

Abstract
Microtubule stabilizing agents (MSAs) comprise a class of drugs that bind to microtubule (MT) polymers and stabilize them against disassembly. Several of these agents are currently in clinical use as anticancer drugs, whereas others are in various stages of development. Nonetheless, there is insufficient knowledge about the molecular modes of their action. Recent studies from our laboratory utilizing hydrogen-deuterium exchange in combination with mass spectrometry (MS) provide new information on the conformational effects of Taxol and discodermolide on microtubules isolated from chicken erythrocytes (CET). We report here a comprehensive analysis of the effects of epothilone B, ixabepilone (IXEMPRATM), laulimalide, and peloruside A on CET conformation. The results of our comparative hydrogen-deuterium exchange MS studies indicate that all MSAs have significant conformational effects on the C-terminal H12 helix of α-tubulin, which is a likely molecular mechanism for the previously observed modulations of MT interactions with microtubule-associated and motor proteins. More importantly, the major mode of MT stabilization by MSAs is the tightening of the longitudinal interactions between two adjacent αβ-tubulin heterodimers at the interdimer interface. In contrast to previous observations reported with bovine brain tubulin, the lateral interactions between the adjacent protofilaments in CET are particularly strongly stabilized by peloruside A and laulimalide, drugs that bind outside the taxane site. This not only highlights the significance of tubulin isotype composition in modulating drug effects on MT conformation and stability but also provides a potential explanation for the synergy observed when combinations of taxane and alternative site binding drugs are used.
Keywords: Anticancer Drug, Mass Spectrometry (MS), Microtubules, Protein Conformation, Protein Drug Interactions, Hydrogen-Deuterium Exchange, Microtubule Stabilizing Agents (MSAs)
Introduction
Microtubules (MTs)4 are major components of the cytoskeleton and play an important role in multiple cellular processes. The dynamic nature of MTs is particularly crucial when cells enter mitosis. At this stage in the cell cycle, the MT network is reorganized into the mitotic spindle, which in concert with other cellular components, is responsible for finding, attaching, and separating chromosomes. These processes require highly coordinated MT dynamics, with the ability to switch from growing to shrinking and vice versa in response to appropriate intracellular stimuli (1). Therefore, agents that disrupt microtubule dynamics inhibit the ability of cells to successfully complete mitosis, thus, limiting proliferation.
Drugs that inhibit MT dynamics have been used in the clinic as anticancer agents for over 20 years. One class of such drugs is the microtubule stabilizing agents (MSAs, Fig. 1), which bind to the assembled MT and inhibit its depolymerization into the component αβ-tubulin heterodimers. The prototype of this group of drugs, Taxol, is an effective chemotherapeutic agent used extensively in the treatment of ovarian, breast, and lung carcinomas (2). It binds to an internal site in β-tubulin (3–7) and stabilizes the longitudinal and lateral contacts within the MT (8, 9), thereby preventing its depolymerization, which leads to cell cycle arrest and subsequent cell death.
FIGURE 1.

Chemical structures of MSAs.
Despite its proven efficacy in the treatment of several tumor types, Taxol has several drawbacks, which include dose-limiting toxicities, poor aqueous solubility, and inherent and acquired drug resistance (10). This has led to an extensive search for other MSAs that would overcome these limitations. One group of drugs identified in this search was the epothilones. Although these agents have been shown to compete with Taxol binding, their pose in the taxane binding site allows for unique contacts with tubulin (11–14), resulting in a similar but slightly distinct mechanism of action (15). A recent study with bovine brain tubulin (BBT) suggested that epothilone A acts similarly to docetaxel, a synthetic analog of Taxol, in mainly stabilizing the MT longitudinal contacts (16). However, the conformational effects of other epothilones, including epothilone B and its Food and Drug Administration-approved synthetic analog, ixabepilone (IXEMPRATM), remain unknown.
Laulimalide and peloruside A, MSAs of marine origin, have also been identified in the search for novel chemotherapeutic agents. In contrast to the epothilones, these compounds do not compete with Taxol (17, 18) for binding to bovine brain tubulin (BBT) and allow for synergism with the taxane site-binding drugs (19–22). This suggests that laulimalide and peloruside bind to an alternative site. Several possibilities have been proposed for where this alternative site may be. Although computational studies based on NMR suggested that the most likely binding site for laulimalide and peloruside A is in α-tubulin (23), hydrogen-deuterium exchange mass spectrometry (HDX-MS) experiments similar to those utilized in the current study proposed a binding site adjacent to the taxane pocket in the β-tubulin subunit (16). The latter work also proposed a distinct mode for MT stabilization for peloruside A, with relaxation of intradimer contacts and β-β interactions across the lateral interface accompanying binding. The precise binding site for these drugs and the conformational effects of laulimalide, however, still remain to be defined.
An understanding of the molecular mechanisms of MT stabilization by the different MSAs is crucial for determining and predicting effectiveness of current and future drugs in this category. As of yet, however, there is insufficient knowledge on this matter.
In the present study we provide a comprehensive analysis of the conformational effects of four MSAs, epothilone B (EpoB), ixabepilone (Ixa), laulimalide (LML), and peloruside A (PelA) on the conformation of MTs isolated from chicken erythrocytes (CET), which complements our previous reports with Taxol and discodermolide (8). The results of our comparative HDX-MS studies indicate that all MSAs have significant conformational effects on the C-terminal H12 helix of α-tubulin that can lead to modulation of MT interactions with microtubule-associated and motor proteins. The major mode of MT stabilization of all the examined MSAs is the tightening of longitudinal contacts between adjacent dimers in the protofilament. Interactions between adjacent protofilaments, on the contrary, are differentially stabilized by the MSAs. This difference appears to be related to the binding modes of the drugs in CET, which are also characterized in the present study, and is, therefore, consistent with the previously observed synergistic activities of different MSA combinations (19–22). Furthermore, we find a significant deviation between the binding modes and the stabilizing activities of MSAs in CET as compared with those reported in BBT (16). As the major difference between the two sources of tubulin is the isotype composition, our results emphasize the importance of tubulin isotype content on drug binding and MT stabilization.
EXPERIMENTAL PROCEDURES
Materials
Tubulin was isolated from the marginal bands of chicken erythrocytes and from bovine brain as previously described (24, 25). BBT was stored in 0.1 m MES, 1 mm EGTA, 0.5 mm MgCl2, and 3 m glycerol, pH 6.6, in liquid nitrogen. Phosphocellulose-purified CET was stored in a nucleotide-free buffer (50 mm MES, 1 mm EGTA, 0.2 mm MgCl2, pH 6.8) at −80 °C. This tubulin contains a single α- and β-isotype, α1 and βVI, whose amino acid sequences are 95 and 83% identical to the corresponding human isotypes, respectively. Purity was 99%, as evaluated by SDS/PAGE and Coomassie staining, and isotype content was confirmed by high resolution isoelectric focusing (not shown). Purified tubulin was fully functional as assessed by measuring its ability to polymerize at 37 °C in the presence of equimolar Taxol, and its morphology was normal, as determined by negative-staining transmission electron microscopy after assembly (not shown).
Taxol and [3H]Taxol were obtained from the Drug Development Branch, National Cancer Institute, Bethesda, MD. Epothilone B was kindly provided by Professor Samuel J. Danishevsky, Memorial Sloan Kettering Cancer Center and Columbia University, New York, NY. Ixabepilone was supplied by Bristol-Myers Squibb Co. Peloruside A was isolated and purified from the marine sponge Mycale hentscheli (26). Laulimalide was isolated from Cacospongia mycofijiensis collected in the Kingdom of Tonga (27). Porcine stomach pepsin was purchased from Sigma. All drugs were stored as 5 mm solutions in DMSO at −20 °C. Deuterium oxide (99.9%) was obtained from Cambridge Isotope Laboratories. Tris(2-carboxyethyl)phosphine (0.5 m), guanidinium hydrochloride, formic acid, and trifluoroacetic acid were from Pierce. GMPCPP (100 mm) was purchased from Jena Bioscience. Acetonitrile was purchased from Fisher. All other reagents were of the highest purity available.
