

Abstract
Protein dynamics are crucial for realizing the catalytic power of enzymes, but how enzymes have evolved to achieve catalysis is unknown. The light-activated enzyme protochlorophyllide oxidoreductase (POR) catalyzes sequential hydride and proton transfers in the photoexcited and ground states, respectively, and is an excellent system for relating the effects of motions to catalysis. Here, we have used the temperature dependence of isotope effects and solvent viscosity measurements to analyze the dynamics coupled to the hydride and proton transfer steps in three cyanobacterial PORs and a related plant enzyme. We have related the dynamic profiles of each enzyme to their evolutionary origin. Motions coupled to light-driven hydride transfer are conserved across all POR enzymes, but those linked to thermally activated proton transfer are variable. Cyanobacterial PORs require complex and solvent-coupled dynamic networks to optimize the proton donor-acceptor distance, but evolutionary pressures appear to have minimized such networks in plant PORs. POR from Gloeobacter violaceus has features of both the cyanobacterial and plant enzymes, suggesting that the dynamic properties have been optimized during the evolution of POR. We infer that the differing trajectories in optimizing a catalytic structure are related to the stringency of the chemistry catalyzed and define a functional adaptation in which active site chemistry is protected from the dynamic effects of distal mutations that might otherwise impact negatively on enzyme catalysis.
Keywords: Enzyme Catalysis, Enzyme Mechanisms, Molecular Dynamics, NAD, NADH, Enzyme Dynamics, Hydrogen Tunneling
Introduction
Currently, one of the most challenging questions in biology is how enzymes have evolved to optimize the dynamic processes that enable their extraordinary rate enhancements (1–8). The role of protein motions and mechanisms of coupling to active site chemistry and solvent dynamics have been debated extensively (1, 5–10), but how the intrinsic motions of enzyme molecules have been affected by millions of years of evolutionary pressure (and the influence these motions have on catalysis) remains an open question. An evolutionary perspective of protein dynamics can be acquired only by considering functional differences between enzymes from species that span the evolutionary time scale. Hence, we have now studied this problem in the light-activated enzyme protochlorophyllide oxidoreductase (POR3; EC 1.3.1.33), which is an excellent system for relating the effects of motions to catalysis in the context of proton and hydride transfer chemistry (11, 12).
POR catalyzes the light-dependent trans-addition of hydrogen across the C17–C18 double bond of the D-ring of protochlorophyllide (Pchlide) to produce chlorophyllide, an essential step in the synthesis of chlorophyll, the most abundant pigment on Earth (Fig. 1) (11, 13). The reaction involves a highly endergonic (ground state to excited state) light-driven hydride transfer from the pro-S face of the nicotinamide ring of NADPH to C17 of the Pchlide molecule (12, 14), followed by an exergonic (ground state) proton transfer from a conserved Tyr residue to C18 (Fig. 1) (15). In POR from Thermosynechococcus elongatus, the hydride and proton transfer reactions occur sequentially by dynamically coupled nuclear tunneling (12). Fast dynamic searches within the lifetime of the Pchlide excited state are required to form degenerate “tunneling-ready” configurations for hydride transfer (12). Ultrafast measurements are consistent with the need for fast motions before the hydride transfer step, as prior excitation with a laser pulse leads to a more efficient conformation of the active site and an enhancement in the catalytic efficiency of the enzyme (16). By contrast, proton transfer from the ground state potential energy surface is linked to a different network of motions, including solvent dynamics (9, 12, 17). Proton transfer is therefore reliant on an extended network of molecular motions coupled to solvent dynamics, whereas hydride transfer is linked only to localized motions within the enzyme active site. Following hydride and proton transfer, the catalytic cycle is completed by a series of ordered product release and cofactor binding steps, which are also linked to major conformational changes in the enzyme (18).
FIGURE 1.

Scheme for the light-activated reduction of Pchlide catalyzed by POR. Upon illumination, a hydride is transferred from NADPH (blue) to C17 of Pchlide (green), followed by proton transfer from Tyr-193 (red) to C18.
