

Abstract
Myosin heavy chain (MHC) isoforms are principal determinants of work capacity in mammalian ventricular myocardium. The ventricles of large mammals including humans normally express ∼10% α-MHC on a predominantly β-MHC background, while in failing human ventricles α-MHC is virtually eliminated, suggesting that low-level α-MHC expression in normal myocardium can accelerate the kinetics of contraction and augment systolic function. To test this hypothesis in a model similar to human myocardium we determined composite rate constants of cross-bridge attachment (fapp) and detachment (gapp) in porcine myocardium expressing either 100% α-MHC or 100% β-MHC in order to predict the MHC isoform-specific effect on twitch kinetics. Right atrial (∼100% α-MHC) and left ventricular (∼100% β-MHC) tissue was used to measure myosin ATPase activity, isometric force, and the rate constant of force redevelopment (ktr) in solutions of varying Ca2+ concentration. The rate of ATP utilization and ktr were approximately ninefold higher in atrial compared with ventricular myocardium, while tension cost was approximately eightfold greater in atrial myocardium. From these values, we calculated fapp to be ∼10-fold higher in α- compared with β-MHC, while gapp was 8-fold higher in α-MHC. Mathematical modeling of an isometric twitch using these rate constants predicts that the expression of 10% α-MHC increases the maximal rate of rise of force (dF/dtmax) by 92% compared with 0% α-MHC. These results suggest that low-level expression of α-MHC significantly accelerates myocardial twitch kinetics, thereby enhancing systolic function in large mammalian myocardium.
Keywords: myosin heavy chain isoforms, cross-bridge cycling kinetics, ventricular myocardium
the ability of mammalian myocardium to perform work depends on a multitude of factors that modulate contractility both acutely and chronically. Beat-to-beat variations in work capacity can be mediated by several mechanisms, including the Frank-Starling response to variations in ventricular end-diastolic volume, as well as phosphorylation of myofibrillar and Ca2+ handling proteins. In contrast, chronic alterations in myocardial work capacity are determined in large part by the level of gene expression of myosin heavy chain (MHC) isoforms. The expression and distribution of the MHC isoforms in myocardium play a crucial role in determining the intrinsic rate of ATP turnover and thus are important determinants of myocyte shortening velocity (Vmax) (44) and power output (21, 22, 28).
Cardiac myosin is a hexamer consisting of two heavy chain isoforms, α and β (24), and four associated light chains. The MHC isoforms are distinguished primarily by the nucleotide turnover rate of the globular head (S1) region of the thick filament, which converts the energy released from the hydrolysis of ATP into force generation and myofilament sliding (16). Comparatively, α-MHC is a faster mechanoenzyme resulting in greater Vmax and power generation (44), while β-MHC cycles ATP at a slower rate and is more efficient at intermediate loads since less ATP is required to maintain a given force (1, 25, 42). While the mechanism underlying the difference in cycling kinetics between MHC isoforms is unclear, it is likely that chemomechanical transduction is regulated at least in part by coordinated action of the hyperflexible surface loops of myosin, namely loop 1 (ATPase loop) and loop 2 (actin binding loop). Together, the discrete regions of S1 are thought to mediate variations in the rates of the multiple transitions that comprise the cross-bridge cycle and thereby contribute to the different velocities of the MHC isoforms. Following the characterization of protein and transcript levels for α- and β-MHC in human myocardium, the relationship between MHC isoform distribution and contractile function has attracted considerable interest. α-MHC accounts for ∼10% of the total myosin expressed in healthy human hearts, while in failing ventricles α-MHC is virtually absent (36) because of greatly reduced α MHC mRNA (33, 37). Such changes during disease appear to be detrimental to contractile function. Furthermore, recent studies have associated mutations in the α-MHC gene with some cases of hypertrophic (HCM) and dilated (DCM) cardiomyopathy (11), suggesting that mutant α-MHC molecules may also lead to myocardial dysfunction and ultimately to heart failure. Taken together, these results indicate that either reduced expression of or mutations in α-MHC contribute to myocardial dysfunction in disease and highlight the crucial role α-MHC plays in the maintenance of function in normal human myocardium.
From a functional standpoint, however, the impact of a shift in MHC isoform expression during human heart failure remains elusive, presumably because experimentation in human myocardium is complicated by the onset of multiple compensatory mechanisms in disease, such as alterations in the expression levels of Ca2+ handling proteins and differences in the level of protein phosphorylation. However, recent studies have suggested that the decrease in α-MHC expression is maladaptive in response to severe cardiovascular stress and that isoform redistribution to virtually all β-MHC may facilitate the development of heart failure (29) due to depression of contraction at both maximal and submaximal levels of activation. In this regard, the rate constant of force redevelopment (ktr) is depressed at all levels of activation in rat myocardium lacking α-MHC, implying that the presence of α-MHC contributes to function in wild-type hearts even under resting conditions (46). Consistent with these results, others have shown improved contractile function in vivo due partly to reexpression of α-MHC by various therapeutic strategies (30, 32), while α-myosin motor protein gene transfer resulted in Ca2+-independent positive inotropy in failing human cardiomyocytes (20). Thus, while increased expression of β-MHC during heart failure may be an important mechanism to reduce tension cost in diseased myocardium, it is reasonable to suppose that the concomitant loss of α-MHC is at least partly responsible for the related functional deficit.
The aim of the present study was to predict the effect of low-level α-MHC expression on contraction kinetics in porcine myocardium, which expresses levels of α-MHC in the ventricle similar to those in human myocardium. To assess the contribution of the myosin isoforms to twitch kinetics in the ventricles of large mammals, we measured parameters of contractile function (i.e., ATPase turnover, ktr, and isometric force) in atrial and ventricular porcine myocardium and used these measurements to estimate the rate constants of cross-bridge attachment (fapp) and detachment (gapp). The rate constants were then used to simulate an isometric twitch in order to predict the functional effects of low-level expression of α-MHC in myocardium. Our results predict that even a small percentage of α-MHC expression (∼10%) dramatically accelerates the kinetics of force development, as manifested by a ∼90% increase in the maximal rate of rise of force (dF/dtmax). Collectively, these data suggest that the loss of α-MHC, as is typically seen in human heart failure, would significantly depress the contractile properties of myocardium and thereby impair systolic function.
MATERIALS AND METHODS
Experimental Animals
Myocardium used in this study was obtained from male and female adolescent pigs (Yorkshire, n = 4) weighing 45 ± 3 kg. Experimental animals were sedated with a combination of (in mg/kg) 1,000 ketamine, 0.8 atropine, and 30 acepromazine followed by infusion of 250 thiopental sodium and 1.5 α-chloralose via intravenous catheterization of the superficial ear vein. Animals were artificially ventilated with supplemental oxygen, and arterial pH, O2, and Pco2 were continuously monitored to ensure maintenance of physiological oxygenation and acid-base balance. The heart was exposed by bilateral thoracotomy with transsternotomy, and euthanasia was performed by removal of the heart followed by immediate freezing in liquid nitrogen. All animal usage, anesthesia, and surgery were conducted under the strict guidelines and approval of the University of Wisconsin Medical School Animal Care and Use Committee.