Drug Displacement Studies
Three separate experiments were done as described previously (8, 25) with the following modifications; tubulin and [3H]Taxol concentrations were increased to 3 μm, 0.3 mm GTP was used, and the total reaction volume was reduced to 160 μl.
Binding of Peloruside A and Laulimalide to CET
Bovine brain and chicken erythrocyte tubulin were thawed and centrifuged at 55,000 rpm and 4 °C for 20 min using a Beckman TLA100.3 rotor to remove protein aggregates. Protein concentration in the resulting supernatants was determined using a Pierce® BCA Protein Assay kit. Tubulin concentration was brought to 30 μm with 0.1 m MEM buffer (0.1 m MES, 1 mm EGTA, and 0.5 mm MgCl2, pH 6.9) and supplemented with 3 m glycerol and 1 mm GTP. To 100 μl of 30 μm BBT or 30 μm CET, 33 μm Taxol and 33 μm peloruside A, laulimalide, or epothilone B were added. Duplicates of each sample were incubated for 30 min at 37 °C and centrifuged for 20 min at 55,000 rpm in a prewarmed Beckman TLA100.3 rotor. The microtubule pellets were washed twice with 100 μl of 0.1 m MEM buffer supplemented with 3 m glycerol to remove nonspecifically bound drug. The final pellets were resuspended in 100 μl of cold 0.1 m MEM buffer, pH 6.9, and extracted 3 times with 200 μl of CH2Cl2. The organic layers, containing drugs, were combined, dried overnight, and resuspended in 200 μl of 70% (v/v) methanol. Drug content was determined by mass spectrometry using direct infusion into a 12-T Varian IonSpec FT-ICR MS (Varian Inc.).
HDX LC-MS System and Peptide Identification
A Shimadzu HPLC with two LC-10AD pumps was used to generate a fast gradient with 30 μl/min flow rate, optimized for best sequence coverage. Solvent A was 5% acetonitrile in H2O, 0.2% formic acid, and 0.01% trifluoroacetic acid, whereas solvent B consisted of 95% acetonitrile in H2O, 0.2% formic acid, and 0.01% trifluoroacetic acid. All components of the set-up, including tubing, injector, and column were submerged in an ice bath at all times to reduce back exchange.
To identify the peptides generated during the digestion step, CET preincubated in the presence of GMPCPP, a very slowly hydrolyzable analog of GTP (28), was treated with pepsin in an aqueous buffer solution. Exactly 20 μl of chilled tubulin digest was injected onto a 1.0-mm inner diameter × 50 mm C8 column (Waters Inc.). After desalting for 5 min with 5% B, the peptides were eluted at 30 μl/min with a 5–10% gradient for 0.01 min, 10–40% for 10 min, 40–50% for 1 min, and 50–95% for 1 min. The effluent was infused into a 12-T Varian IonSpec FT-ICR MS (Varian Inc.). For peptide identification 30-s fractions were collected into a 96-well plate by coupling the HPLC with TriVersa NanoMate (Advion Inc.). Each fraction was spiked with an internal standard, and the mass spectra were collected by coupling chip-based infusion of the TriVersa NanoMate with the FT-ICR MS. The MS spectra were submitted to the ProteinProspector server (MSNonspecific) to identify CET peptides based on their exact monoisotopic masses, with an error threshold set at 2 ppm, which was the instrument accuracy after internal calibration. Isobaric peptides were differentiated by their MS/MS fragmentation patterns, as obtained with the linear trap quadrupole mass spectrometer (Thermo Electron Corp.). The extent of deuterium incorporation of each peptic peptide was determined by FT-ICR MS from the centroid mass difference between deuterated and nondeuterated samples.
HDX/MS Experiments
All experiments were done in triplicate exactly as described in our previous studies with Taxol and discodermolide (8). Briefly, to map drug-induced alterations in deuterium incorporation into CET, after preincubation with GMPCPP, CET was further incubated with DMSO (control) or drug, subjected to HDX for 30 min, quenched with cold pH 2.5 denaturing phosphate buffer (0.5 m ammonium phosphate, 100% w/v guanidinium hydrochloride, 2.5 mm Tris(2-carboxyethyl)phosphine, pH 2.5) in a chilled ice bath, and immediately digested with equimolar pepsin in solution, pH 2.1, for 5 min. The resulting peptides were then separated and analyzed as above.
Data Analysis and Presentation
The MS distribution for each peptide was fitted to a Gaussian curve, and the centroid value (xc) was determined using OriginPro8. Changes in deuterium incorporation (ΔHDX) were defined as the difference between the xc values of the GMPCPP-stabilized MTs in the presence of drug and in the absence of drug (control). Average changes in deuterium incorporation (ΔHDX) ± S.D. were determined from three separate experiments. Significance was determined based on an independent two-sample t test, assuming equal sample size and equal variance, with degrees of freedom = 2n − 2, using Equation 1,
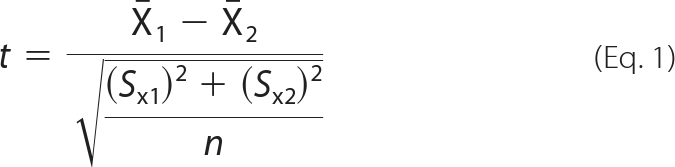 |
where X̄1 is the mean xc for drug experiments, X̄2 is the mean xc for control experiments, S is the S.D., and n is the number of experiments (n = 3 for all peptides). Based on a combination of instrument accuracy and precision of data analysis, the significance was set at p < 0.05. Thus, any change in deuterium incorporation with p < 0.05 was considered significant even if the absolute average value for ΔHDX was low. Peptides that exhibited significant changes in deuterium incorporation were mapped onto the tubulin dimer structure (PDB code 1JFF) and onto a structure of a microtubule protofilament pair previously constructed in our laboratory (9). Molecular representations of tubulin in all figures were generated using Pymol.
Docking Simulations
Docking simulations were conducted with the program Autodock using a previously developed protein structure model of chicken erythrocyte tubulin dimer built by the M4T method (9). The initial chemical structures of the four drugs (peloruside A, laulimalide, epothilone B, and ixabepilone) used to seed the docking simulations were generated using the DS Visualizer 2.0 software package. Each drug was docked using Autodock's Lamarckian-Genetic Algorithm minimization routine, with each run consisting of 100 separate trials. Blind docking to the α-subunit was used to obtain control values for the intermolecular energies based on the assumption that none of the drugs binds significantly to the α-subunit, which was suggested by the results of the HDX experiments (see “Results”). Three docking runs were carried out for each drug in the β-subunit; 1) a subunit-wide blind docking, 2) a site-directed docking in the Taxol pocket, and 3) a site-directed docking in an alternative binding site, as suggested by HDX data from this study and a previous study (16). For each run, the top 100 poses were filtered to exclude configurations making excessive primary contacts, as defined by the LPC software server (29), with residues that were not protected by ligand binding, as determined by HDX data. The binding energies (ΔGbind = RTln Kd) are reported in kcal/mol at 298.15 K. Affinity, as used in the text, is a relative term and refers to the difference between the binding energies of the conformations being compared.
RESULTS
Peptide Identification and Rapid Exchange Maps
CET is composed of only one α- and one β-tubulin isotype and has limited posttranslational modifications (30), which makes it ideal to study using MS, as it eliminates any ambiguity in the assignment of measured masses and potential conformational differences between various tubulin isotypes. Moreover, with the exception of the hypervariable C terminus, chicken βVI is nearly 90% identical both to the most abundant mammalian brain β-tubulin isotype (βII) and to human βI, the major isotype in non-neuronal tissue (supplemental Fig. S1).