From an evolutionary perspective, POR was introduced into plant cells during the primary symbiosis of an ancestral cyanobacterium, reflecting the transformation from anoxygenic to oxygenic photosynthesis (13, 19). Among prokaryotic phototrophs, which also possess a light-independent Pchlide reductase, POR is found only in cyanobacteria, suggesting that light-dependent chlorophyll biosynthesis was established prior to eukaryotic photosynthesis (19). Taken together with the experimental accessibility of the hydride and proton transfer reactions, this makes the POR enzymes attractive for studying the evolution of enzymes in relation to motions that are linked to catalysis. Here, our approach has been to analyze the dynamics of hydride and proton transfer in different POR enzymes from cyanobacteria and higher plants and to attempt to relate the dynamic profiles of each enzyme to the evolution of this enzyme family.
EXPERIMENTAL PROCEDURES
Sample Preparation
The genes encoding POR from Gloeobacter violaceus and POR B from Arabidopsis thaliana were amplified from genomic DNA using the following primers: 5′-CCG CAT ATG GCT GAA CAG ACG GTA ATC ATC-3′ and 5′-CCG AGA TCT TTA GGC AAG TCC GAC CAG TCC-3′ for G. violaceus POR and 5′-CCG CAT ATG GAC GGC AAG AAA ACG TTG AGG-3′ and 5′-CCG AGA TCT TTA GGC CAA GCC CAC GAG CTT-3′ for A. thaliana POR B. The resulting PCR products were cloned into the pET9-His expression plasmid, and His-tagged protein was overproduced in Escherichia coli and purified as described previously (17). Recombinant POR from T. elongatus and Synechocystis sp. PCC6803 was overexpressed in E. coli and purified as described (17). Pchlide was purified as described (17). Deuterated pro-S-NADPH (NADP2H) was purified and analyzed for chemical and isotopic purity as described (20). Solvent isotope effects were measured using deuterated buffer systems made with D2O (Goss Scientific). For solvent isotope effect measurements, POR was deuterated by exchange into a deuterated buffer system containing 50 mm Tris (pH 7.5), 100 mm NaCl, and 1 mm DTT.
Laser Photoexcitation Measurements
Absorption transients at the stated wavelength were measured using laser photoexcitation of dark-assembled enzyme-NADPH-Pchlide ternary complexes. Samples were excited at 450 nm using an optical parametric oscillator of a Q-switched Nd:YAG laser (Brilliant B, Quantel) in a cuvette of 1-cm path length as described previously (12). Rate constants were measured at the stated temperature from the average of at least five time-dependent absorption measurements by fitting to a single exponential function. The formation of an intermediate with an absorbance band at 696 nm represents an initial hydride transfer reaction from NADPH, and the subsequent slower exponential decay in absorbance at 696 nm represents the proton transfer reaction (12). Slower kinetically resolved absorbance changes at 670 nm on the millisecond time scale represent a series of ordered product release and cofactor rebinding events (18). For studies of the temperature dependence of rate constants, data were fitted to the Eyring equation. Buffer conditions for cryogenic studies of hydride and proton transfer were as described previously (12). For viscosity studies, glycerol solutions were prepared by weight, and calculation of solution viscosity was as described (9). The effect of viscosity on the observed rate was fitted to Equation 1 (9, 21) to describe the contribution of the protein friction to the total friction of the system,
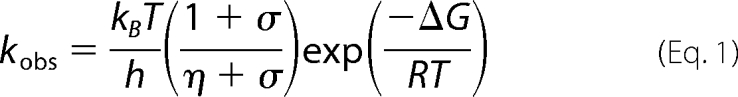 |
where σ, in units of viscosity, is the contribution of the protein friction, and η is the absolute viscosity.
