

Abstract
Tumor progression involves the ability of cancer cells to communicate with each other and with neighboring normal cells in their microenvironment. Microvesicles (MV) derived from human cancer cells have received a good deal of attention because of their ability to participate in the horizontal transfer of signaling proteins between cancer cells and to contribute to their invasive activity. Here we show that MV may play another important role in oncogenesis. In particular, we demonstrate that MV shed by two different human cancer cells, MDAMB231 breast carcinoma cells and U87 glioma cells, are capable of conferring onto normal fibroblasts and epithelial cells the transformed characteristics of cancer cells (e.g., anchorage-independent growth and enhanced survival capability) and that this effect requires the transfer of the protein cross-linking enzyme tissue transglutaminase (tTG). We further demonstrate that tTG is not sufficient to transform fibroblasts but rather that it must collaborate with another protein to mediate the transforming actions of the cancer cell-derived MV. Proteomic analyses of the MV derived from MDAMB231 and U87 cells indicated that both these vesicle preparations contained the tTG-binding partner and cross-inking substrate fibronectin (FN). Moreover, we found that tTG cross-links FN in MV from cancer cells and that the ensuing MV-mediated transfers of cross-linked FN and tTG to recipient fibroblasts function cooperatively to activate mitogenic signaling activities and to induce their transformation. These findings highlight a role for MV in the induction of cellular transformation and identify tTG and FN as essential participants in this process.
Keywords: cancer progression, extracellular matrix, oncosomes, shedding vesicles, tumor microenvironment
The release of microvesicles (MV) or oncosomes from different types of high-grade or aggressive forms of human cancer cells into their surroundings is becoming increasingly recognized as a feature of tumor biology (1–3), but how these structures are generated and their importance in cancer progression are only just beginning to be appreciated. Of particular interest are the recent findings that at least some of the MV generated by aggressive brain, breast, and prostate cancer cells involve the direct budding or shedding of vesicles from their plasma membranes. These membrane-bound structures range in size up to ~2.0 microns in diameter and contain a variety of cell-surface receptors, intracellular signaling proteins, and cytoskeletal components, as well as RNA transcripts encoding mitogenic factors, depending upon their cellular origin (4–8). Interestingly, they are taken up by recipient cancer cells where the transferred cargo can promote the activation of survival and mitogenic signaling proteins, including protein kinase B (PKB/AKT) and ERK (5, 7, 9).
The finding that MV provide a means for cancer cells to share intracellular proteins and genetic information with each other has begun to shed some light on a poorly understood but potentially important mechanism underlying the onset and development of human cancers. However, numerous questions regarding MV biogenesis and function need to be addressed to understand more thoroughly the significance of this unconventional form of cell communication in oncogenesis. Here we describe findings that suggest a unique role for MV in cancer progression and identify the proteins that are essential to this function.
Results
Analysis of serum-starved cultures of the highly aggressive human breast cancer cell line MDAMB231 by scanning electron microscopy (SEM) (Fig. 1A, Left) or by fluorescent microscopy performed on cells stained for F-actin (Fig. 1A, Right) showed that MV ranging from ~0.2–2.0 microns in diameter were present on the surface of ~35% of these cells (Fig. 1B). MV also were detected on ~25% of serum-deprived U87 human glioma cells, and their formation was induced in HeLa cervical carcinoma cells by EGF stimulation (Fig. 1 B and C). In contrast, MV were not detected on the surface of normal NIH 3T3 fibroblasts cultured under serum-starved or EGF-stimulated conditions, indicating that some cell types may not generate MV. Moreover, we determined that MV were actively shed from these cancer cells, as demonstrated by time-lapse images of the release of a GFP-labeled MV from the plasma membrane of an MDAMB231 cell transfected with an EGFP-labeled plasmid (pEGFP) encoding the plasma membrane targeting sequence (GFP-PM) of the Lyn tyrosine kinase (Fig. 1D and Movie S1), as well as through the detection of MV containing GFP in the culturing medium collected from transfectants expressing only pEGFP by immunoblot (Fig. 1E) and FACS (Fig. S1 A and B) analysis.
Fig. 1.