The CET peptides generated by peptic digest were identified as described under “Experimental Procedures.” Isobaric peptides, those with identical masses but different sequences, were differentiated by their MS/MS fragmentation patterns (supplemental Fig. S2). The resulting map gave 91 and 93% coverage of 413 and 409 amino acids for α- and β-tubulin, respectively (supplemental Table S1).
During the HDX experiments, however, sequence coverage was reduced due to the broadening of some mass peaks caused by partial deuterium incorporation. The working peptide map consisted of 30 α- and 24 β-tubulin peptides, corresponding to 74 and 73% sequence coverage, respectively (supplemental Fig. S3). Fig. 2 summarizes the effects of four MSAs, EpoB, Ixa, LML, and PelA, on deuterium labeling per amino acid of individual peptides of α- and β-tubulin. Total ΔHDX values for each peptide shown in Fig. 2 correspond to the difference between the centroid values (xc) of the mass distributions of the peptides from GMPCPP-stabilized MTs in the presence and absence (control) of drug (supplemental Fig. S4). These ΔHDX values are also listed in the supplemental Table S2, where the corresponding significance (p value) is also indicated.
FIGURE 2.

Drug-induced alterations in deuteration referenced against GMPCPP-stabilized chicken erythrocyte microtubules for α-tubulin (a) and β-tubulin (b). Data indicate the mean ± S.D. of three separate experiments. Differences in deuteration (ΔHDX) per amino acid are expressed in mmu. Peptides are labeled with the corresponding amino acid numbers in the sequences of α- (a) and β-tubulin (b).
Region-specific Alterations in Deuterium Incorporation (HDX):Interdimer Interface
A schematic of all interfaces involved in MT stabilization is presented in Fig. 3. Fig. 4 maps the drug effects on deuterium incorporation in the vicinity of the interdimer interface, a region of contact between the adjacent αβ-tubulin dimers along the length of the protofilament. The stabilizing activity of the MSAs was strongest on this region.
FIGURE 3.

Schematic representation of interfaces involved in microtubule stabilization. In a, two adjacent protofilaments of a previously constructed chicken tubulin model (9) are shown in gray. The α- and β-tubulin subunits are dark and light gray, respectively. Directionality and orientation of the MT protofilaments are indicated in the upper left corner; IN refers to the inside of the MT, OUT to the outside, (+) to the plus end (GTP-cap), and (−) to the minus end (MT organizing center). In b, the structure of the tubulin heterodimer is enlarged (PDB code 1JFF), with several key secondary structures labeled and highlighted in red. Orientation of the dimer is indicated on the left.
FIGURE 4.

Mapping the local HDX alterations on the interdimer interface of the tubulin dimer (PDB code 1JFF). Peptides are color-coded as follows; tan = missing fragment; green = no change in deuterium incorporation (ΔHDX = 0); yellow, orange, and red = significant reduction in deuterium incorporation (ΔHDX < 0) with weak, medium, and strong effects, respectively. The conformational effects of ixabepilone (a), epothilone B (b), peloruside A (c), and laulimalide (d) are illustrated. In a, tubulin subunits are separated into three distinct regions, labeled 1–3, with each region in α-tubulin interacting with the corresponding one in β-tubulin. For clarity, only those peptides that are significantly different from panel a are shown in color in panels b–d. Secondary structure designations are based on Löwe et al. (6). The directional coordinates are shown in a, adjacent to each α- and β-tubulin component of the interface, with designations as indicated in Fig. 3.
Both the α- and β-tubulin sides of the interface were strongly protected by all four drugs. For instance, regions 2 and 3 of both α- and β-tubulin were stabilized to the same extent by all four drugs (Figs. 4 and 2 and supplemental Table S2).
With the exception of the peptide α344–351 (αH10-S9 loop), all other peptides on the α-tubulin side of region 2 were unaffected by MSAs. Of the corresponding residues in β-tubulin, two were not detected by MS after deuterium incorporation, and one, β166–180 (S5-loop), was generally unaffected by drugs (Fig. 2). Region 3, composed of peptide α319–335 (αloop-H10), as well as peptides β208–212 and β216–229 (βH6 helix and βloop-H7 respectively) exhibited a significant reduction in labeling, suggesting the tightening of contacts in this region due to drug binding.
The only part of the interdimer interface that exhibited significant differences between the MSA effects is marked as region 1. Compared with ixabepilone, peloruside A only differed in its effects on the peptide β66–73 (βS2-loop-H2), which it protected from deuterium incorporation to a significantly greater extent (Fig. 4c). The same effect on this peptide was observed with EpoB. However, in the presence of EpoB, peptides α249–254 (αloop-H8) and β10–20 (βloop-H1) were less protected than in the presence of Ixa. Finally, LML appeared to have the strongest stabilizing activity on the interdimer interface via its overwhelming allosteric effects on peptide α120–134 (αH3-loop-S4) and the corresponding β-tubulin peptide β74–100 (βH2-loop-S3-loop) in addition to the aforementioned β66–73 (βS2-loop-H2).
Exchangeable Nucleotide Binding Site
The exchangeable nucleotide binding site (E-site) in β-tubulin was protected by all four drugs, with differential effects on the specific peptides. For example, although EpoB and Ixa led to significant reduction in labeling of β133–150 (βS4-loop-H4), which contains residues that come in direct contact with the GMPCPP phosphates, the effects of PelA and LML were statistically insignificant on this region (supplemental Table S2). However, α249–254 (αloop-H8), which includes αGlu-254, involved in the hydrolysis of the E-site nucleotide phosphate (31), was strongly protected by all four drugs, which suggests that despite its accessibility to the solvent, the hydrolysis is unlikely to occur as long as the catalytic residue (i.e. αGlu-254) is unavailable. Part of peptide β168–187 (βT5 loop), involved in the interactions with the nucleotide ribose, was slightly deprotected by ixabepilone and unaffected by the rest of the ligands. Residues that contact the nucleotide base, however, were strongly protected by all ligands. These include β212–230 and β4–20, both of which are parts of the taxane binding site.
Intradimer Interface
As previously shown with Taxol and discodermolide (8) and in bovine brain tubulin (16), the region between the α- and β-tubulin subunits within a heterodimer was the least protected from deuterium incorporation, with the exception of several residues located close to the outside of the MT (Fig. 5). Specifically, peptide β341–353 (βloop-S9) was very strongly protected by all MSAs, in contrast to the adjacent β251–265 (βH8-loop), which was significantly deprotected by all drugs. The corresponding residues on the opposite side of the intradimer interface exhibited a similar trend. α400–408 (αloop-H11′) and α181–189 (αloop-H5), both of which interact with βH8-loop, were unaffected by EpoB, deprotected by PelA and LML, and only slightly protected by Ixa. Weak, but significant stabilization of the contacts between α214–227 (αloop-H7) and β316–323 (βS8-loop) was induced by all four MSAs. Although Ixa only weakly stabilized the interactions between α92–116 (αloop-H3) and β152–166 (βH4-loop-S5) and between α68–77 (αS2-loop-H2) and β240–246 (βH7-H8 loop), the remainder of the drugs induced enhanced stabilization in these regions. All three drugs, PelA, LML, and EpoB, strengthened the former contacts via their effects on the α-tubulin face of the intradimer region. EpoB was the only drug to further stabilize the latter contacts, also via its enhanced stabilization on the α-tubulin side of the interface (α68–77, αS2-loop-H2). The stabilizing activities of all four MSAs were almost identical on the β-tubulin side of the intradimer interface, with the exception of enhanced stabilization of peptide β341–353 by PelA and LML.