Sequence Analyses
Multiple sequence alignments were carried out using Multalign software and the Blosum 62 algorithm. The resulting patterns of sequence conservation observed across the cyanobacterial and plant PORs were mapped onto the coordinates of the POR homology model (22) and subsequently displayed visually using the PyMOL Molecular Graphics System (DeLano Scientific, Palo Alto, CA). The maximum likelihood phylogenetic tree was inferred PHYML, with the LG amino acid replacement model, a four-category discrete γ-distribution, BIONJ start tree, and NNI plus SPR tree topology search. One-thousand maximum likelihood bootstrap replicates were performed with the same PHYML parameters. The tree was rooted with G. violaceus based on prior phylogenetic analyses (23, 24).
RESULTS AND DISCUSSION
Different Patterns of Structural Conservation in Cyanobacterial and Plant POR Enzymes
The phylogeny of POR enzymes has an evolutionary history with distinct cyanobacterial and eukaryotic lineages, with the cyanobacterial species G. violaceus branching off first or basal to the other cyanobacterial and plant species (supplemental Fig. S1) (23, 24). In addition, POR isoforms from the same plant are more closely related (data not shown), indicating independent duplication events (13). Although there are many highly conserved features across all POR enzymes (Fig. 2A), including the glycine-rich Rossmann fold and Tyr-X-X-X-Lys catalytic motif, there are several clearly differentiated regions between cyanobacterial and plant PORs (supplemental Fig. S2), such as the additional four residues at position 281 (numbering in T. elongatus POR) in the cyanobacterial PORs. Spatial mapping of the sequence conservation within each group of POR enzymes onto the three-dimensional model of the enzyme (22) reveals a much higher level of overall conservation among the plant PORs (Fig. 2B). Hence, it is clear that evolutionary pressures have resulted in variable patterns of structural conservation among the different POR enzymes.
FIGURE 2.

Spatial conservation of POR sequences in cyanobacterial and plant enzymes. A, residues conserved across all PORs (shown in red) are mapped onto the homology model of the enzyme from Synechocystis sp. PCC6803 (22). B, differential patterns of conservation observed in cyanobacterial (cyan) and plant (red) PORs mapped onto the surface of the POR homology model (22). Those residues showing complete conservation across all PORs are not highlighted.
Highly Conserved Localized Dynamics Control Hydride Transfer
To understand how these structural differences have influenced motions coupled to the hydride and proton transfers, we studied the catalytic mechanism and associated protein dynamics of different PORs from a variety of organisms. These included PORs from thermophilic (T. elongatus) and mesophilic (Synechocystis sp. PCC6803) cyanobacteria, a plant POR (A. thaliana POR B), and the basal cyanobacterial species (G. violaceus), which has features that are common to both cyanobacterial and plant PORs (supplemental Fig. S2). The time-dependent spectral changes following laser photoactivation confirmed that the kinetic mechanism of hydride and proton transfer is sequential (i.e. hydride precedes proton transfer) in each enzyme (supplemental Fig. S3). Dynamic information was obtained from an analysis of the temperature dependence of the kinetic isotope effects associated with hydride and proton transfer (4, 6, 8, 12, 25–29). Also, the solvent viscosity dependence of reaction rates provides further information on the nature of motions linked to the reaction chemistry (9, 21, 30–32). We used these approaches to interrogate the nature of the motions coupled to hydride and proton transfer in each of the identified POR enzymes.