Distinct types of human cancer cells generate MV. (A) MDAMB231 cells were analyzed by SEM (Left) and immunofluorescent microscopy (IF) using rhodamine-conjugated phalloidin to detect F-actin (Right). Some of the largest MV are indicated by arrows. (B) Quantification of MV production by various cell lines cultured under serum-starved or EGF-stimulated conditions. Cells generating MV were detected by labeling the samples with rhodamine-conjugated phalloidin. The data shown represent the mean ± SD from three independent experiments. (C) Images of cells from the experiment performed in B. Some of the MV are denoted by arrows. (D) MDAMB231 cells transiently expressing a GFP-tagged form of the plasma membrane-targeting sequence (GFP-PM) from the Lyn tyrosine kinase were subjected to live-imaging fluorescent microscopy. Shown are a series of time-lapse images of a transfectant taken at 2-min intervals. The arrow indicates an MV that forms and is shed from a cell. (E) Serum-deprived MDAMB231 cells that were mock transfected or transfected with pEGFP were lysed, and the MV shed into the medium by the transfectants were isolated and lysed as well. The WCL and the MV lysates then were immunoblotted with antibodies against GFP, the MV marker flotillin-2, and the cytosolic-specific marker IκBα.
Although previously MV have been reported to share their cargo with cells, we were interested in seeing whether MV might be capable of conferring some of the transformed characteristics of the donor cancer cells onto normal (nontransformed) recipient cells. Thus, we isolated MV constitutively shed by MDAMB231 breast cancer cells and U87 brain tumor cells from their serum-free culturing medium (Fig. 2A) and added them to cultures of nontransformed NIH 3T3 fibroblasts. MV generated by either of these cancer cell lines were capable of stimulating the activities of the signaling protein kinases AKT and ERK in the recipient fibroblasts (Fig. 2B), similar to observations when cancer cell-derived MV were incubated with other cancer cells or endothelial cells (5, 7, 9). Moreover, when NIH 3T3 fibroblasts were incubated with MV derived from MDAMB231 cells or U87 cells, they exhibited two phenotypes characteristic of cancer cells, namely an enhanced survival capability (Fig. 2C) and an ability to grow under low-serum conditions (Fig. 2D). We then asked whether the cancer cell-derived MV, when added to normal cells, could induce cellular transformation as read out by anchorage-independent growth (i.e., colony formation in soft agar). Fig. 2 E and F shows that although the control NIH 3T3 fibroblasts failed to form colonies in soft agar, sustained treatment of fibroblasts with MV collected from either MDAMB231 cells or U87 cells conferred on NIH 3T3 fibroblasts the ability to grow under anchorage-independent conditions. MDAMB231 cell-derived MV similarly promoted the survival (Fig. S2A) and aberrant growth (Fig. S2B) of the normal human mammary epithelial cell line MCF10A. Thus, the continuous MV-mediated transfer of cargo from cancer cells to normal cells is indeed capable of endowing normal cells with the characteristics induced by oncogenic transformation.
Fig. 2.

MDAMB231 and U87 cancer cell-derived MV are capable of transforming normal fibroblasts. (A) WCL of serum-starved MDAMB231 and U87 cells, as well as lysates of the MV shed by these cells, were immunoblotted with antibodies against the MV markers actin and flotillin-2, the cytosolic-specific marker IκBα, and the activated (phospho)-EGF receptor. (B–D) Multiple sets of serum-deprived NIH 3T3 fibroblasts were incubated with serum-free medium, medium containing 2% CS, or medium supplemented with intact MV derived from either MDAMB231 or U87 cells as indicated. (B) One set of cells was lysed after being exposed to the various culturing conditions for the indicated lengths of time and then was immunoblotted with antibodies that recognize the activated and total forms of AKT and ERK. Two additional sets of fibroblasts were evaluated for their abilities to undergo serum deprivation-induced cell death (C) and to grow in low serum (2% CS) (D). For the growth assays, the culturing medium (including the MV) was replenished daily. (E) NIH 3T3 fibroblasts incubated with or without MV derived from MDAMB231 or U87 cells were subjected to anchorage-independent growth assays. The soft agar cultures were re-fed (including adding freshly prepared MV) every third day. NIH 3T3 cells expressing Cdc42 F28L were used as a positive control for these experiments. (F) Images of the resulting colonies that formed in E. The data shown in C–E represent the mean ± SD from at least three independent experiments.