FIGURE 5.

Mapping the local HDX alteration on the intradimer interface of the tubulin dimer (PDB code 1JFF). Peptides are colored according to the code in Fig. 4. In addition, blue = significant increase in deuterium incorporation (ΔHDX > 0). The conformational effects of ixabepilone (a), peloruside A and laulimalide (b), and epothilone B (c) are illustrated. In b the asterisk marks peptide β341–353, which is strongly protected by all drugs but significantly more so by peloruside A and laulimalide. For clarity, only regions significantly different from panel a are shown in color in panels b and c. Secondary structure designations are based on Löwe et al. (6) The directional coordinates are shown in a above each α- and β-tubulin component of the interface, with designations as indicated in Figs. 3 and 4.
Non-exchangeable GTP Binding Site
In contrast to the results obtained with bovine brain tubulin (16), the non-exchangeable GTP binding site in α-tubulin (N-site), particularly the residues involved in the interactions with the nucleotide base and phosphates (6), was protected to a similar extent by all four ligands (Fig. 2, supplemental Table S2). This includes α-tubulin peptides α214–227, α225–230, and α137–153 (αH6-loop-H7, αH7 helix, and αT4 loop, respectively). Peptide α225–230 (αH7 helix) was particularly strongly protected by PelA and LML, suggesting a unique stabilizing effect of these two drugs on the regions interacting with the nucleotide base. Peptide α181–189 (αH5 helix), which contains a single residue shown to interact with the GTP ribose (6), was not significantly affected by EpoB and Ixa, but PelA and LML induced a small increase in deuterium incorporation. However, because the majority of the contacts made with the nucleotide ribose involve the peptide α170–180 (αT5 loop), which was not detected by MS in our experiments, conclusions about the effects of the drugs on the ribose-interacting regions of α-tubulin cannot be made. The only portion of the N-site that was significantly deprotected by all four drugs was peptide β251–265 (βH8-loop), which results from a concomitant protection of the adjacent region in β-tubulin, represented by peptide β341–353 (βloop-S9).
Interprotofilament Region
The lateral interface, a region of interaction between two adjacent protofilaments, was differentially affected by the MSAs (Fig. 6). The interactions between the α-tubulin subunits were weakly (ΔHDX/aa ≤50 millimass units (mmu)) but significantly (p < 0.05) stabilized by all four MSAs. The M-loop, represented by peptide α272–289, was significantly protected from deuterium incorporation by all four drugs. The corresponding residues on the opposite side of the interface, α37–49 of the H1-S2 loop, were similarly protected by EpoB, LML, and PelA but almost not at all by Ixa. The H3 helix, which makes additional contacts with the M-loop, was equally stabilized by all four drugs (peptide α120–134).
FIGURE 6.

Expanded view of the local HDX alterations induced by peloruside A and laulimalide (a), epothilone B (b), and ixabepilone (c) on the lateral interprotofilament interface of a previously constructed chicken tubulin model (9), which assumes a B-form lattice. The interactions between adjacent protofilaments are shown as if viewed down the length of the protofilaments with the upper portion corresponding to the inside and the lower portion corresponding to the outside of the microtubule. The peptides are color-coded based on the extent of protection from deuterium incorporation as in Figs. 4 and 5. In addition, portions of β-tubulin that comprise the alternative binding site (16) are highlighted in black. Parts of H1-S2 loop, H3 helix, and the M-loop involved in lateral contacts are indicated in parentheses in a. The labels are omitted in b and c for clarity. In a peloruside A (blue) is shown bound to the alternative site in β-tubulin, whereas in b and c epothilone B (green) and ixabepilone (magenta), respectively, are shown bound to the taxane site. All drugs have been docked into the CET MT based on computational simulations.
Overall, the interprotofilament interface between the β-tubulin subunits was significantly more stabilized than that between the α-tubulin subunits, albeit to a different extent by each drug. For this reason, the B-form lattice, in which there are β-β and α-α lateral interactions as opposed to α-β contacts found in the A-form, is the more likely configuration for the chicken erythrocyte MTs under the applied experimental conditions. Thus, the B-form lattice is assumed for CET throughout the paper. Although peptide β52–65 (βH1-S2 loop) was similarly protected by all four MSAs, notable distinctions between the drugs were observed in the effects on peptide β113–118 (βH3 helix) and, most importantly, on peptides β266–280 and β281–293, which constitute the β-tubulin M-loop. The N-terminal portion of the M-loop (β266–280), which is complementary to the βH1-S2 loop, was weakly protected by all drugs, except for ixabepilone, whose effects are negligible as compared with control GMPCPP-MTs. Peloruside A and laulimalide induced the strongest stabilizing effects on the C-terminal portion of the M-loop (β281–293; ΔHDX/aa [PelA] = −135.9 mmu, [LML] = −170.3 mmu) and the complementary βH3 helix (ΔHDX/aa [PelA] = −132.7 mmu, [LML] = −84.86 mmu). Although there was notable stabilization of the C-terminal region of the βM-loop by EpoB (ΔHDX/aa = −46.21 mmu) and Ixa (ΔHDX/aa = −30.81 mmu), the βH3 helix was only weakly protected from deuterium incorporation by Ixa (ΔHDX/aa = −19.98 mmu) and almost not at all by EpoB. In addition to providing insight into the lateral stabilization induced by the binding of the MSAs, these results shed some light on the unique binding modes of these drugs in the β-tubulin subunit of CET (see “Discussion”).
MAP Binding Site
The C-terminal portions of both α- and β-tubulin, including helices H11, H12, and the hypervariable C terminus, are known to play a major role in the interactions between MTs and microtubule-associated proteins (32) and motors (33–35). In our studies, although the H12 helix of β-tubulin was not detected under the experimental conditions, the H12 helix of α-tubulin, represented by peptide α426–438, was found to be consistently and strongly protected from deuterium incorporation by all MSAs (Fig. 2a), including Taxol and discodermolide (8). Because the hypervariable C-terminal tail is highly flexible and most immediately exposed to the solvent, we detected only small effects of the MSAs on the deuterium incorporation of the corresponding β-tubulin peptide, β435–446 (ΔHDX ≈ 11–35 mmu, Fig. 2b).
Changes in HDX Specific to the Drug Binding Sites; Epothilone B and Ixabepilone
For both epothilone B and ixabepilone, the largest reduction in labeling in the data set other than the inter- and intradimer interfaces, was found in peptides β212–230 (βH6-H7 loop) and β231–246 (core helix βH7). These two regions have been previously shown to interact with epothilone A (14). As in the case of Taxol and discodermolide (8), the M-loop, particularly the proximal portion represented by peptide β266–280, represents a region of divergence between EpoB and Ixa. Although EpoB caused a small, yet significant reduction in deuterium incorporation into this region (ΔHDX/aa = −20 mmu), Ixa had no effect. Similarly, although the distal part of the β-tubulin M-loop was protected by both drugs, EpoB imparted significantly greater stability to this region than Ixa (ΔHDX/aa = −46 mmu for EpoB versus −31 mmu for Ixa). These results suggest an important difference in the binding modes of these two drugs in CET (see “Discussion”).
Peloruside A and Laulimalide
Other than the αH7 helix (α225–230), no other residues in α-tubulin previously proposed to have interactions with PelA and LML (23) showed significant alterations in deuteration, as compared with the proposed alternative binding site in β-tubulin (16) or even the taxane site.