Rate constants for the initial light-activated hydride transfers are similar for each of the POR enzymes using either NADPH or NADP2H (Table 1). Eyring plots of the hydride transfer step (Fig. 3) indicate an almost identical temperature dependence (range of −70 °C to +50 °C) with a breakpoint at −27 °C for each enzyme. At the higher temperatures, the temperature-dependent kinetic isotope effect (KIE) (Table 1 and supplemental Table S1) confirmed that hydride tunneling is coupled to a dominant promoting motion along the hydride transfer coordinate (12, 25–29). Conversely, below −27 °C, the KIE was essentially independent of temperature (Table 1 and supplemental Table S1), indicating that promoting motions are “frozen” at these temperatures with a consequent higher energetic cost to hydride tunneling, as reflected in the increased enthalpy of the reaction (12). Remarkably, for the hydride tunneling step, the dynamic profile of each POR enzyme was identical to that we reported previously for hydride tunneling in T. elongatus POR (12), regardless of the evolutionary lineage of the enzyme. The lack of a solvent viscosity dependence on the hydride transfer reaction for all PORs studied herein (see Fig. 5) points to the importance of localized (and not extended) dynamics in the hydride transfer reaction, as we previously inferred for T. elongatus POR (9). The NADPH-binding site is highly conserved in all PORs (Fig. 2A), and single residue changes in this region of the enzyme are known to compromise hydride transfer by substantially affecting the lifetime of the photoexcited state (15, 33, 34). This is consistent with the geometry and active site dynamics being optimally configured in all POR enzymes to enable efficient photochemistry. Evolution has therefore discovered a unique light-dependent solution to overcome a highly unfavorable (2.18 eV) (12) hydride transfer reaction, which will depend on the precise positioning of NADPH and Pchlide in the active site. This stringent geometrical and dynamic solution enables the “difficult” light-driven chemistry in all POR enzymes. The reaction chemistry is controlled by localized dynamics in all PORs through conservation of protein structure in the enzyme active site. The stringent structural requirements of the hydride transfer chemistry are consistent with the known sensitivity of this reaction to minor active site structural perturbations (15, 33, 34).
TABLE 1.
Rate constants, isotope effects, and temperature dependences (ΔΔH‡) of the hydride and proton transfer steps for the various POR enzymes
All rates were measured at 25 °C as described under “Experimental Procedures.”
| T. elongatus | Synechocystis sp. PCC6803 | G. violaceus | Arabidopsis POR B | |
|---|---|---|---|---|
| Hydride transfer | ||||
| KH− (s−1 × 106) | 1.87 ± 0.17 | 1.86 ± 0.18 | 1.86 ± 0.12 | 1.81 ± 0.24 |
| KIE | 1.91 ± 0.17 | 1.90 ± 0.16 | 1.94 ± 0.13 | 1.91 ± 0.23 |
| ΔΔH‡ (kJ mol−1)a | 8.2 ± 0.6 | 7.0 ± 0.3 | 6.9 ± 0.7 | 7.6 ± 0.9 |
| ΔΔH‡ (kJ mol−1)b | (0.4 ± 1.0) | (0.4 ± 1.5) | (0.5 ± 1.2) | (0.3 ± 1.2) |
| Proton transfer | ||||
| kH+ (s−1 × 103) | 27.4 ± 1.0 | 30.4 ± 0.6 | 152.0 ± 5.7 | 123.3 ± 3.7 |
| SIE | 2.01 ± 0.14 | 1.80 ± 0.08 | 1.54 ± 0.07 | 1.50 ± 0.07 |
| ΔΔH‡ (kJ mol−1) | 15.6 ± 3.1 | 17.6 ± 3.7 | 2.6 ± 2.4 | 1.0 ± 1.9 |
FIGURE 3.

Temperature dependence of the rate constants for the hydride transfer step. Eyring plots of ln(kobs/T) versus 1/T for the hydride transfer step with NADPH and pro-S-NADP2H (NADPD) are shown for POR from T. elongatus (A), Synechocystis sp. PCC6803 (B), G. violaceus (C), and A. thaliana POR B (D). Data are shown fitted to the Eyring equation. Activation enthalpies (ΔH‡) and activation entropies (ΔS‡) are shown in Table 1 and supplemental Table S1. The arrows indicate a conserved breakpoint at −27 °C, and error bars were calculated from the average of at least five kinetic traces. The insets show the dependence of ln KIE as a function of 1/T to illustrate how the KIE becomes essentially independent of temperature below the −27 °C breakpoint.
FIGURE 5.

