits the potential fold-increase in ATPase rate of imposing N-domain proximity. Of note, the specific activity of human coilNMC (0.7 min−1) is similar to the activity of yeast NMC (1.8 min−1) but almost 10-fold slower than yeast coilNMC (6.1 min−1). These results demonstrate that N-domain proximity has a large impact on ATPase rates, but that other species-dependent factors also influence the ATPase activity of the purified protein.
The ATPase activity of Hsp90 is affected by the binding of co-chaperones including Aha1 that stimulates ATPase activity (25) and Sba1 that inhibits ATP turnover (24). We investigated how these two ATPase-modulating co-chaperones were affected by enforced Hsp90 N-domain proximity using coilNMC. We monitored ATPase rates as a function of both Aha1 and Sba1 concentration (Fig. 5, A and B). In both cases we observe stronger apparent Kd with coilNMC than with NMC (0.3 versus 1.4 μm for Aha1 and 0.8 versus 1.4 μm for Sba1). Both Aha1 and Sba1 influenced the rate of ATPase activity of coilNMC and NMC Hsp90 in the same direction. Sba1 reduced ATPase activity and Aha1 stimulated ATPase activity. However, the magnitude of the ATPase changes was very different for coilNMC and NMC. The ATPase activator Aha1 had a larger fold increase for NMC than for coilNMC, while the ATPase inhibitor Sba1 had a larger fold decrease for coilNMC than NMC. The net effect of these changes causes the ATPase rate of coilNMC to resemble that of NMC when bound to these co-chaperones (Fig. 5C).
FIGURE 5.

Imposing N-domain proximity has distinct biochemical impacts on the ATPase modifying co-chaperones Aha1 and Sba1. A, Aha1 co-chaperone has a stronger apparent affinity for coilNMC compared with NMC. B, apparent affinity for the Sba1 co-chaperone is only modestly stronger for coilNMC compared with NMC. C, presence of saturating levels of either Sba1 or Aha1 reduces the difference in ATPase rate between coilNMC and NMC Hsp90. Error bars represent statistical propagation of errors based on Vmax determined from titration experiments in A and B and Fig. 4A.
To examine the impact of enforced N-domain proximity on Hsp90 function in cells, we examined the coilNMC ability to support viability in yeast. Plasmid swap experiments demonstrate that coilNMC supports robust yeast growth as the sole Hsp90 in yeast (Fig. 6A). Consistent with this observation, coilNMC expresses to similar levels as NMC in yeast (Fig. 6B). Of note, coilNMC migrates slower in SDS-PAGE than NMC, demonstrating that the coiled-coil fusion is stable when expressed in yeast. These observations demonstrate that enforced N-domain proximity is compatible with the essential function of Hsp90 in yeast.
FIGURE 6.

Enforced N-domain proximity of Hsp90 is compatible with essential function in yeast. A, plasmid swap experiments demonstrate that coilNMC is able to support yeast viability as the sole Hsp90 when the wild-type copy is selected against using FOA. Cells were plated in 5-fold dilutions grown at 25 °C for 3 days. B, Western blotting demonstrates that coilNMC expresses to similar levels as NMC in yeast. To control for loading a separate gel was run and stained with Coomassie and a region corresponding to about 60–80 kDa is shown. C, GR activity in yeast cells induced with the GR agonist deoxycorticosterone with NMC or coilNMC as the sole Hsp90. The coilNMC construct supports GR activity that is reduced relative to NMC. D, maturation of the v-Src kinase in yeast cells with NMC or coilNMC as the sole Hsp90. The coilNMC construct supports v-Src activity albeit at reduced levels relative to NMC. The Hsp90 inhibitor radicicol blocks both GR and v-Src activation demonstrating that these processes are Hsp90-dependent.
To further explore the functional impacts of enforced N-domain proximity in yeast cells, we analyzed the ability of coilNMC to mature two well-established Hsp90 clients whose activity requires ATPase activity by Hsp90. For both the glucorticoid receptor (Fig. 6C) and the v-src kinase (Fig. 6D), we find that coilNMC does support maturation of these clients. Importantly, the activity of both GR and v-Src is eliminated in the presence of the Hsp90-specific inhibitor radicicol, which demonstrates that activation is Hsp90-dependent. The activity level of both GR and v-Src is reduced in yeast with coilNMC relative to NMC Hsp90 indicating that the efficiency of client maturation is reduced when the N-domains are tethered with the coiled-coil.
DISCUSSION
Our results utilizing a coiled-coil to enforce N-domain proximity in Hsp90 clearly demonstrate the central mechanistic role that N-domain organization plays in the function of the protein. Enforcing N-domain proximity caused numerous changes in all aspects of Hsp90 ATPase activity that we monitored: apparent binding to ATP, ATPase activity, binding to co-chaperones, and chaperone-influenced ATPase activity. These impacts appear to be general as human Hsp90 also demonstrated elevated ATPase activity when appended with a coiled-coil. In both yeast and human Hsp90, our results are consistent with N-terminal dimerization being rate-limiting for ATP hydrolysis. Thus, our results strongly support the N-terminal dimerization model as an evolutionary conserved central mechanism in the ATPase-driven conformational cycle of Hsp90.