In β-tubulin, peptides 281–293 (βM-loop-H9 helix), 301–313 (βH9-S8 loop), and 331–340 (βH10 helix) represent the regions most strongly protected from deuterium incorporation by PelA and LML. As previously shown with bovine brain tubulin (16), these residues constitute an alternative binding site in β-tubulin adjacent to the taxane pocket, which is most likely to be involved in the binding of both PelA and LML to MTs. However, strong protection of the taxane binding pocket (6) by these two drugs suggests that the alternative site might not be the only candidate for the binding location of PelA and LML. Specifically, peptides β212–230 (βH6-H7 loop), β231–246 (βH7-loop), β21–31 (βH1), and the proximal portion of the M-loop (β266–280) exhibited significant reductions in deuterium incorporation, with ΔHDX values comparable with those of the taxane site binding drugs, epothilone B, and ixabepilone (Fig. 2; supplemental Table S2).
Drug Binding Modes
None of the MSAs used for the current studies was able to inhibit the binding of [3H]Taxol to CET (Fig. 7a). In BBT, epothilone B and ixabepilone inhibited [3H]Taxol binding, each to a different extent, whereas peloruside A and laulimalide did not (Fig. 7b). In fact, the amount of bound [3H]Taxol in the BBT pellet was increased by 40% in the presence of PelA or LML. Although it has previously been shown that PelA and LML do not compete with Taxol for binding to BBT (17, 18), our results are the first to indicate their ability to enhance Taxol binding to the MTs.
FIGURE 7.

Drug displacement with [3H]Taxol in CET (a) and BBT (b). Displacement analyses were performed as described under “Experimental Procedures.” MTs (3 μm) were assembled in the presence of different concentrations of nonradioactive drug (Taxol, discodermolide, epothilone B, ixabepilone, peloruside A, or laulimalide) and 0.3 mm GTP before the addition of 3 μm [3H]Taxol. Percent inhibition of [3H]Taxol binding to the MTs was determined based on the amount of tritiated drug found in the pellet relative to control (0 μm unlabeled drug, DMSO only).
To determine whether these drugs bound to an alternative site simultaneously with Taxol or to the taxane site with weaker affinity, Taxol and peloruside A or laulimalide or epothilone B were extracted from the MT pellet composed of bound drugs and either CET or BBT (supplemental Fig. S5). At a 1:1:1 ratio of Taxol:peloruside A:CET, both drugs were detected by direct infusion Fourier transform-MS. Similarly, in the presence of laulimalide, both Taxol and laulimalide were detected. Extracts from BBT pellets also contained both Taxol and peloruside A or laulimalide, as previously reported (17, 18). These results suggest that peloruside A and laulimalide bind to an alternative site but do not exclude the possibility that in the case of CET in the absence of Taxol, these drugs may also bind to the taxane site. The control samples with epothilone B, both in BBT and CET, yielded expected results that were consistent with those from the [3H]Taxol displacement experiments. At a ratio of 1:1:1 of Taxol:epothilone B:CET, the vast majority of organic content extracted from the MT pellet was represented by Taxol, with only trace amounts of epothilone B. The major compound found in the BBT extract, on the other hand, was epothilone B, with only trace amounts of Taxol. Therefore, epothilone B binds to the taxane pocket of both bovine brain and chicken erythrocyte MTs, with stronger affinity than Taxol for the former and weaker affinity for the latter. Analogous results were obtained with ixabepilone (data not shown), suggesting the same binding mode as epothilone B.
Computational Docking of MSAs; Epothilone B and Ixabepilone
Based on a combination of the local HDX profiles (Fig. 2) and the drug extraction experiments, the most likely binding site for EpoB and Ixa is the taxane pocket in β-tubulin. Docking simulations suggested that the taxane pocket is the preferred binding site for both of these drugs, with mean binding energies significantly lower at this site as compared with the alternative binding pocket in β-tubulin and to the α-tubulin subunit (supplemental Table S3).
In addition, these simulations suggest that a significant difference exists between the flexibilities of the two ligands within the binding pocket. Although EpoB assumes a rather static conformation at the taxane site, Ixa retains a fair degree of flexibility (Fig. 8). In fact, three major poses for Ixa were obtained from 100 top-scoring conformations (Fig. 8a), whereas EpoB assumed only one predominant pose (Fig. 8b). These findings are consistent with the HDX data, where EpoB was significantly more protective of the β-tubulin M-loop than Ixa.
FIGURE 8.

Binding sites and poses of ixabepilone (magenta) (a) and epothilone B (green) (b) in β-tubulin. The β-tubulin subunit is shown in gray (adapted from PDB code 1JFF). The residues in the taxane and the alternative binding sites are color-coded based on the extent of protection from deuterium incorporation by the corresponding drug, as in Figs. 4–6. Secondary structure designations are based on Löwe et al. (6). Orientation displayed in the right-most β-tubulin subunit of a applies to all other structural representations, with directional designations as defined in Fig. 3. In a, the three most common poses of ixabepilone (magenta), labeled 1–3, are shown in the taxane binding pocket. In b, the single most likely pose of epothilone B (green) in the taxane site is shown. The table in c lists the root mean square deviations for the two most divergent poses of each drug (Maximum RMSD). The table in d lists the minimum binding energies (ΔGbind = RT lnKd) of the three ixabepilone poses and of epothilone B for the taxane and the alternative sites. ΔGbind in α-tubulin represents a control nonspecific binding value for each drug.
The pose of EpoB in the CET taxane binding site was different from the previously proposed orientation of EpoA in BBT (14). The implications for this finding are detailed under “Discussion.”
Peloruside A and Laulimalide
A site in α-tubulin, which corresponds to the taxane binding pocket in β-tubulin, has been proposed as a possible binding site for PelA and LML based on computational docking experiments (23). Our deuterium incorporation data, however, suggested that this site in α-tubulin is an unlikely candidate for the binding of PelA and LML. This was further supported by docking simulations, in which the binding energies obtained for these drugs in the α-tubulin subunit were unfavorable compared with those in other candidate binding sites (supplemental Table S3).
A separate study, utilizing HDX-MS, has localized PelA to a binding site in β-tubulin, adjacent to the taxane pocket but closer to the outside of the MT (16). This site, defined by the H9 helix (β286–293), H9-H9′ loop (β294–301), H9′-S8 loop (β302–313), and the H10 helix (β332–340), was very strongly protected from deuterium incorporation by PelA and LML but not nearly as much by EpoB and Ixa (Fig. 2b). Co-extraction of Taxol and LML or Taxol and PelA from the same CET pellet suggests the existence of an alternative binding site for PelA and LML, which based on the aforementioned results of our HDX experiments is most likely the β-tubulin site proposed by Huzil et al. (16).
It is of note, however, that the taxane site also exhibited decreased labeling in the presence of PelA and LML, albeit to a lesser extent than the alternative β-tubulin site. Because the degree of protection of the residues in the taxane pocket was about equivalent for all four MSAs, it could not be excluded as a possible binding site for PelA and LML.
Flexible ligand docking simulations of PelA and LML in CET predicted a similar affinity of the former drug for the taxane and the alternative sites and suggested that LML would preferentially bind to the taxane pocket. Analogous simulations in BBT yielded the same result for PelA as with CET but with LML bound preferentially to the alternative site (supplemental Table S4). This difference between the predicted LML binding energies in CET and BBT can only be attributed to the sequence differences in the binding sites, as all other conditions were identical for the two docking simulations.
In summary, based on the combination of HDX experiments and docking simulations, a single binding site for PelA and LML could not be determined. Instead, we found that it was equally likely for these drugs to bind in the taxane site and the alternative site in β-tubulin adjacent to it (Fig. 9).
FIGURE 9.