Evolution of dynamics in the hydride and proton transfer reactions catalyzed by POR. A, the degree of solvent-coupled dynamics required for the hydride and proton transfer steps for PORs that span the evolutionary spectrum. The relative levels of solvent-coupled dynamics were obtained by fitting the viscosity dependence of the hydride and proton transfer rates (supplemental Fig. S5) to Equation 1. The different POR enzymes are denoted by the following abbreviations: T. e., T. elongatus BP-1; Syn., Synechocystis sp. PCC6803; G. v., G. violaceus; and A. t., A. thaliana POR B. B, scheme illustrating the twin-track approach for the evolution of dynamics in POR. The light-driven hydride transfer step is controlled by localized dynamics in all PORs through conservation of protein structure across evolutionary history (“cliff-edge” effect). Conversely, the dynamics required for proton transfer have evolved gradually, and the chemistry exhibits less reliance on complex dynamic networks in plant PORs.
Diverse Dynamic Regimes Control Proton Transfer
In contrast to the hydride transfer reaction, significant differences exist in the dynamic control of the subsequent proton transfer reaction. Proton transfer is faster (in both H2O and 2H2O buffers) (supplemental Fig. S3), and there is a smaller accompanying solvent isotope effect (SIE) for Arabidopsis and Gloeobacter PORs compared with the other cyanobacterial enzymes (Table 1), consistent with a narrower reaction barrier that facilitates quantum mechanical tunneling. Eyring plots (Fig. 4) indicate that different models for dynamically coupled hydrogen tunneling (25, 26) are required to explain the significant differences observed in the temperature dependence of the proton transfer reactions. The SIE is temperature-dependent in the thermophilic and mesophilic PORs (Fig. 4 and supplemental Table S2), consistent with a proton tunneling reaction dependent on fast (subpicosecond) promoting motions coupled to the reaction coordinate (8, 12, 26, 28). The need for compression of the energy barrier for proton tunneling implies a less than optimal structural configuration for this reaction in the cyanobacterial POR enzymes. By contrast, the rate of proton transfer for the plant and Gloeobacter enzymes is less dependent on temperature, and there is no measurable temperature dependence on the SIE (Fig. 4 and supplemental Table S2). This suggests that the active site is optimally configured (or evolved) for proton transfer, as there is no detectable promoting motion and consequent reliance on barrier compression to support a high probability of tunneling (4, 28).
FIGURE 4.

Temperature dependence of the rate constants for the proton transfer step. Eyring plots of ln(kobs/T) versus 1/T for the proton transfer step in H2O and D2O are shown for POR from T. elongatus (A), Synechocystis sp. PCC6803 (B), G. violaceus (C), and A. thaliana POR B (D). Data are shown fitted to the Eyring equation. Activation enthalpies (ΔH‡) and activation entropies (ΔS‡) are shown in Table 1 and supplemental Table S2. Error bars were calculated from the average of at least five kinetic traces. The insets show the dependence of ln KIE as a function of 1/T.
Solvent fluctuations are important in controlling the dynamics of protein systems, and these can be explored through studies of the dependence of reaction rates on solvent viscosity (9, 21, 30–32). We found major differences in the viscosity dependence of the proton transfer reaction for each of the POR enzymes, reflecting different extents of solvent control on the internal protein dynamics for this reaction (Fig. 5A and supplemental Fig. S4). The rate constant for proton transfer in the cyanobacterial enzymes is very sensitive to changes in the solvent viscosity, suggesting that a network of long-range solvent-coupled protein motions is required for proton transfer (9). In contrast, in the Gloeobacter enzyme, the solvent viscosity of the rate constant for proton transfer is significantly diminished, revealing a less dominant “slaving” effect of solvent dynamics on protein motions and reaction chemistry. Moreover, in the POR enzyme from Arabidopsis, the rate constant for proton transfer is not dependent on solvent, suggesting a small or no role for a network of long-range solvent-coupled protein motions. We infer that the faster proton transfers observed in the plant POR enzymes are reliant only on localized motions in the active site.