Our results with yeast and human Hsp90 also reveal that many aspects of the ATPase-driven conformational cycle are not solely due to the presence or absence of N-terminal dimerization. Yeast Hsp90 had previously been observed to populate N-terminal dimerized conformations more often than human Hsp90 (12) and the slow ATPase activity of human Hsp90 had been attributed to this property. In its simplest form, the N-terminal dimerization model predicts that ATPase activity is proportional to the probability of forming the N-terminal dimerized conformation. From our results, it is apparent that other factors make large contributions to the ATPase rate of Hsp90. Enforced N-domain proximity with human Hsp90 results in an ATPase rate that is about 10-fold slower than enforced N-domain proximity with yeast Hsp90 (0.7 versus 6.1 min−1). This difference suggests that human Hsp90 is a slower ATPase than yeast Hsp90 in part due to factors independent from N-terminal dimerization. These other factors likely include slight geometric or dynamic differences in the active site due to sequence differences between human and yeast Hsp90.
This dichotomy of N-terminal dimerization playing a central but not completely deterministic role in Hsp90 function is also borne out by our analysis of the Aha1 and Sba1 co-chaperones. Thus, enforced N-domain proximity impacts the apparent binding affinity of Hsp90 for both co-chaperones, but once bound to co-chaperones, the impacts of the coiled-coil on ATPase activity compared with wild-type Hsp90 are dampened by the presence of either the ATPase stimulating (Aha1) or inhibiting (Sba1) co-chaperone. These findings are consistent with a model where binding of both co-chaperones preferentially populates N-terminal dimerized conformations, but where structural details of the active site are distinct for each co-chaperone bound conformation. Thus, once co-chaperones are bound in the context of a dimerized N-terminal conformation, structural details of the co-chaperone bound conformation determine the efficiency of ATP turnover.
Enforced N-domain proximity is compatible with biological activity indicating that the transition from fully open to closed conformations is not required for function and instead that more detailed conformational changes are the driving force for the maturation of clients in vivo (Fig. 7). Hsp90 is a very flexible protein as evidenced by its ability to populate dramatically distinct open and closed conformations (11–13, 19). The three ordered domains of Hsp90 from the open and closed conformations observed by x-ray crystallography (11, 19) can be readily superimposed and much of the flexibility of Hsp90 is due to flexible linkers that separate these domains. In particular, a stretch of about 30 acid rich amino acids between the N-terminal and middle domains provides an abundance of conformational flexibility. Thus, appending the coiled-coil to the N-domain enforces proximity, but does not prevent conformational sampling allowable by inherent flexibility. That this limited conformational sampling in coilNMC is able to support viability indicates that the maturation of clients in vivo is readily accomplished with relatively small conformational changes compared with those accessible to the wild-type protein.
FIGURE 7.

Models of the Hsp90 conformational cycle and the impact of appending a dimeric coiled-coil to the N terminus. A, for wild-type Hsp90, ATP binding and hydrolysis lead to large conformational rearrangements. B, our observations with coil-NMC demonstrate that imposing N-domain proximity dramatically decreases the magnitude of these conformational rearrangements, yet remains compatible with function in yeast.
Acknowledgments
We thank Ben Roscoe and Ryan Hietpas for helpful discussions and comments, Kim Crowley and the University of Massachusetts Ultracentrifugation Facility for assistance in collecting analytical ultracentrifugation data and Tom Irving and Liang Guo and the Advanced Photon Source General User Program (GUP-11364) for a bright light and assistance in obtaining and analyzing small-angle x-ray scattering data.
This work was supported, in whole or in part, by National Institutes of Health Grant R01-GM083038-01A and by American Cancer Society Grant RSG-08-17301-GMC.
- SAXS
- small-angle x-ray scattering
- AMPPNP
- adenylyl-imidodiphosphate
- FOA
- 5-fluoroorotic acid
- AUC
- analytical ultracentrifugation
- coilNMC
- coiled-coil at the N terminus
- NMCcoil
- coiled-coil at the C-terminal end.