Predicted binding sites of laulimalide and peloruside A. The β-tubulin subunit is shown in gray (adapted from PDB code 1JFF). The residues in the taxane and the alternative binding sites are color-coded based on the extent of protection from deuterium incorporation by the corresponding drug, as in Figs. 4–6. Secondary structure designations are based on Löwe et al. (6). Directional coordinates shown in b apply to a, and the designations are as indicated in Fig. 3. The poses of laulimalide (lime) (a) and peloruside A (green) (b) are shown in both the taxane and the alternative sites. The table in c lists the average binding energies (ΔGbind = RTln Kd) of laulimalide and peloruside A for each site. ΔGbind in α-tubulin represents a control nonspecific binding value for each drug.
DISCUSSION
Our studies have focused on four microtubule stabilizing agents, epothilone B, ixabepilone, peloruside A, and laulimalide. The former two compounds have been previously shown to bind at the taxane pocket on the inside of the β-tubulin subunit (11, 12, 14), which was further confirmed by our HDX experiments and supported by docking simulations. These results in combination with differential effects of EpoB and Ixa on the binding of [3H]Taxol to CET as compared with BBT (Fig. 7) suggest that although in BBT these drugs bind with a stronger affinity to the taxane pocket, their affinity for the same site in CET is weaker than that of Taxol. This highlights the importance of tubulin isotype composition on the interactions with MSAs.
Although EpoB and Ixa differ only in one atom (Fig. 1), in contrast to EpoB, Ixa appears to retain a significantly greater flexibility within the binding pocket as evidenced by a smaller reduction in labeling of the M-loop and supported by the larger value of max RMSD for 100 top-scoring poses obtained from docking simulations (Fig. 8c; 10.3 for Ixa versus 3.2 for EpoB). Such a difference in the binding modes of these two ligands could account for the disparities between the conformational effects.
Although the predicted binding site for EpoB was in the taxane pocket, as previously shown, the binding pose of EpoB obtained from the flexible ligand docking simulations was different from that previously obtained for EpoA in mammalian brain tubulin (14). Moreover, one of the residues that makes primary contacts with EpoB was Phe-270. Ovarian cancer cell lines containing a tubulin mutation F270V were shown to have resistance to Taxol but remained largely sensitive to EpoB (36). On the basis of this finding one would conclude that EpoB does not interact with Phe-270. To explore whether EpoB loses its affinity for the taxane site when Phe-270 is mutated to Val, we docked EpoB and Ixa into the mutant CET structure. The results suggested that this mutation does not affect the binding energies of these drugs (supplemental Fig. S6). However, the simulated pose of EpoB in the taxane binding pocket is substantially different from that of the wild type structure, such that it orients itself more closely to what has been proposed for EpoA (14). Because a change in one amino acid in the binding site may have such global effects on ligand conformation, it is not surprising that in CET, whose amino acid sequence differs from BBT in several locations within the binding site (most notably, positions 231 and 275), the pose of EpoB may differ significantly from the predicted EpoA conformation in BBT.
Unlike EpoB and Ixa, peloruside A and laulimalide have been proposed to bind to an alternative site outside the taxane pocket (16, 23), as they do not compete with Taxol for binding (17, 18) and synergize with the taxane site binding drugs in promoting tubulin assembly (20, 21) and inducing cell death (19, 22). Our work suggests several possibilities for the binding modes of peloruside A and laulimalide.
One option is that PelA and LML bind exclusively to the alternative site close to the outside of β-tubulin and adjacent to the taxane pocket. This, however, is not consistent with the docking simulations with CET in which PelA showed similar affinities for both sites and LML preferentially bound to the taxane pocket. Although the results with BBT were similar for PelA, LML preferentially bound to the alternative β-tubulin site. This difference between the binding of LML to CET and BBT can most likely be attributed to the differences in amino acids within the two available binding pockets. Two most notable substitutions, from CET to BBT, are A296S in the alternative site and A275S in the taxane site. Residues 296 and 275 in CET were mutated in Autodock to the corresponding amino acids in BBT to examine the effects on LML binding. Although mutating Ala-275 to a Ser led to a significant reduction in LML affinity for the taxane pocket, mutating Ala-296 to a Ser resulted in a mild increase in LML affinity for the alternative binding site (supplemental Table S4). These same mutations had little effect on the binding energies of PelA (see supplemental Table S3 for values). Although this shift in binding energies for LML was not large enough to bring it up to the results obtained with BBT, it was in the right direction. It is, therefore, likely that the complete combination of the amino acid differences in the binding sites of CET and BBT is responsible for the obtained docking results of laulimalide in the corresponding wild type chicken tubulin.
Because neither PelA nor LML was able to inhibit the binding of [3H]Taxol to CET, an alternative possibility is that in the presence of Taxol both drugs preferentially bind to the alternative site, whereas in the absence of Taxol and in the presence of excess PelA or LML, the drugs bind to both sites. Although there is no available binding stoichiometry data for either ligand with CET, it has previously been shown that LML binds in a 1:1 ratio to BBT even when it is added in excess (18). However, stoichiometry, although informative, does not provide information on the specific binding site of a ligand. In other words, laulimalide may still preferentially bind in the taxane pocket when a higher affinity taxane site ligand is absent. In the presence of a competitive taxane inhibitor, however, laulimalide may bind to an alternative site that may be made allosterically favorable by the binding of the taxane at its site. In fact, the reverse of this allosteric effect appears to play a role in the MSA interactions with BBT, because in the presence of PelA and LML the binding of [3H]Taxol to MTs is significantly enhanced (Fig. 7b). This observation may explain the synergistic activities of PelA and LML seen with the taxane site drugs in inducing BBT polymerization (21). The lack of enhancement of [3H]Taxol binding to CET further emphasizes the role of tubulin isotypes in determining drug binding and interactions and further complicates the determination of the binding sites of PelA and LML.
The ability to co-extract Taxol and PelA or LML from the same CET pellet (supplemental Fig. S5) does not exclude any of the aforementioned possibilities. One way to determine the specific binding sites of PelA and LML would be to utilize photoaffinity-labeled analogues of these drugs with substitutions at multiple positions to obtain labeled tubulin segments corresponding to drug-interacting regions both in the presence and in the absence of Taxol. At the present time, however, such analogues are not available.
Consistent with the distinction between the binding modes of the taxane site compounds, EpoB and Ixa, and the alternative site drugs, PelA and LML, there are also significant differences between their modes of MT stabilization (Table 1). Specifically, one such difference is apparent on the lateral interface, a region of interactions between the adjacent protofilaments in the MT. Although PelA and LML are very strongly protective of this region, especially at the interface between the adjacent β-tubulin subunits, EpoB and Ixa exhibit significantly weaker effects. This is in contrast to the findings with Pel A in BBT (16). The most likely reason for this inconsistency is that BBT is composed of several β-tubulin isotypes that could differentially interact with and be affected by PelA. This in fact is expected based on multiple studies showing that β-tubulin isotypes purified by immunoaffinity from BBT exhibit inherent differences in assembly properties, MT dynamics, and drug interactions (37–42). Therefore, because the reported results for BBT are averaged for all component isotypes, the effect of one can counteract that of another, leading to the observed lack of stabilization of lateral interactions by PelA. Another possibility is that the differences between the sequences of chicken βVI-tubulin and all of the BBT β-tubulin components are significant enough to result in opposite effects of PelA on lateral stabilization. Regardless of the reasons, enhanced stabilization by PelA and LML of contacts between the C-terminal portion of the M-loop and the H3 helix of the adjacent β-tubulin subunits is one way in which the conformational effects of these drugs are complementary to those of the taxane site ligands and can at least in part account for the aforementioned synergistic effects between these two groups of MSAs.
TABLE 1.
Summary of microtubule-stabilizing trends for MSAs

# The values are based on Khrapunovich-Baine et al. (8) and cannot be compared with the remainder of the drugs quantitatively, as the methods employed for data analysis were different; p values are based on a two-tailed Student's t test assuming unequal sample size (n = number of peptides per interface) and equal variance, with degrees of freedom (df) = n1 + n2 − 2.