We then asked what MV-associated protein(s) is responsible for mediating the transfer of transforming capability. We initially considered the EGF receptor as a possible candidate protein, because activated forms of this receptor can be shared between brain cancer cells via MV (5, 6, 9). However, it is unlikely that the EGF receptor accounts for the similar transforming abilities associated with the MV derived from MDAMB231 breast cancer cells and U87 glioblastoma cells (Fig. 2 C–E), because activated EGF receptors cannot be detected in the MV shed from U87 cells (Fig. 2A). This notion was supported further by the finding that the anchorage-independent growth advantage imparted to NIH 3T3 cells by U87 cell-derived MV is insensitive to treatment with the EGF receptor tyrosine kinase inhibitor AG1478 (Fig. S2C).
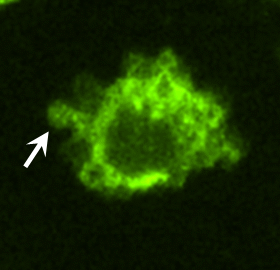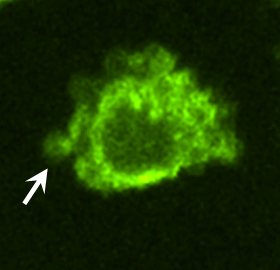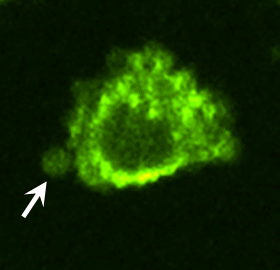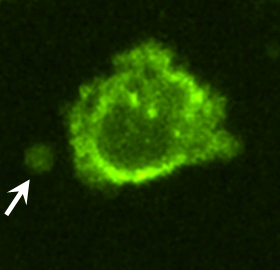
To identify proteins potentially involved in the transforming actions of these MV, proteomic screens were carried out. Proteins common to MV derived from MDAMB231 cells and U87 cells are listed in Fig. S3. Notably, among the MV-associated proteins was tissue transglutaminase (tTG), a protein cross-linking enzyme that has been linked to the chemoresistance and aberrant cell growth exhibited by some cancer cells (10–15) and that is secreted from cells by an unknown mechanism (15–17). We confirmed by immunoblot analysis that tTG is a component of MV derived from MDAMB231 and U87 cells (Fig. 3A) and demonstrated that the MV on the surfaces of MDAMB231 cells were detectable when immunostained with a tTG antibody (Fig. 3B, Upper) but not when stained with only the secondary antibody (Fig. 3B, Lower Left). Likewise, the MV generated by U87 cells and HeLa cervical carcinoma cells stimulated with EGF also contained tTG (Fig. 3C). These findings, when coupled with the fact that a GFP-tagged form of tTG is incorporated more efficiently than GFP alone into MV shed by MDAMB231 (Fig. 3D), demonstrate that tTG is targeted to MV generated by distinct types of cancer cells and in response to specific cell-culturing conditions.
Fig. 3.

MV shed by cancer cells contain tTG. (A) WCL of serum-starved MDAMB231 and U87 cells, as well as lysates of the MV shed by these cells, were immunoblotted with several antibodies, including one against tTG. (B) (Upper Left) MDAMB231 cells immunostained with a tTG antibody. (Upper Right) The boxed area was enlarged, and arrows indicated certain MV. (Lower) An MDAMB231 cell costained with only the secondary antibody (Left) and with rhodamine-conjugated phalloidin to label the MV (Right). (C) Images of serum-starved U87 glioma cells and HeLa cervical carcinoma cells that were left untreated or were stimulated with EGF for 15 min as indicated and then were immunostained with a tTG antibody. Pronounced MV are indicated by arrows. (D) WCL of MDAMB231 cells ectopically expressing either GFP only or GFP-tTG, as well as lysates of the MV shed by these transfectants into their culturing medium, were immunoblotted with antibodies against GFP, the MV marker flotillin-2, and the cytosolic-specific marker IκBα. (E) Fluorescent images of permeabilized and nonpermeabilized samples of MDAMB231 cells stained with antibodies against tTG, the intracellular protein Ras homolog enriched in brain (Rheb), and DAPI to label nuclei. (F) WCL of serum-starved MDAMB231 cells and intact MV generated by these cells treated with or without the tTG inhibitors T101 (cell impermeable) or MDC (cell permeable), were assayed for transamidation activity as read out by the incorporation of BPA into casein. The samples then were immunoblotted with antibodies against tTG, flotillin-2, and IκBα.