Solutions
The compositions of solutions used for mechanical experiments were calculated with the computer program of Fabiato (13) and the stability constants (corrected to pH 7 and 20°C) listed by Godt and Lindley (19). Relaxing solution (pH 7.0) used for bathing skinned trabeculae contained (in mmol/l) 100 KCl, 20 imidazole, 7 MgCl2, 2 EGTA, and 4 ATP. In addition, pCa 9.0 solution contained (in mmol/l) 5.86 ATP, 6.53 MgCl2, 20 EGTA, 48.23 μM CaCl2, and 100 N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES); pCa 4.5 solution contained (in mmol/l) 5.89 ATP, 6.01 MgCl2, 20 EGTA, 19.85 CaCl2, and 100 BES; and preactivating solution contained 0.5 EGTA. In addition, pCa 9.0, 4.5, and preactivating solutions contained (in mmol/l) 5 NaN3 (sodium azide), 0.01 leupeptin, 0.01 P1-P5-di(adenosine-5′)pentaphosphate (A2P5), and 10 phospho(enol)pyruvic acid (PEP). The ionic strength of all solutions was adjusted to 200 mmol/l with potassium propionate. Solutions containing a range of free Ca2+ concentrations ([Ca2+]free; i.e., pCa 6.2–5.4) were prepared by mixing appropriate volumes of solutions of pCa 9.0 and pCa 4.5. Pyruvate kinase (0.91 mg/ml) and lactate dehydrogenase (0.31 mg/ml) were added to all pCa and preactivating solutions before mechanical experiments. Solutions used for measurements of ktr were as previously described (46).
Experimental Protocols
Tissue preparation.
To prepare skinned myocardial preparations for measurements of ATP utilization, isometric force, and ktr, subendocardial tissue sections from frozen atria and ventricles that had been rapidly frozen in liquid nitrogen were homogenized in relaxing solution for ∼2 s with a Polytron homogenizer, which yielded multicellular preparations with dimensions of 100–250 μm × 600–900 μm. The homogenate was centrifuged at 120 g for 60 s, and the resulting pellet was washed with fresh relaxing solution and resuspended in relaxing solution containing 1% Triton X-100. After a 30-min permeabilization period, the skinned preparations were washed with fresh relaxing solution and then dispersed in relaxing solution in a glass petri dish. The dish was kept on ice at all times except during the selection of preparations for mechanical experiments. This protocol yields a large number of high-quality, robust preparations, and previous work from this laboratory has found no difference in mechanical measurements between trabeculae or homogenized skinned myocardium frozen rapidly in liquid nitrogen (39, 46, 48).
ATPase experiments.
While immersed in relaxing solution, a single multicellular preparation was attached to the arms of a high-speed length controller (model 312B, Aurora Scientific) and a force transducer (model 403A, Aurora Scientific) by fixing each end to a stainless steel trough with a 0.5-mm length of 4-0 monofilament secured in place with loops of 10-0 monofilament. This procedure has been optimized to virtually eliminate compliance at the ends and leave ∼750–1,000 μm of the preparation exposed to the solution, which allowed accurate measurement of sarcomere length via the diffraction pattern obtained with a He-Ne laser, described below.
To calculate cross-bridge attachment and detachment rate constants (6) three variables were measured: ATPase activity per volume of preparation [proportional to fg/(f + g)], tension cost (proportional to g/Funi, where Funi is the force generated per cross bridge), and the rate constant of force redevelopment (ktr = f + g). Measurements of preparation dimensions were collected during each experiment to allow calculations of sample volume.
ATPase activity, isometric force, and ktr were recorded in solutions of varying [Ca2+] at 20°C and a mean sarcomere length of 2.2 μm. The active force at each pCa was measured as the difference between total steady-state force and resting force determined by applying a 20% slack step to the preparation in solution of pCa 9.0. Active force was normalized to the cross-sectional area of the preparation. A He-Ne laser (wavelength 632.8 nm) was directed toward the mounted preparation, and the resulting diffraction pattern was projected onto a screen. Measurements of the distance between the 0th- and 1st-order lines were then used to calculate sarcomere length, a method that is accurate to within 0.1 μm. In addition, careful monitoring of sarcomere length during each experiment quickly revealed compliance in the attachments. If the change in sarcomere length upon activation with Ca2+ was >0.1 μm, the preparation was discarded.
ATP consumption was measured with an NADH-coupled enzyme system (18, 49), as previously used by this laboratory in rat myocardium (31). ADP production during ATP hydrolysis was measured as a change in NADH concentration, as shown in the reaction scheme below:
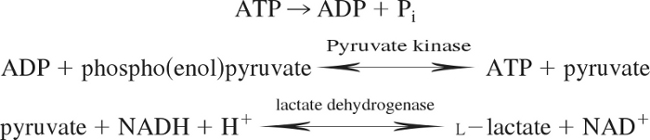 |
NADH absorbs UV light at a wavelength of 339 nm, whereas NAD+ does not, and thus ATP consumption can be measured as the change in absorbance at 339 nm (18, 49). The experimental chamber consisted of a 15-μl well enclosed on two sides by quartz windows. Near-UV light was directed to the skinned myocardial preparation in the experimental chamber, and the transmitted beam was split and directed to two photodiodes: one to record the transmitted light at 339 nm (for determination of NADH concentration) and the other to correct the signal for photobleaching and to determine nonspecific absorbance at a reference wavelength other than 339 nm. NADH (70 mmol/l) was injected into the measuring chamber before incubation of the skinned myocardial preparations, while calibration of the ATPase signal was performed with 25-nl injections of 10 mmol/l ADP into the measuring chamber. During experiments, solution in the measuring chamber was continually stirred via motor-driven displacement of a latex membrane on the bottom of the chamber. Recording of the two signals was necessary to minimize noise that may arise from irregularities due to continuous mixing. The rate of NADH consumption was taken as the ATPase activity and was determined by linear regression analysis of the measured absorption trace.
Ca2+ sensitivity and kinetics of force redevelopment.
Active force at each pCa was measured as the difference between total steady-state force and passive force determined by applying a 20% slack step to the preparation in solution of pCa 9.0. Force-pCa relationships were derived by expressing submaximal force (P) at each pCa (range 6.1–5.4) as a fraction of maximum force (Po) determined at pCa 4.5 (P/Po). The force required for half-maximal activation (pCa50) was determined by fitting force-pCa data with the Hill equation P/Po = [Ca2+]nH/(knH + [Ca2+]nH), where nH is the Hill coefficient and k is the [Ca2+] required for pCa50. ktr was estimated in pig atrial and ventricular myocardial preparations expressing nearly 100% α-MHC or 100% β-MHC with a modified experimental protocol originally described by Brenner and Eisenberg (7). After a 1-min incubation in preactivating solution, each preparation was transferred to solutions ranging from maximal [Ca2+]free (pCa 4.5) to submaximal [Ca2+]free (pCa 6.1–5.4). Once steady-state force had developed, the preparation was rapidly slackened (<2 ms) by 20% of its original length, resulting in a reduction in force to near zero. After a brief period (10 ms) of unloaded shortening, the preparation was rapidly restretched to its original length and force redeveloped to the original steady-state level. The apparent rate constant of force redevelopment (ktr) was estimated by linear transformation of the half-time of force redevelopment, i.e., ktr = 0.693/t1/2, as described previously (46).