** p < 0.01.
* p < 0.05.
† p < 0.15 (assigned for trend recognition).
Although there is a clear difference between the extent of stabilization of β-β lateral contacts induced by the two groups of drugs, all MSAs appear to have significant effects. It has been proposed that a destabilizing effect on lateral contacts by nucleotide hydrolysis could happen via a conformational change of H3 that is transmitted through the T3, γ-phosphate-sensing loop (43). The stabilizing activity of the MSAs, therefore, corrects this hydrolysis-induced destabilizing effect, especially in the case of PelA and LML.
Because α-tubulin always contains unhydrolyzed GTP and the extra eight residues in the S9-S10 loop stabilize the M-loop, it has been proposed that lateral contacts between α-subunits should be intrinsically strong (43). This explains why there is very little stabilization of the α-α lateral contacts by all MSAs, including Taxol and discodermolide (8).
Similarly, interactions at the intradimer interface between the α- and β-tubulin subunits of a heterodimer are intrinsically strong, as tubulin is never found in its monomeric state in vivo. Thus, the effects of MSAs on this region are not expected to be as notable as those at other interfaces, which is precisely what the HDX experiments suggested. Nevertheless, we found that all MSAs have some stabilizing activity at the intradimer interface, with all drugs having very similar conformational effects. This again contrasts with the findings reported for BBT (16), where taxane site drugs appeared to have stronger stabilizing activity than PelA. As discussed above, these differences between the conformational effects of the drugs on CET as compared with BBT are most likely due to the differences in tubulin isotype composition and/or sequence divergence. Consistent with our previous reports for Taxol (9), deprotection of βH8-loop (β251–265) in addition to very strong protection of βH10 helix (β331–340) and βloop-S9 (β341–353) leads to the straightening of the dimer in the direction of H10 upon binding of the MSAs, including discodermolide (8). This effect is directly connected to the reduction in labeling of peptides β231–246 and β212–230 due to the binding of EpoB, Ixa, and discodermolide. In the case of PelA and LML, this straightening in the direction of βH10 helix is most likely the result of direct interactions between these compounds with the H10 helix in the alternative site. It has been proposed that straightening of the tubulin dimer promotes assembly of the MT lattice (44, 45). Thus, the aforementioned conformational effects of the MSAs on the intradimer interface are part of the overall mechanism of MT stabilization by these agents.
Of all regions, the most stabilized by the MSAs was the interdimer interface, where contacts between adjacent αβ-tubulin heterodimers within the protofilament are made. In the current study the alternative site-binders, especially laulimalide, had significantly stronger stabilizing effects at the interdimer interface than the taxane site drugs. In previous work we reported the same effect of Taxol and discodermolide (8). Longitudinal stabilization via the interdimer interface, therefore, is the major mode of molecular action for the MSAs.
Finally, we found that all MSAs, including Taxol and discodermolide (8), very strongly stabilize the H12 helix of α-tubulin (α426–438), which has previously been shown to bind motor proteins, kinesin, and dynein (33–35) as well as other microtubule regulatory proteins, such as tau and MAP2 (32). Moreover, several studies have demonstrated that the binding site and affinity of tau are altered in the presence of MSAs (46, 47) and that a mutation in the αH12 helix leads to a significant reduction in the rate of ATP hydrolysis in kinesin (34). The βH12 helix and the H11 helices of both α- and β-tubulin have also been implicated in the binding of endogenous proteins (32–35, 46, 47). The peptides corresponding to these regions were either not detected under the experimental conditions utilized in our studies, or the signals were suppressed beyond detectable levels after deuterium incorporation. Nevertheless, the considerable conformational effects of the MSAs on the αH12 helix provide one potential mechanism by which this class of drugs modulates MT interactions with endogenous proteins.
The results obtained in the present study are important for understanding the molecular modes of microtubule stabilization induced by MSAs. Although all compounds in this class of drugs have the strongest stabilizing activity on the longitudinal interactions at the interdimer interface and the weakest at the interface between α- and β-tubulin subunits in a heterodimer (intradimer), there is a clear distinction between the conformational effects on the lateral interactions of the taxane site-binding drugs and the drugs that have an alternative binding site. Most notably, peloruside A and laulimalide, which belong to the latter group, very strongly stabilize lateral contacts, suggesting a distinct mode of MT stabilization that is complementary to that of the taxane site drugs and consistent with the synergy observed when these two groups of drugs are used together (19–22). The fact that the opposite effects were observed with peloruside A in BBT (16) and that there are apparent differential effects of MSAs on the [3H]Taxol binding to CET as compared with BBT highlights the importance of tubulin isotype composition in determining drug interactions and conformational effects on MTs. Furthermore, these results in combination with the established association of βIII-tubulin isotype with more aggressive and drug-resistant cancers (48–52) emphasize the need for future comparative HDX studies with human isotypically homogeneous tubulin samples. These experiments will allow for a more accurate evaluation of the differential effects of MSAs on the conformation and stability of MTs composed of different tubulin isotypes. The results of such studies could potentially have significant implications for personalized cancer therapy based on tubulin isotype profiling.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant CA124898 (NCI) (to S. B. H.). This work was also supported by the National Foundation for Cancer Research (to S. B. H.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S4 and Figs. S1–S6.
- MT
- microtubule
- MSA
- microtubule stabilizing agent
- HDX
- hydrogen-deuterium exchange
- CET
- chicken erythrocyte tubulin
- BBT
- bovine brain tubulin
- EpoB
- epothilone B
- Ixa
- ixabepilone
- PelA
- peloruside A
- LML
- laulimalide
- GMPCPP
- guanosine-5′-[(α,β)-methylene]triphosphate
- FT-ICR MS
- Fourier transform ion cyclotron resonance mass spectrometer
- mmu
- millimass units
- aa
- amino acids.