REFERENCES
- 1. Borkovich K. A., Farrelly F. W., Finkelstein D. B., Taulien J., Lindquist S. (1989) Mol. Cell. Biol. 9, 3919–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Whitesell L., Lindquist S. L. (2005) Nat. Rev. Cancer. 5, 761–772 [DOI] [PubMed] [Google Scholar]
- 3. Pearl L. H., Prodromou C. (2001) Adv. Protein Chem. 59, 157–186 [DOI] [PubMed] [Google Scholar]
- 4. Pratt W. B., Toft D. O. (1997) Endocr. Rev. 18, 306–360 [DOI] [PubMed] [Google Scholar]
- 5. Whitesell L., Mimnaugh E. G., De Costa B., Myers C. E., Neckers L. M. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 8324–8328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Taldone T., Sun W., Chiosis G. (2008) Bioorg. Med. Chem. 15, 2225–2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Panaretou B., Prodromou C., Roe S. M., O'Brien R., Ladbury J. E., Piper P. W., Pearl L. H. (1998) EMBO J. 17, 4829–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wayne N., Lai Y., Pullen L., Bolon D. N. (2010) J. Biol. Chem. 285, 234–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prodromou C., Roe S. M., O'Brien R., Ladbury J. E., Piper P. W., Pearl L. H. (1997) Cell 90, 65–75 [DOI] [PubMed] [Google Scholar]
- 10. Stebbins C. E., Russo A. A., Schneider C., Rosen N., Hartl F. U., Pavletich N. P. (1997) Cell 89, 239–250 [DOI] [PubMed] [Google Scholar]
- 11. Shiau A. K., Harris S. F., Southworth D. R., Agard D. A. (2006) Cell 127, 329–340 [DOI] [PubMed] [Google Scholar]
- 12. Southworth D. R., Agard D. A. (2008) Mol. Cell 32, 631–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krukenberg K. A., Förster F., Rice L. M., Sali A., Agard D. A. (2008) Structure 16, 755–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prodromou C., Panaretou B., Chohan S., Siligardi G., O'Brien R., Ladbury J. E., Roe S. M., Piper P. W., Pearl L. H. (2000) EMBO J. 19, 4383–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hessling M., Richter K., Buchner J. (2009) Nat. Struct. Mol. Biol. 16, 287–293 [DOI] [PubMed] [Google Scholar]
- 16. Mickler M., Hessling M., Ratzke C., Buchner J., Hugel T. (2009) Nat. Struct. Mol. Biol. 16, 281–286 [DOI] [PubMed] [Google Scholar]
- 17. Richter K., Muschler P., Hainzl O., Buchner J. (2001) J. Biol. Chem. 276, 33689–33696 [DOI] [PubMed] [Google Scholar]
- 18. Weikl T., Muschler P., Richter K., Veit T., Reinstein J., Buchner J. (2000) J. Mol. Biol. 303, 583–592 [DOI] [PubMed] [Google Scholar]
- 19. Ali M. M., Roe S. M., Vaughan C. K., Meyer P., Panaretou B., Piper P. W., Prodromou C., Pearl L. H. (2006) Nature 440, 1013–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prodromou C., Roe S. M., Piper P. W., Pearl L. H. (1997) Nat. Struct. Biol. 4, 477–482 [DOI] [PubMed] [Google Scholar]
- 21. Richter K., Reinstein J., Buchner J. (2002) J. Biol. Chem. 277, 44905–44910 [DOI] [PubMed] [Google Scholar]
- 22. McLaughlin S. H., Ventouras L. A., Lobbezoo B., Jackson S. E. (2004) J. Mol. Biol. 344, 813–826 [DOI] [PubMed] [Google Scholar]
- 23. McLaughlin S. H., Smith H. W., Jackson S. E. (2002) J. Mol. Biol. 315, 787–798 [DOI] [PubMed] [Google Scholar]
- 24. Richter K., Walter S., Buchner J. (2004) J. Mol. Biol. 342, 1403–1413 [DOI] [PubMed] [Google Scholar]
- 25. Panaretou B., Siligardi G., Meyer P., Maloney A., Sullivan J. K., Singh S., Millson S. H., Clarke P. A., Naaby-Hansen S., Stein R., Cramer R., Mollapour M., Workman P., Piper P. W., Pearl L. H., Prodromou C. (2002) Mol. Cell 10, 1307–1318 [DOI] [PubMed] [Google Scholar]
- 26. McLaughlin S. H., Sobott F., Yao Z. P., Zhang W., Nielsen P. R., Grossmann J. G., Laue E. D., Robinson C. V., Jackson S. E. (2006) J. Mol. Biol. 356, 746–758 [DOI] [PubMed] [Google Scholar]
- 27. Retzlaff M., Hagn F., Mitschke L., Hessling M., Gugel F., Kessler H., Richter K., Buchner J. (2010) Mol. Cell 37, 344–354 [DOI] [PubMed] [Google Scholar]
- 28. Meyer P., Prodromou C., Liao C., Hu B., Mark Roe S., Vaughan C. K., Vlasic I., Panaretou B., Piper P. W., Pearl L. H. (2004) EMBO J. 23, 511–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Havranek J. J., Harbury P. B. (2003) Nat. Struct. Biol. 10, 45–52 [DOI] [PubMed] [Google Scholar]
- 30. Marqusee S., Robbins V. H., Baldwin R. L. (1989) Proc. Natl. Acad. Sci. U.S.A. 86, 5286–5290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nørby J. G. (1988) Methods Enzymol. 156, 116–119 [DOI] [PubMed] [Google Scholar]
- 32. Svergun D. I. (1991) J. Appl. Cryst. 24, 485–492 [Google Scholar]
- 33. Nathan D. F., Lindquist S. (1995) Mol. Cell. Biol. 15, 3917–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wayne N., Bolon D. N. (2007) J. Biol. Chem. 282, 35386–35395 [DOI] [PubMed] [Google Scholar]
- 35. Cunningham C. N., Krukenberg K. A., Agard D. A. (2008) J. Biol. Chem. 283, 21170–21178 [DOI] [PMC free article] [PubMed] [Google Scholar]