Quantification of MHC expression.
After each mechanical experiment, the myocardial preparation was cut free at the points of attachment and placed in 10 μl of SDS sample buffer (62.5 mmol/l Tris, 75 mmol/l dithiothreitol, 25% glycerol, 25 mmol/l sodium dodecyl sulfate, and 0.01% bromophenol blue) and stored at −80°C until subsequent analysis of MHC isoform content by SDS-PAGE. Before loading on SDS-PAGE, protein concentration was determined using the RC DC Protein Assay (Bio-Rad Laboratories, Hercules, CA). The assay is based on the Lowry (35) assay and has been modified to be reducing agent compatible (RC) and detergent compatible (DC). MHC isoforms were separated with the use of 6% acrylamide gels, which were stained with a modified silver stain technique that reproducibly detects 2 ng of protein per band and exhibits linearity between 2 and 70 ng of protein per band (17, 51). The relative proportions of α- and β-MHC isoforms were then determined by densitometric analysis of silver-stained gels using an imaging densitometer and LaserPix software (Bio-Rad Laboratories). The proportions of MHC isoforms were estimated by expressing the area under the peak for each isoform as a fraction of the total area for the two isoforms. To ensure linearity of staining and the accuracy of measurements, all samples were serially diluted and run in parallel lanes next to MHC standards from myocardium expressing 100% α-MHC or 100% β-MHC.
Determination of level of phosphorylation of myofibrillar proteins.
To test whether variations in the basal level of phosphorylation contribute to altered kinetics, myofibrillar proteins from atrial and ventricular myocardium were separated by SDS-PAGE using 12.5% Tris·HCl Criterion gels (Bio-Rad). To determine the level of protein phosphorylation, different volumes (1, 2, 3, and 4 μl) from each myocardial preparation were loaded on the same gel. Phosphoproteins were detected with a Pro-Q Diamond staining protocol (Molecular Probes), followed by staining with SYPRO Ruby (Molecular Probes) for the determination of total protein content. Briefly, gels were 1) fixed in 50% methanol-10% glacial acetic acid for 1.5 h with solution changes every 30 min, 2) washed with double-distilled H2O six times for 5 min each, 3) stained with Pro-Q Diamond stain for 1.5 h, and 4) destained with Pro-Q Diamond destaining solution (Molecular Probes) overnight. For the determination of myofibrillar protein content, the gels were stained with SYPRO Ruby (Molecular Probes) for 3 h and destained with SYPRO Ruby destaining solution (10% methanol-7% glacial acetic acid) for 2 h with solution changes every 30 min. Immediately after each staining protocol, gels were imaged with a UVP BioImaging System (UVP, Upland, CA) and analyzed with LabWorks 4.5 (UVP). The area of protein and phosphoprotein bands was multiplied by mean optical density and plotted versus volume loaded, and a first-order linear regression was fitted to the data points to determine the slope of the relationship between optical density and volume loaded.
Data analysis.
All data are reported as means ± SE. An unpaired t-test (P < 0.05) was performed to test for the significance of effects of altered MHC content on ATP utilization. Comparisons of steady-state force and ktr at different levels of Ca2+ activation for different MHC compositions were done with a one-way analysis of variance (ANOVA) with a Tukey post hoc test for significance (P < 0.05).
RESULTS
Electrophoretic Analysis of Myofibrillar Proteins in Porcine Atrial and Ventricular Myocardium
Estimation of the rate constants of cross-bridge attachment and detachment required the use of myocardium containing very high levels of the MHC isoform of interest (i.e., 100% α-MHC or 100% β-MHC), since the presence of even a small amount of contaminating isoform (i.e., >5%) could unduly influence the derivation of rate constants of cross-bridge cycling. For this reason we used preparations isolated from atrial and ventricular myocardium, which expressed ∼100% α-MHC and ∼100% β-MHC, respectively (Fig. 1). Additional gels stained for total protein content (SYPRO Ruby) and phosphoprotein levels (Pro-Q Diamond) revealed that the basal levels of phosphorylation of cardiac myosin binding protein-C (MyBP-C), troponin (Tn)T, TnI, or myosin light chain (MLC)-2 were not significantly different in atrial or ventricular myocardium (Fig. 2).
Fig. 1.

Myosin heavy chain (MHC) composition of pig atrial and ventricular myocardium. MHC isoform expression was determined with 6% SDS-PAGE. This representative gel shows pig ventricular myocardium (lane 1) expressing ∼100% β-MHC and pig atrial myocardium expressing ∼100% α-MHC (lane 4). Lanes 2 and 3 show combinations of atria and ventricle at various dilutions.
Fig. 2.

Determination of total protein content and basal phosphorylation in porcine atrial and ventricular myocardium. A: myofibrillar protein phosphorylation was measured with SYPRO-Ruby staining for total protein content (left) and Pro-Q Diamond staining for protein phosphorylation (right), as shown in this representative SDS-PAGE at loading concentrations of 3 and 4 μl. Volumes of 3, 4, 6, and 8 μl were loaded in successive lanes for each sample preparation. cMyBP-C, cardiac myosin binding protein-C; TnT, troponin T; TnI, troponin I; ALC-1 and VLC-1, atrial and ventricular essential light chain, respectively; ALC-2 and VLC-2, atrial and ventricular regulatory light chain, respectively. B–E: levels of protein phosphorylation in atrial and ventricular porcine myocardium were compared for MyBP-C (B), TnT (C), TnI (D), and myosin light chain 2 (MLC-2) (E). Regression lines for optical band intensity vs. protein load were determined for both Pro-Q Diamond- and SYPRO-Ruby (not shown)-stained gels, and the ratio of regression line slopes was calculated. AU, arbitrary units.
Steady-State Mechanical Properties of Porcine Atrial and Ventricular Myocardium
Table 1 summarizes the maximal Ca2+-activated force, the steepness of the force-pCa relationship (Hill coefficient nH), and calcium sensitivities of force in porcine atrial and ventricular myocardium. No differences were measured in maximal isometric force or nH derived from force-pCa relationships. In addition, no sex differences were found in mechanical measurements. We did observe a difference in the calcium sensitivity of force (pCa50) between atrial and ventricular preparations, a result that could be due in part to variability of the tissue-specific MLC isoforms to modulate force since changes in other proteins that could account for such a shift were not evident (e.g., MyBP-C, TnT, TnI, or MLC-2). While the different light chain isoforms of atrial and ventricular myocardium would be expected to modulate force to some degree, they are unlikely to account for the ninefold difference in ATPase turnover observed here.
Table 1.