REFERENCES
- 1. Rusan N. M., Fagerstrom C. J., Yvon A. M., Wadsworth P. (2001) Mol. Biol. Cell 12, 971–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rowinsky E. K. (1997) Annu. Rev. Med. 48, 353–374 [DOI] [PubMed] [Google Scholar]
- 3. Ganesh T., Guza R. C., Bane S., Ravindra R., Shanker N., Lakdawala A. S., Snyder J. P., Kingston D. G. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 10006–10011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Geney R., Sun L., Pera P., Bernacki R. J., Xia S., Horwitz S. B., Simmerling C. L., Ojima I. (2005) Chem. Biol. 12, 339–348 [DOI] [PubMed] [Google Scholar]
- 5. Li Y., Poliks B., Cegelski L., Poliks M., Gryczynski Z., Piszczek G., Jagtap P. G., Studelska D. R., Kingston D. G., Schaefer J., Bane S. (2000) Biochemistry 39, 281–291 [DOI] [PubMed] [Google Scholar]
- 6. Löwe J., Li H., Downing K. H., Nogales E. (2001) J. Mol. Biol. 313, 1045–1057 [DOI] [PubMed] [Google Scholar]
- 7. Nogales E., Wolf S. G., Downing K. H. (1998) Nature 391, 199–203 [DOI] [PubMed] [Google Scholar]
- 8. Khrapunovich-Baine M., Menon V., Verdier-Pinard P., Smith A. B., 3rd, Angeletti R. H., Fiser A., Horwitz S. B., Xiao H. (2009) Biochemistry 48, 11664–11677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xiao H., Verdier-Pinard P., Fernandez-Fuentes N., Burd B., Angeletti R., Fiser A., Horwitz S. B., Orr G. A. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 10166–10173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Orr G. A., Verdier-Pinard P., McDaid H., Horwitz S. B. (2003) Oncogene 22, 7280–7295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bode C. J., Gupta M. L., Jr., Reiff E. A., Suprenant K. A., Georg G. I., Himes R. H. (2002) Biochemistry 41, 3870–3874 [DOI] [PubMed] [Google Scholar]
- 12. Bollag D. M., McQueney P. A., Zhu J., Hensens O., Koupal L., Liesch J., Goetz M., Lazarides E., Woods C. M. (1995) Cancer Res. 55, 2325–2333 [PubMed] [Google Scholar]
- 13. Giannakakou P., Gussio R., Nogales E., Downing K. H., Zaharevitz D., Bollbuck B., Poy G., Sackett D., Nicolaou K. C., Fojo T. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 2904–2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nettles J. H., Li H., Cornett B., Krahn J. M., Snyder J. P., Downing K. H. (2004) Science 305, 866–869 [DOI] [PubMed] [Google Scholar]
- 15. Kowalski R. J., Giannakakou P., Hamel E. (1997) J. Biol. Chem. 272, 2534–2541 [DOI] [PubMed] [Google Scholar]
- 16. Huzil J. T., Chik J. K., Slysz G. W., Freedman H., Tuszynski J., Taylor R. E., Sackett D. L., Schriemer D. C. (2008) J. Mol. Biol. 378, 1016–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gaitanos T. N., Buey R. M., Díaz J. F., Northcote P. T., Teesdale-Spittle P., Andreu J. M., Miller J. H. (2004) Cancer Res. 64, 5063–5067 [DOI] [PubMed] [Google Scholar]
- 18. Pryor D. E., O'Brate A., Bilcer G., Díaz J. F., Wang Y., Wang Y., Kabaki M., Jung M. K., Andreu J. M., Ghosh A. K., Giannakakou P., Hamel E. (2002) Biochemistry 41, 9109–9115 [DOI] [PubMed] [Google Scholar]
- 19. Clark E. A., Hills P. M., Davidson B. S., Wender P. A., Mooberry S. L. (2006) Mol. Pharm. 3, 457–467 [DOI] [PubMed] [Google Scholar]
- 20. Gapud E. J., Bai R., Ghosh A. K., Hamel E. (2004) Mol. Pharmacol. 66, 113–121 [DOI] [PubMed] [Google Scholar]
- 21. Hamel E., Day B. W., Miller J. H., Jung M. K., Northcote P. T., Ghosh A. K., Curran D. P., Cushman M., Nicolaou K. C., Paterson I., Sorensen E. J. (2006) Mol. Pharmacol. 70, 1555–1564 [DOI] [PubMed] [Google Scholar]
- 22. Wilmes A., Bargh K., Kelly C., Northcote P. T., Miller J. H. (2007) Mol. Pharm. 4, 269–280 [DOI] [PubMed] [Google Scholar]
- 23. Pineda O., Farràs J., Maccari L., Manetti F., Botta M., Vilarrasa J. (2004) Bioorg. Med. Chem. Lett. 14, 4825–4829 [DOI] [PubMed] [Google Scholar]
- 24. Murphy D. B., Wallis K. T. (1983) J. Biol. Chem. 258, 8357–8364 [PubMed] [Google Scholar]
- 25. Xia S., Kenesky C. S., Rucker P. V., Smith A. B., 3rd, Orr G. A., Horwitz S. B. (2006) Biochemistry 45, 11762–11775 [DOI] [PubMed] [Google Scholar]
- 26. West L. M., Northcote P. T., Battershill C. N. (2000) J. Org. Chem. 65, 445–449 [DOI] [PubMed] [Google Scholar]
- 27. Field J. J., Singh A. J., Kanakkanthara A., Halafihi T., Northcote P. T., Miller J. H. (2009) J. Med. Chem. 52, 7328–7332 [DOI] [PubMed] [Google Scholar]
- 28. Hyman A. A., Salser S., Drechsel D. N., Unwin N., Mitchison T. J. (1992) Mol. Biol. Cell 3, 1155–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sobolev V., Sorokine A., Prilusky J., Abola E. E., Edelman M. (1999) Bioinformatics 15, 327–332 [DOI] [PubMed] [Google Scholar]
- 30. Rüdiger M., Weber K. (1993) Eur. J. Biochem. 218, 107–116 [DOI] [PubMed] [Google Scholar]
- 31. Nogales E., Downing K. H., Amos L. A., Löwe J. (1998) Nat. Struct. Biol. 5, 451–458 [DOI] [PubMed] [Google Scholar]
- 32. Al-Bassam J., Ozer R. S., Safer D., Halpain S., Milligan R. A. (2002) J. Cell Biol. 157, 1187–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mizuno N., Toba S., Edamatsu M., Watai-Nishii J., Hirokawa N., Toyoshima Y. Y., Kikkawa M. (2004) EMBO J. 23, 2459–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uchimura S., Oguchi Y., Hachikubo Y., Ishiwata S., Muto E. (2010) EMBO J. 29, 1167–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Uchimura S., Oguchi Y., Katsuki M., Usui T., Osada H., Nikawa J., Ishiwata S., Muto E. (2006) EMBO J. 25, 5932–5941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Giannakakou P., Sackett D. L., Kang Y. K., Zhan Z., Buters J. T., Fojo T., Poruchynsky M. S. (1997) J. Biol. Chem. 272, 17118–17125 [DOI] [PubMed] [Google Scholar]
- 37. Banerjee A., Roach M. C., Trcka P., Ludueña R. F. (1990) J. Biol. Chem. 265, 1794–1799 [PubMed] [Google Scholar]
- 38. Banerjee A., Roach M. C., Trcka P., Luduena R. F. (1992) J. Biol. Chem. 267, 5625–5630 [PubMed] [Google Scholar]
- 39. Derry W. B., Wilson L., Khan I. A., Luduena R. F., Jordan M. A. (1997) Biochemistry 36, 3554–3562 [DOI] [PubMed] [Google Scholar]
- 40. Lu Q., Luduena R. F. (1993) Cell Struct. Funct. 18, 173–182 [DOI] [PubMed] [Google Scholar]
- 41. Lu Q., Luduena R. F. (1994) J. Biol. Chem. 269, 2041–2047 [PubMed] [Google Scholar]
- 42. Panda D., Miller H. P., Banerjee A., Ludueña R. F., Wilson L. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 11358–11362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nogales E., Whittaker M., Milligan R. A., Downing K. H. (1999) Cell 96, 79–88 [DOI] [PubMed] [Google Scholar]
- 44. Bennett M. J., Chik J. K., Slysz G. W., Luchko T., Tuszynski J., Sackett D. L., Schriemer D. C. (2009) Biochemistry 48, 4858–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nogales E., Wang H. W. (2006) Curr. Opin. Struct. Biol. 16, 221–229 [DOI] [PubMed] [Google Scholar]
- 46. Kar S., Florence G. J., Paterson I., Amos L. A. (2003) FEBS Lett. 539, 34–36 [DOI] [PubMed] [Google Scholar]
- 47. Makrides V., Massie M. R., Feinstein S. C., Lew J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 6746–6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Katsetos C. D., Dráberová E., Legido A., Dumontet C., Dráber P. (2009) J. Cell. Physiol. 221, 505–513 [DOI] [PubMed] [Google Scholar]
- 49. Katsetos C. D., Herman M. M., Mörk S. J. (2003) Cell Motil. Cytoskeleton 55, 77–96 [DOI] [PubMed] [Google Scholar]
- 50. Lee K. M., Cao D., Itami A., Pour P. M., Hruban R. H., Maitra A., Ouellette M. M. (2007) Histopathology 51, 539–546 [DOI] [PubMed] [Google Scholar]
- 51. Sève P., Dumontet C. (2008) Lancet Oncol. 9, 168–175 [DOI] [PubMed] [Google Scholar]
- 52. Kavallaris M. (2010) Nat. Rev. Cancer 10, 194–204 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.