These differences point to considerable variation in the energy and conformational space explored by the active site proton donor (a tyrosine residue) (15) and the Pchlide substrate in different POR enzymes. Sequence analysis shows that the Pchlide-binding site is very well conserved in plant PORs but is more diverse in the cyanobacterial POR enzymes (Fig. 2). Hence, the less stringent geometrical and dynamic constraints on proton transfer compared with the highly endergonic hydride transfer have resulted in an optimal configuration for proton transfer in plant POR enzymes, whereas the cyanobacterial POR enzymes require significant protein rearrangements to ensure that an optimal proton donor-acceptor distance is achieved. This more dominant dynamic control of the proton transfer chemistry in cyanobacterial PORs also complements slower motions associated with product release and substrate binding in these enzymes. Laser experiments on the millisecond time scale indicate major conformational change associated with release of the chlorophyllide and NADP+ products from T. elongatus POR (18), but these are not observed in comparable studies with the Gloeobacter and Arabidopsis enzymes (supplemental Fig. S5 and supplemental Table S3).
Conclusions
Our analysis suggests that there is evolutionary pressure to minimize extensive and complex networks of protein motions and solvent dynamics in enzymes and that optimization of the reaction chemistry is best achieved by employing more localized active site dynamics. A twin-track approach for the evolution of POR in relation to dynamics exists (Fig. 5B). When the chemical requirements are stringently related to structure (for the highly endergonic light-driven hydride transfer), an optimal configuration of the reaction geometry has been discovered early in evolution, which prevents further significant optimization through evolutionary pressure (Fig. 5B). However, when stringency is less (proton transfer), multiple structural and dynamic solutions are possible, and optimization of the reaction chemistry is achieved by evolution to minimize the reliance on complex dynamic networks and coupling to solvent dynamics (Fig. 5B). As G. violaceus is basal to all POR enzymes (supplemental Fig. S1) and has features of both the cyanobacterial and plant PORs (supplemental Fig. S2), this indicates that the evolutionary origins of the two distinct dynamic profiles precede the endosymbiosis event that gave rise to plant PORs. Subsequent evolutionary pressures as a consequence of the stringency of the chemistry catalyzed have resulted in the differential evolutionary trajectories of POR in plants and the other cyanobacteria. That the dynamic “reach” from the active site is minimized in plant enzymes is reassuring for the design of bio-inspired catalysts, as a requirement for complex networks would surely compromise attempts to engineer new catalysts from first principles.
Supplementary Material
This work was supported by the Biotechnology and Biological Sciences Research Council, United Kingdom.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5, Tables S1–S3, and additional references.
- POR
- protochlorophyllide oxidoreductase
- Pchlide
- protochlorophyllide
- KIE
- kinetic isotope effect
- SIE
- solvent isotope effect.
REFERENCES
- 1. Eisenmesser E. Z., Bosco D. A., Akke M., Kern D. (2002) Science 295, 1520–1523 [DOI] [PubMed] [Google Scholar]
- 2. Benkovic S. J., Hammes-Schiffer S. (2003) Science 301, 1196–1202 [DOI] [PubMed] [Google Scholar]
- 3. Garcia-Viloca M., Gao J., Karplus M., Truhlar D. G. (2004) Science 303, 186–195 [DOI] [PubMed] [Google Scholar]
- 4. Masgrau L., Roujeinikova A., Johannissen L. O., Hothi P., Basran J., Ranaghan K. E., Mulholland A. J., Sutcliffe M. J., Scrutton N. S., Leys D. (2006) Science 312, 237–241 [DOI] [PubMed] [Google Scholar]
- 5. Henzler-Wildman K., Kern D. (2007) Nature 450, 964–972 [DOI] [PubMed] [Google Scholar]