Steady-state mechanical properties in porcine atrial and ventricular myocardium
| % α-MHC | n | Ca2+-Activated Force, mN/mm2 | nH | pCa50 |
|---|---|---|---|---|
| 100 (atria) | 10 | 23.2 ± 1.9 | 4.17 ± 0.2 | 5.81 ± 0.01 |
| 0 (ventricle) | 10 | 21.4 ± 1.2 | 4.35 ± 0.3 | 5.71 ± 0.01* |
Data are means ± SE for n cardiac preparations. MHC, myosin heavy chain; Ca2+-activated force, difference between maximal force generated at pCa 4.5 and rest force generated at pCa 9.0; pCa50, pCa required for half-maximal force generation; nH, Hill coefficient for total force-pCa relationship.
Significantly different from atria (P < 0.05).
A modified release-restretch protocol (7) was used to determine ktr in steadily Ca2+-activated skinned myocardium expressing ∼100% α- or ∼100% β-MHC (Table 2, Fig. 3); ktr is the sum of the rate constants (fapp + gapp) describing the transition of cross bridges from non-force-generating states to force-generating states (5). As in previous studies on rat myocardium (15, 31, 43, 46) expression of the α-MHC isoform significantly increased maximal ktr compared with preparations expressing ∼100% β-MHC (22.4 ± 1.2 s−1 for α-MHC and 2.5 ± 0.7 s−1 for β-MHC), although the between-isoform differences measured in porcine myocardium were much larger than in rat myocardium, presumably because of the greater species-specific disparity in the rate constants for the MHC isoforms.
Table 2.
Kinetics of cross-bridge cycling in porcine atrial and ventricular myocardium
| % α-MHC | n | ktr, s−1 | ATPase, pmol·s−1·mm−3 | Tension Cost, pmol·s−1·mN−1·mm−1 | fapp | gapp |
|---|---|---|---|---|---|---|
| 100 (atria) | 10 | 22.4 ± 1.2 | 550.5 ± 26.2 | 23.73 ± 0.8 | 14.69 | 7.71 |
| 0 (ventricle) | 10 | 2.5 ± 0.7* | 64.4 ± 12.3* | 3.00 ± 0.4* | 1.51 | 0.99 |
Data are means ± SE for n cardiac preparations. ktr, Rate constant of force redevelopment in response to a rapid slack-restretch maneuver; ATPase, rate of ATP hydrolysis; tension cost, ratio of ATPase to isometric force; fapp, derived rate constant of cross-bridge attachment; gapp, derived rate constant of cross-bridge detachment.
Significantly different from atria (P < 0.05).
Fig. 3.

Rate constant of force redevelopment (ktr) in porcine atrial and ventricular myocardium. A representative trace of ktr as measured in maximal Ca2+ concentration ([Ca2+]) (pCa 4.5) in pig atrial (∼100% α-MHC, 22.4 ± 1.2 s−1) and ventricular (∼100% β-MHC, 2.5 ± 0.7 s−1) myocardium. Data are means ± SE. P, submaximal force; Po, maximal force.
Kinetics of Cross-Bridge Cycling in Porcine Atrial and Ventricular Myocardium
The effect of MHC content on ATPase activity is summarized in Table 2 and Fig. 4. Linear regression analysis of the absorbance of NADH revealed that atrial myocardium expressing ∼100% α-MHC exhibited maximal rates of ATP utilization that were approximately ninefold higher than ventricular preparations expressing ∼100% β-MHC (551 ± 26 pmol·s−1·mm−3 for α-MHC and 64 ± 12 pmol·s−1·mm−3 for β-MHC, respectively). Qualitatively, this finding is consistent with the ktr data from atrial and ventricular preparations in that myocardium expressing higher levels of α-MHC exhibit greater rates of cross-bridge cycling due to faster nucleotide turnover by myosin S1.
Fig. 4.

Rate of ATP utilization in porcine atrial and ventricular myocardium. The rate of ATP utilization was measured in skinned myocardium from atrial (●, 100% α-MHC, n = 10) and ventricular (○, 0% β-MHC, n = 10) preparations and plotted against relative force (P/Po). Forces recorded at submaximal free Ca2+ (P) were expressed relative to the maximal force measured at pCa 4.5 (Po). Data are means ± SE.
From measurements of steady-state isometric force and ATPase activity in each preparation we calculated tension cost (Fig. 5), which is defined as ATPase rate divided by isometric force and is directly proportional to gapp, the rate constant of cross-bridge detachment (6). Tension cost was approximately eightfold higher in preparations expressing ∼100% α-MHC compared with those expressing ∼100% β-MHC (23.73 ± 0.8 pmol·s−1·mm−1·mN−1 for α-MHC vs. 3.00 ± 0.4 pmol·s−1·mm−1·mN−1 for β-MHC). Qualitatively, this finding is consistent with previous studies from this lab (31) and others (43) in rat myocardium and indicates that β-MHC is a more economical isoform, i.e., less ATP is utilized to maintain a given isometric force at intermediate levels of activation. Furthermore, the rate of ATP utilization by β-MHC in pig myocardium was reduced to a greater extent than previously observed in rat myocardium (31, 40), while the difference in nucleotide turnover between pig and rat myocardium expressing purely α-MHC was much smaller. Such a large difference in turnover kinetics between MHC isoforms in pig myocardium would suggest that low-level expression of α-MHC in larger mammals such as humans would contribute to an even greater acceleration of the rate of force development and relaxation during the cardiac cycle.
Fig. 5.

Tension cost in porcine atrial and ventricular myocardium. The rate of ATP utilization was measured in skinned myocardium from atrial (●, 100% α-MHC, n = 10) and ventricular (○, 0% α-MHC, n = 10) preparations and plotted against Ca2+-activated isometric force. The slope of the line is equal to tension cost. Force and ATPase were measured simultaneously in solutions of varying free [Ca2+] (pCa 4.5, 5.4–6.2). Data are means ± SE.
Rate Constants of Cross-Bridge Attachment and Detachment and Simulations of Isometric Twitch Kinetics
To model the effects of MHC content on twitch kinetics, the rate constants of cross-bridge attachment (fapp) and detachment (gapp) were calculated for each isoform assuming a two-state cross-bridge model originally described by Brenner (5). This model assumes that myosin cross bridges are either attached to the thin filament and generating force or detached and not generating force. We calculated the rate constants of cross-bridge cycling at maximal activation, using data obtained from measurements of isometric force [proportional to f/(f + g)], the rate constant of force redevelopment (ktr = f + g), and ATPase activity [proportional to fg/(f + g)]. Together, these values can be used to solve a system of four equations with four unknowns, as previously described (31):
| (1) |
| (2) |
| (3) |
| (4) |
Derivations revealed that fapp was ∼10-fold greater in α-MHC than in β-MHC, while gapp was ∼8-fold greater in α-MHC. The values were as follows: 14.69 and 7.71 s−1 (α-MHC) and 1.51 and 0.99 s−1 (β-MHC) for fapp and gapp, respectively.
These rate constants were then used to model the myocardial twitch in response to a standardized Ca2+ transient, calculated as the difference between two exponential functions:
where ki represents variables that can be altered to vary the duration of the Ca2+ transient and Camax corresponds to the maximally activated state of the thin filament and is arbitrarily set to 1. The model considers only the contribution of the MHC isoforms to force development without the effect of cooperative recruitment of cross bridges or any other nonlinearity. Twitch simulations were performed with expression levels of 0%, 10%, 20%, or 30% α-MHC, all of which were modeled with the same Ca2+ transient. The mathematical model is in the form of one ordinary differential equation and one algebraic equation, which are solved by the standard fourth-order Runge-Kutta method.
Calculations based on these rate constants predict that the kinetics of the isometric twitch are significantly accelerated as low-level expression of α-MHC on a mostly β-MHC background progressively increases (Fig. 6). Our model predicts that dF/dtmax for 10% α-MHC/90% β-MHC is ∼92% faster than for 100% β-MHC (Fig. 7), while increases in α-MHC from 0 to 20% and 0 to 30% result in increases of 186% and 280%, respectively, in dF/dtmax. Moreover, the greatest reductions in the time required to reach peak force were observed for α-MHC expression in the range of 0–10% (Table 3, Fig. 8).
Fig. 6.

A: model predictions of twitch time courses in myocardium with variable ratios of α-MHC and β-MHC. y-Axis indicates the fraction of cross bridges bound; x-axis indicates time in seconds. Indigo line indicates the Ca2+ pulse (arbitrary units) that was used in each of the simulations. Traces represent 100% β-MHC (0% α-MHC), 90% β-MHC (10% α-MHC), 80% β-MHC (20%-α MHC), and 70% β-MHC (30% α-MHC) of the total MHC content. B: model predictions of normalized twitch time courses in myocardium with variable ratios of α-MHC and β-MHC. Force simulations are expressed relative to the maximal force calculated in A. Indigo line indicates the Ca2+ pulse (arbitrary units) that was used in each of the simulations. Traces represent 100% β-MHC (0% α-MHC), 90% β-MHC (10% α-MHC), 80% β-MHC (20% α-MHC), and 70% β-MHC (30% α-MHC) of the total MHC content.
Fig. 7.

Model predictions for the rates of force development and relaxation in myocardium with variable expression of α-MHC. Maximal rate of force development (dF/dtmax) is shown on y-axis; x-axis is time in seconds. Traces represent 100% β-MHC (0% α-MHC), 90% β-MHC (10% α-MHC), 80% β-MHC (20% α-MHC), and 70% β-MHC (30% α-MHC) of the total MHC content.
Table 3.
Effect of low-level expression of α-MHC on time required to reach peak force
| % α-MHC | Time Required to Reach Peak Force, ms | % Decrease in Time to Peak Force Compared with 0% α-MHC | Fold Decrease in Time to Peak Force Compared with 0% α-MHC |
|---|---|---|---|
| 0 | 120.0 | 1 | |
| 10 | 85.4 | 28.8 | 0.71 |
| 20 | 74.4 | 38.0 | 0.62 |
| 30 | 69.4 | 42.1 | 0.58 |
Data were obtained from model predictions of twitch time courses in myocardium with variable ratios of α-MHC and β-MHC. Fold decrease in time to peak force from 0% α-MHC was computed as the ratio of the time to peak force from 10%, 20%, and 30% α-MHC to 0% α-MHC.
Fig. 8.

Effect of α-MHC expression on the time required to reach peak force in mammalian myocardium. Data were obtained from model predictions of twitch time courses in myocardium with variable ratios of α-MHC and β-MHC. Each data point is taken from a single time value obtained from the derived time required to reach peak force; therefore no error bars are present. Fold decrease in time to peak force from 0% α-MHC was computed as the ratio of the time to peak force from 10%, 20%, and 30% α-MHC to 0% α-MHC. Porcine myocardium (●) is shown compared with values from rat myocardium (○) taken from previous work by the authors (31).
DISCUSSION
In the present study we assessed the effect of low-level α-MHC expression on contraction kinetics in porcine myocardium, a model relevant to human hearts because of similarities in mechanical properties such as heart rate, stroke volume, and mean arterial pressure (26). From measurements of ATP utilization, ktr, and isometric force, we derived fapp and gapp for both α- and β-MHC and subsequently modeled the effect of low-level of α-MHC expression on myocardial twitch kinetics. Compared with previous results from rat myocardium (31), we observed a much larger difference in cross-bridge cycling kinetics between α- and β-MHC, a result that is presumably due to a greater difference in ATPase between the MHC isoforms of myocardium from large mammals. As a result, simulations of isometric twitch kinetics predicted that expression of ∼10% α-MHC on a predominantly β-MHC background would dramatically increase the maximal rate of rise of twitch force (dF/dtmax) compared with myocardium expressing only β-MHC. These results indicate that the presence of α-MHC plays an important role in in vivo pressure development in the ventricles of large mammals, and when considered in the context of human myocardial disease, it is reasonable to suppose that the contractile deficit observed during such conditions is associated at least in part with the loss or absence of α-MHC.
Porcine Myocardium As a Model for the Effect of Low-Level α-MHC Expression
To predict the functional effects of low-level expression of α-MHC in the present study, it was necessary to first determine fapp and gapp for α-MHC and β-MHC in myocardial preparations expressing either ∼100% α- or ∼100% β-MHC. Porcine atrial myocardium expressed ∼100% α-MHC, while ventricular preparations expressed nearly ∼100% β-MHC. These expression levels provide a reasonable framework for the estimation of the rate constants of cross-bridge cycling: the levels of contaminating MHC isoform here (at most on the order of 1–2%) would be unlikely to influence the large variations in measurements of ATPase and ktr, and thus would not be expected to influence derivations of fapp and gapp.
A potentially important consideration for the present study is the impact of the tissue-specific light chain isoforms on cross-bridge cycling kinetics. Atrial and ventricular myocardium contains different essential and regulatory light chain isoforms (designated MLC-1a, MLC-2a, MLC-1v, and MLC-2v, respectively) that presumably modulate contractile function through changes in force production (50) and shortening velocity (34), although the extent to which they influence function in the present study is unclear. Previous studies have found greater unloaded shortening velocities (Vo) 1) in the mouse ventricle, where MLC-1v was replaced with MLC-1a (14), and 2) in the mouse atria, where MLC-2a was replaced with MLC-2v (8). On the basis of these data, it would appear that the contribution to Vo of the tissue-specific light chains may be ordered as MLC-1a > MLC-1v and MLC-2v > MLC-2a, yet the degree to which endogenous light chains of the atria alter velocity compared with those of the ventricle is uncertain. In this regard, rat atrial myocardium displayed higher Vmax and Vo (68% and 52% higher, respectively) but similar ATPase activity compared with ventricular myocardium, a result attributed to different light chains since heavy chain composition was similar in both preparations (4). It should be noted, however, that variability in Vo between the atria and the ventricle due to endogenous light chain isoforms is relatively modest compared with the large differences in cross-bridge cycling kinetics (9- to 10-fold greater in atria) we observed. This would imply that while light chains may modulate force generation and Vo, such changes would be relatively small and the dramatic difference in cycling kinetics seen here is largely a result of substantial disparity in ATP turnover between the MHC isoforms.