- 6. Yahashiri A., Howell E. E., Kohen A. (2008) ChemPhysChem 9, 980–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nagel Z. D., Klinman J. P. (2009) Nat. Chem. Biol. 5, 543–550 [DOI] [PubMed] [Google Scholar]
- 8. Bandaria J. N., Cheatum C. M., Kohen A. (2009) J. Am. Chem. Soc. 131, 10151–10155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heyes D. J., Sakuma M., Scrutton N. S. (2009) Angew. Chem. Int. Ed. Engl. 48, 3850–3853 [DOI] [PubMed] [Google Scholar]
- 10. Pisliakov A. V., Cao J., Kamerlin S. C., Warshel A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 17359–17364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heyes D. J., Hunter C. N. (2005) Trends Biochem. Sci. 30, 642–649 [DOI] [PubMed] [Google Scholar]
- 12. Heyes D. J., Sakuma M., de Visser S. P., Scrutton N. S. (2009) J. Biol. Chem. 284, 3762–3767 [DOI] [PubMed] [Google Scholar]
- 13. Masuda T., Takamiya K. (2004) Photosynth. Res. 81, 1–29 [DOI] [PubMed] [Google Scholar]
- 14. Heyes D. J., Heathcote P., Rigby S. E., Palacios M. A., van Grondelle R., Hunter C. N. (2006) J. Biol. Chem. 281, 26847–26853 [DOI] [PubMed] [Google Scholar]
- 15. Menon B. R., Waltho J. P., Scrutton N. S., Heyes D. J. (2009) J. Biol. Chem. 284, 18160–18166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sytina O. A., Heyes D. J., Hunter C. N., Alexandre M. T., van Stokkum I. H., van Grondelle R., Groot M. L. (2008) Nature 456, 1001–1004 [DOI] [PubMed] [Google Scholar]
- 17. Durin G., Delaunay A., Darnault C., Heyes D. J., Royant A., Vernede X., Hunter C. N., Weik M., Bourgeois D. (2009) Biophys. J. 96, 1902–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heyes D. J., Sakuma M., Scrutton N. S. (2007) J. Biol. Chem. 282, 32015–32020 [DOI] [PubMed] [Google Scholar]
- 19. Suzuki J. Y., Bauer C. E. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 3749–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pudney C. R., Hay S., Sutcliffe M. J., Scrutton N. S. (2006) J. Am. Chem. Soc. 128, 14053–14058 [DOI] [PubMed] [Google Scholar]
- 21. Ansari A., Jones C. M., Henry E. R., Hofrichter J., Eaton W. A. (1992) Science 256, 1796–1798 [DOI] [PubMed] [Google Scholar]
- 22. Townley H. E., Sessions R. B., Clarke A. R., Dafforn T. R., Griffiths W. T. (2001) Proteins 44, 329–335 [DOI] [PubMed] [Google Scholar]
- 23. Nelissen B., Van de Peer Y., Wilmotte A., De Wachter R. (1995) Mol. Biol. Evol. 12, 1166–1173 [DOI] [PubMed] [Google Scholar]
- 24. Gupta R. S., Mathews D. W. (2010) BMC Evol. Biol. 10, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuznetsov A. M., Ulstrup J. (1999) Can. J. Chem. 77, 1085–1096 [Google Scholar]
- 26. Knapp M. J., Rickert K., Klinman J. P. (2002) J. Am. Chem. Soc. 124, 3865–3874 [DOI] [PubMed] [Google Scholar]
- 27. Knapp M. J., Klinman J. P. (2002) Eur. J. Biochem. 269, 3113–3121 [DOI] [PubMed] [Google Scholar]
- 28. Johannissen L. O., Hay S., Scrutton N. S., Sutcliffe M. J. (2007) J. Phys. Chem. B 111, 2631–2638 [DOI] [PubMed] [Google Scholar]
- 29. Masgrau L., Basran J., Hothi P., Sutcliffe M. J., Scrutton N. S. (2004) Arch. Biochem. Biophys. 428, 41–51 [DOI] [PubMed] [Google Scholar]
- 30. Fenimore P. W., Frauenfelder H., McMahon B. H., Parak F. G. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 16047–16051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fenimore P. W., Frauenfelder H., McMahon B. H., Young R. D. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 14408–14413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Samuni U., Roche C. J., Dantsker D., Friedman J. M. (2007) J. Am. Chem. Soc. 129, 12756–12764 [DOI] [PubMed] [Google Scholar]
- 33. Lebedev N., Karginova O., McIvor W., Timko M. P. (2001) Biochemistry 40, 12562–12574 [DOI] [PubMed] [Google Scholar]
- 34. Menon B. R., Davison P. A., Hunter C. N., Scrutton N. S., Heyes D. J. (2010) J. Biol. Chem. 285, 2113–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.