Effect of MHC Isoforms in Porcine Myocardium on Rate of Rise of Force Development
The present study extends previous work from this laboratory (31) in which rat myocardium was used to assess the effect of low-level expression of α-MHC on contraction kinetics. Our simulations indicated that incremental, 10% expressions of α-MHC progressively accelerated the maximal rate of rise of force. Furthermore, a slight disproportionality in the acceleration of dF/dtmax was observed as expression of α-MHC increased from 0% to 10% (∼18% increase in dF/dtmax). These results suggested that in the ventricles of large mammals, in which the differences in turnover kinetics between α- and β-MHC are expected to be much larger, the presence of low-level expression of α-MHC might disproportionately accelerate contraction kinetics to a greater degree.
In this regard, simulations in porcine myocardium indicated that low-level expression of α-MHC (∼10%) accelerated dF/dtmax by ∼92% compared with myocardium expressing only β-MHC. Such a difference in contraction kinetics is likely due to the large difference in the rate of cross-bridge turnover between the MHC isoforms in porcine myocardium and functionally would be manifested in a substantial disparity between mechanical parameters from preparations expressing either 100% α-MHC or 100% β-MHC. Indeed, measurements of ATPase and ktr in porcine atrial myocardium expressing ∼100% α-MHC were nearly 10-fold greater than in ventricular preparations expressing ∼100% β-MHC. Consequently, we estimated an ∼10-fold difference between fapp and an ∼8-fold difference in gapp between α- and β-MHCs. These results indicate that low-level expression of α-MHC contributes dramatically to ventricular pressure development and relaxation in porcine myocardium. In a functional context, rapid cross-bridge turnover would increase dF/dtmax in response to a Ca2+ transient and enhance power generation during systole, while an accelerated rate of cross-bridge detachment at the end of systole would enhance force relaxation and facilitate ventricular filling, thus increasing stroke volume in the subsequent beat.
Furthermore, the rate of rise of force was nonlinear (Fig. 6B) over the iterations of α-MHC expression (i.e., 10%, 20%, and 30%) simulated here, with the greatest increase in rate observed as a small population of α-MHC cross bridges (∼10%) is compared with myocardium expressing only β-MHC (Table 3, Fig. 8). Considered in the context of a three-state cross-bridge model in which cross bridges transition from a noncycling population to a cycling population (10), it is assumed that α-and β-MHC cross bridges would transition into the cycling pool at similar rates upon activation. Once in the cycling pool, the rate at which cross bridges transition from non-force-generating to force-generating states and back again would depend in large part on the rate constants governing these transitions, namely f and g. Thus, in response to a limited-duration Ca2+ transient, α-MHC cross bridges would cycle between states at a faster rate and would presumably be the dominant determinant of the rate of rise of force due to a larger population of activated cross bridges. The effect would yield greater peak twitch force and accelerate the rate of rise of force progressively for populations including α-MHC, but most rapidly as α-MHC cross bridges are initially titrated into the total population compared with 100% β-MHC, as seen in Fig. 8.
Implied Effects of Altered MHC Expression on Rate of Rise of Pressure and Timing of Contraction in Myocardium
Our simulations of isometric twitch kinetics predicted that myocardium with greater expression of α-MHC has a greater peak twitch force in response to a Ca2+ transient, presumably as a direct consequence of a larger number of attached cross bridges. At the whole organ level, this would indicate that low-level expression of α-MHC on a predominantly β-MHC background would accelerate the maximal rate of pressure development (±dP/dtmax), thus facilitating both systolic ejection and diastolic filling. Conversely, a reduction in α-MHC content would lead to a decrease in ±dP/dtmax and a concomitant functional deficit, which to some degree would be compensated by an increase in sympathetic tone and reexpression of atrial essential MLC (MLC-1a) in the ventricles of human failing myocardium (14, 27, 41). While these observations suggest an important role for α-MHC expression in human myocardium, an extension of the present results to humans is premature since the ATP turnover rates for human α- and β-MHCs are not known for certain. However, it has been shown that force development and ATPase were significantly lower in ventricular compared with atrial human myocardium, a finding presumably related to MHC isoform distribution (38). In light of this observation, our model would predict that expression of α-MHC on the order of 10% would dramatically increase the rate of force development and enhance dP/dtmax in human ventricles compared with myocardium expressing only β-MHC, and such an effect would likely remain proportionally similar at more physiological temperatures.
In the context of heart failure, our results suggest that the associated contractile deficit is at least partly attributable to reduced levels of α-MHC. However, it should be noted that the reversion to β-MHC during conditions of disease may be beneficial in some respects. Myocardium expressing predominantly β-MHC appears to be more resistant to decreases in power output resulting from the accumulation of metabolites such as Pi and H+ during conditions of ischemia (23) in addition to an energetic benefit gained in terms of reduced tension cost with higher levels of β-MHC (31, 43). These factors would certainly benefit diseased myocardium, particularly in light of the proposed inefficient sarcomeric ATP utilization characterizing specific types of human heart failure (2, 12). Despite these improvements, however, the energetic benefit gained from a reversion to β-MHC is most likely outweighed by the associated reduction in α-MHC that occurs during disease (33, 36, 37). It is apparent that low-level α-MHC plays a critical role in the ability of myocardium to match pressure development with increasing hemodynamic load, and our simulations imply that the absence of α-MHC would reduce twitch force and impair contraction kinetics.
In addition, the outcome of our simulations would also be affected by alterations in calcium transients that occur during heart failure, which are characterized by reduced amplitude and prolonged duration (3). A reduced calcium load in the sarcoplasmic reticulum (SR) would decrease peak twitch force and slow the rate of rise of force development because of a lower driving force for calcium release, while diminished levels of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) during failure would impair relaxation because of persistent levels of calcium in the cytoplasm. It is important to note that while such changes in the calcium transient would uniformly impact twitch force and kinetics, our model would still predict that the presence of α-MHC in the ventricles would increase peak twitch force and accelerate force development and relaxation compared with myocardium expressing only β-MHC.
Furthermore, stimulation frequency would also be expected to influence the outcome of our twitch simulations. Normal myocardium exhibits increased contractility at higher stimulation frequencies (positive force frequency response), in large part because of the effect of sympathetic input on contractile function. At high heart rates, our simulations would predict that the increase in twitch force would be greater in ventricles expressing low levels of α-MHC compared with those expressing only β-MHC, and in this regard it is possible that the distribution of myosin isoforms across the ventricular wall plays an important role in optimizing contraction. Recent work by Stelzer et al. (47) has shown that transmural variation of MHC expression is an important component of the temporal pattern of force development in porcine left ventricle. It has been shown that electrical activation proceeds from endocardium to epicardium (9, 45), yet the timing of fiber shortening is synchronized across the two regions. Stelzer and colleagues found that a ∼10% variation in the expression of α-MHC between the epicardium and the endocardium (13% α-MHC and 3% α-MHC, respectively) contributed to faster rates of stretch activation and force redevelopment in the absence of differences in total protein content or phosphorylation status of other myofilament proteins. This finding is important in the context of our model because the loss of α-MHC during conditions of human heart failure not only would lead to an overall decrease in the rate of force development during systole but also would cause a normalization of the kinetics of force development transmurally, leading to dyssnchronous contraction across the ventricular wall.
Limitations of the Model
As can be seen in Fig. 1, there is a small amount of β-MHC in the atrial preparation shown in lane 4. It is possible that such levels could influence our mechanical measurements, leading to distortions in our derivations of fapp and gapp since these calculations assume that the myocardial preparations express only the isoform of interest without contaminating levels of the other. However, the level of contaminating isoform in our preparations was very low (∼2%), and distortions in the calculations due to such levels would be small.
Conclusions
Our mechanical data in porcine myocardium show significantly greater cross-bridge cycling kinetics in α-MHC compared with β-MHC. Simulations based on these measurements predict that low-level expression of α-MHC (∼10%) on a predominantly β-MHC background significantly accelerates myocardial twitch kinetics in the ventricles of large mammals and would be at least partly responsible for greater myocardial power generation in the face of increasing hemodynamic loads. This finding is important in the context of human myocardium, where the expression of α-MHC dramatically decreases as the heart transitions from healthy to disease states (33, 36, 37). While the developmental pattern of heart failure is multifactorial and complex, it is clear that the underlying contractile dysfunction is associated in part with a redistribution of the MHC isoforms to nearly 100% β-MHC and the absence of α-MHC is at least partly responsible for the slower kinetics and decreased contractile force, thereby impairing the rate of force development during systole as well as relaxation during diastole.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grant HL-61635 (to R. L. Moss).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
REFERENCES
- 1. Alpert NR, Mulieri LA. Heat, mechanics, and myosin ATPase in normal and hypertrophied heart muscle. Fed Proc 41: 192–198, 1982 [PubMed] [Google Scholar]
- 2. Ashrafian H, Redwood C, Blair E, Watkins H. Hypertrophic cardiomyopathy: a paradigm for myocardial energy depletion. Trends Genet 19: 263–268, 2003 [DOI] [PubMed] [Google Scholar]
- 3. Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 85: 1046–1055, 1992 [DOI] [PubMed] [Google Scholar]
- 4. Bottinelli R, Canepari M, Cappelli V, Reggiani C. Maximum speed of shortening and ATPase activity in atrial and ventricular myocardia of hyperthyroid rats. Am J Physiol Cell Physiol 269: C785–C790, 1995 [DOI] [PubMed] [Google Scholar]
- 5. Brenner B. The cross-bridge cycle in muscle. Mechanical, biochemical, and structural studies on single skinned rabbit psoas fibers to characterize cross-bridge kinetics in muscle for correlation with the actomyosin-ATPase in solution. Basic Res Cardiol 81, Suppl 1: 1–15, 1986 [DOI] [PubMed] [Google Scholar]
- 6. Brenner B. Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc Natl Acad Sci USA 85: 3265–3269, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brenner B, Eisenberg E. Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc Natl Acad Sci USA 83: 3542–3546, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buck SH, Konyn PJ, Palermo J, Robbins J, Moss RL. Altered kinetics of contraction of mouse atrial myocytes expressing ventricular myosin regulatory light chain. Am J Physiol Heart Circ Physiol 276: H1167–H1171, 1999 [DOI] [PubMed] [Google Scholar]
- 9. Buckberg GD, Mahajan A, Jung B, Markl M, Hennig J, Ballester-Rodes M. MRI myocardial motion and fiber tracking: a confirmation of knowledge from different imaging modalities. Eur J Cardiothorac Surg 29, Suppl 1: S165–S177, 2006 [DOI] [PubMed] [Google Scholar]
- 10. Campbell K. Rate constant of muscle force redevelopment reflects cooperative activation as well as cross-bridge kinetics. Biophys J 72: 254–262, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carniel E, Taylor MR, Sinagra G, Di Lenarda A, Ku L, Fain PR, Boucek MM, Cavanaugh J, Miocic S, Slavov D, Graw SL, Feiger J, Zhu XZ, Dao D, Ferguson DA, Bristow MR, Mestroni L. Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation 112: 54–59, 2005 [DOI] [PubMed] [Google Scholar]
- 12. Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, McKenna WJ, Ostman-Smith I, Clarke K, Watkins H. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol 41: 1776–1782, 2003 [DOI] [PubMed] [Google Scholar]
- 13. Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol 157: 378–417, 1988 [DOI] [PubMed] [Google Scholar]
- 14. Fewell JG, Hewett TE, Sanbe A, Klevitsky R, Hayes E, Warshaw D, Maughan D, Robbins J. Functional significance of cardiac myosin essential light chain isoform switching in transgenic mice. J Clin Invest 101: 2630–2639, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fitzsimons DP, Patel JR, Moss RL. Role of myosin heavy chain composition in kinetics of force development and relaxation in rat myocardium. J Physiol 513: 171–183, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Geeves MA, Holmes KC. Structural mechanism of muscle contraction. Annu Rev Biochem 68: 687–728, 1999 [DOI] [PubMed] [Google Scholar]
- 17. Giulian GG, Moss RL, Greaser M. Improved methodology for analysis and quantitation of proteins on one-dimensional silver-stained slab gels. Anal Biochem 129: 277–287, 1983 [DOI] [PubMed] [Google Scholar]
- 18. Glyn H, Sleep J. Dependence of adenosine triphosphatase activity of rabbit psoas muscle fibres and myofibrils on substrate concentration. J Physiol 365: 259–276, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. J Gen Physiol 80: 279–297, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herron TJ, Devaney E, Mundada L, Arden E, Day S, Guerrero-Serna G, Turner I, Westfall M, Metzger JM. Ca2+-independent positive molecular inotropy for failing rabbit and human cardiac muscle by alpha-myosin motor gene transfer. FASEB J 24: 415–424, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Herron TJ, Korte FS, McDonald KS. Loaded shortening and power output in cardiac myocytes are dependent on myosin heavy chain isoform expression. Am J Physiol Heart Circ Physiol 281: H1217–H1222, 2001 [DOI] [PubMed] [Google Scholar]
- 22. Herron TJ, McDonald KS. Small amounts of alpha-myosin heavy chain isoform expression significantly increase power output of rat cardiac myocyte fragments. Circ Res 90: 1150–1152, 2002 [DOI] [PubMed] [Google Scholar]
- 23. Hinken AC, McDonald KS. Beta-myosin heavy chain myocytes are more resistant to changes in power output induced by ischemic conditions. Am J Physiol Heart Circ Physiol 290: H869–H877, 2006 [DOI] [PubMed] [Google Scholar]
- 24. Hoh JF, McGrath PA, Hale PT. Electrophoretic analysis of multiple forms of rat cardiac myosin: effects of hypophysectomy and thyroxine replacement. J Mol Cell Cardiol 10: 1053–1076, 1978 [DOI] [PubMed] [Google Scholar]
- 25. Holubarsch C, Litten RZ, Mulieri LA, Alpert NR. Energetic changes of myocardium as an adaptation to chronic hemodynamic overload and thyroid gland activity. Basic Res Cardiol 80: 582–593, 1985 [DOI] [PubMed] [Google Scholar]
- 26. Ibrahim Z, Busch J, Awwad M, Wagner R, Wells K, Cooper DK. Selected physiologic compatibilities and incompatibilities between human and porcine organ systems. Xenotransplantation 13: 488–499, 2006 [DOI] [PubMed] [Google Scholar]
- 27. Khalina YN, Bartsch H, Petzhold D, Haase H, Podlubnaya ZA, Shpagina MD, Morano I. Reconstitution of ventricular myosin with atrial light chains 1 improves its functional properties. Acta Biochim Pol 52: 443–448, 2005 [PubMed] [Google Scholar]
- 28. Korte FS, Herron TJ, Rovetto MJ, McDonald KS. Power output is linearly related to MyHC content in rat skinned myocytes and isolated working hearts. Am J Physiol Heart Circ Physiol 289: H801–H812, 2005 [DOI] [PubMed] [Google Scholar]
- 29. Krenz M, Robbins J. Impact of beta-myosin heavy chain expression on cardiac function during stress. J Am Coll Cardiol 44: 2390–2397, 2004 [DOI] [PubMed] [Google Scholar]
- 30. Ladenson PW, Sherman SI, Baughman KL, Ray PE, Feldman AM. Reversible alterations in myocardial gene expression in a young man with dilated cardiomyopathy and hypothyroidism. Proc Natl Acad Sci USA 89: 5251–5255, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Locher MR, Razumova MV, Stelzer JE, Norman HS, Patel JR, Moss RL. Determination of rate constants for turnover of myosin isoforms in rat myocardium: implications for in vivo contractile kinetics. Am J Physiol Heart Circ Physiol 297: H247–H256, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lowes BD, Gilbert EM, Abraham WT, Minobe WA, Larrabee P, Ferguson D, Wolfel EE, Lindenfeld J, Tsvetkova T, Robertson AD, Quaife RA, Bristow MR. Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. N Engl J Med 346: 1357–1365, 2002 [DOI] [PubMed] [Google Scholar]
- 33. Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, Roden RL, Dutcher DL, Robertson AD, Voelkel NF, Badesch DB, Groves BM, Gilbert EM, Bristow MR. Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest 100: 2315–2324, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lowey S, Waller GS, Trybus KM. Skeletal muscle myosin light chains are essential for physiological speeds of shortening. Nature 365: 454–456, 1993 [DOI] [PubMed] [Google Scholar]
- 35. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275, 1951 [PubMed] [Google Scholar]
- 36. Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res 86: 386–390, 2000 [DOI] [PubMed] [Google Scholar]
- 37. Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Invest 100: 2362–2370, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Narolska NA, van Loon RB, Boontje NM, Zaremba R, Penas SE, Russell J, Spiegelenberg SR, Huybregts MA, Visser FC, de Jong JW, van der Velden J, Stienen GJ. Myocardial contraction is 5-fold more economical in ventricular than in atrial human tissue. Cardiovasc Res 65: 221–229, 2005 [DOI] [PubMed] [Google Scholar]
- 39. Patel JR, Fitzsimons DP, Buck SH, Muthuchamy M, Wieczorek DF, Moss RL. PKA accelerates rate of force development in murine skinned myocardium expressing alpha- or beta-tropomyosin. Am J Physiol Heart Circ Physiol 280: H2732–H2739, 2001 [DOI] [PubMed] [Google Scholar]
- 40. Pereira JS, Pavlov D, Nili M, Greaser M, Homsher E, Moss RL. Kinetic differences in cardiac myosins with identical loop 1 sequences. J Biol Chem 276: 4409–4415, 2001 [DOI] [PubMed] [Google Scholar]
- 41. Ritter O, Luther HP, Haase H, Baltas LG, Baumann G, Schulte HD, Morano I. Expression of atrial myosin light chains but not alpha-myosin heavy chains is correlated in vivo with increased ventricular function in patients with hypertrophic obstructive cardiomyopathy. J Mol Med 77: 677–685, 1999 [DOI] [PubMed] [Google Scholar]
- 42. Rossmanith GH, Hamilton AM, Hoh JF. Influence of myosin isoforms on tension cost and crossbridge kinetics in skinned rat cardiac muscle. Clin Exp Pharmacol Physiol 22: 423–429, 1995 [DOI] [PubMed] [Google Scholar]
- 43. Rundell VL, Manaves V, Martin AF, de Tombe PP. Impact of beta-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am J Physiol Heart Circ Physiol 288: H896–H903, 2005 [DOI] [PubMed] [Google Scholar]
- 44. Schiaffino S, Reggiani C. Molecular diversity of myofibrillar proteins: gene regulation and functional significance. Physiol Rev 76: 371–423, 1996 [DOI] [PubMed] [Google Scholar]
- 45. Sengupta PP, Korinek J, Belohlavek M, Narula J, Vannan MA, Jahangir A, Khandheria BK. Left ventricular structure and function: basic science for cardiac imaging. J Am Coll Cardiol 48: 1988–2001, 2006 [DOI] [PubMed] [Google Scholar]
- 46. Stelzer JE, Brickson SL, Locher MR, Moss RL. Role of myosin heavy chain composition in the stretch activation response of rat myocardium. J Physiol 579: 161–173, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stelzer JE, Norman HS, Chen PP, Patel JR, Moss RL. Transmural variation in myosin heavy chain isoform expression modulates the timing of myocardial force generation in porcine left ventricle. J Physiol 586: 5203–5214, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stelzer JE, Patel JR, Olsson MC, Fitzsimons DP, Leinwand LA, Moss RL. Expression of cardiac troponin T with COOH-terminal truncation accelerates cross-bridge interaction kinetics in mouse myocardium. Am J Physiol Heart Circ Physiol 287: H1756–H1761, 2004 [DOI] [PubMed] [Google Scholar]
- 49. Stienen GJ, Roosemalen MC, Wilson MG, Elzinga G. Depression of force by phosphate in skinned skeletal muscle fibers of the frog. Am J Physiol Cell Physiol 259: C349–C357, 1990 [DOI] [PubMed] [Google Scholar]
- 50. VanBuren P, Waller GS, Harris DE, Trybus KM, Warshaw DM, Lowey S. The essential light chain is required for full force production by skeletal muscle myosin. Proc Natl Acad Sci USA 91: 12403–12407, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Warren CM, Greaser ML. Method for cardiac myosin heavy chain separation by sodium dodecyl sulfate gel electrophoresis. Anal Biochem 320: 149–151, 2003 [DOI] [PubMed] [Google Scholar]