As shown in Fig. 3B, tTG frequently was enriched in the membranes of MV, as indicated by the ring-shaped staining patterns detected with a tTG antibody in cells actively forming MV. The same tTG antibody labeled MV that protruded from the plasma membranes of nonpermeabilized MDAMB231 cells (Fig. 3E) and detected tTG on the surfaces MV of individually isolated from MDAMB231 cells by immuno-SEM (Fig S4A). The upper panel in Fig. 3F shows that tTG expressed in whole-cell lysates (WCL) from MDAMB231 cells or in intact MV shed by these cells was enzymatically active as read out by its ability to catalyze the incorporation of biotinylated pentylamine (BPA) into casein. Pretreatment of the intact MDAMB231 cell-derived MV with the cell-permeable tTG inhibitor monodansylcadaverine (MDC) greatly diminished the levels of BPA-labeled casein detected in the assay. Interestingly, the cell-impermeable tTG inhibitor T101 (Fig. S4 B and C) also effectively blocked the cross-linking activity associated with MV derived from MDAMB231 cells (Fig. 3F), suggesting that tTG is predominantly localized and activated on the outer leaflet of MV membranes.
The MV derived from MDAMB231 breast cancer cells were not sensitive to the traditional secretory inhibitors BFA or ExoI, which block Arf GTPase activation (18), as indicated by monitoring MV formation by immunofluorescence staining of vesicle-associated tTG (Fig. S5A). We then considered the possibility that the ability of tTG to cross-link proteins is important for the formation and/or shedding of MV by cancer cells. Immunofluorescent analysis with a tTG antibody revealed that exposing MDAMB231 cells to the tTG inhibitors MDC and T101 had no effect on MV formation (Fig. S5A). The shedding of MV by MDAMB231 cells also did not require tTG enzymatic activity, nor was it affected by ExoI or BFA, as shown by the detection of nearly equivalent amounts of the MV marker flotillin-2 (5) and tTG in MV isolated from the culturing medium of control cells or cells treated with different inhibitors (Fig. S5B, Left and Right). Correspondingly, knocking down tTG in MDAMB231 cells, which depleted the expression of tTG in the MV, caused little change in the amount of MV shed by these cells as read out by the flotillin-2 marker (Fig. S5B, Center). Moreover, tTG mutants defective in their ability to cross-link substrates (tTG C277V) or to bind GTP (tTG R580L) when ectopically expressed in MDAMB231 cells were targeted to MV as efficiently as ectopically expressed wild-type tTG (Fig. S5C). Thus, these results indicate that tTG is not essential for the ability of cancer cells to form or shed MV, nor is the enzymatic activity of tTG needed for its targeting to MV.
We then examined whether tTG might function as MV cargo and be transferred to recipient cells. NIH 3T3 fibroblasts were incubated for 30 min with MV derived from serum-starved cultures of either MDAMB231 cells or U87 cells and then analyzed for tTG expression by immunoblot analysis (Fig. 4A and Fig. S6A) and immunofluorescent microscopy (Fig. S6B). The results from these experiments show that tTG levels were increased significantly in fibroblasts that had been incubated with the cancer cell-derived MV relative to the barely discernible levels of tTG in control fibroblasts.
Fig. 4.

The ability of MDAMB231 cell-derived MV to induce cellular transformation requires the transfer of tTG from MV to recipient cells. (A and B) Extracts of serum-starved NIH 3T3 fibroblasts that were incubated with serum-free medium or serum-free medium supplemented with MDAMB231 cell-derived MV that had been pretreated for 30 min with or without the tTG inhibitor T101 were immunoblotted with tTG and actin antibodies (A) and assayed for transamidation activity as read out by the incorporation of BPA into lysate proteins (B). (C) Cell death assays were performed on fibroblasts maintained in serum-free medium, 2% CS medium, or serum-free medium containing MDAMB231 cell-derived MV. Each culturing medium was left unsupplemented or was supplemented further with the tTG inhibitors T101 (cell impermeable) or MDC (cell permeable) as indicated. (D) Anchorage-independent growth assays were performed on fibroblasts incubated with MDAMB231 cell-derived MV that were untreated or treated with T101, the RGD peptide, or the control RGE peptide. (E) Lysates of NIH 3T3 cells stably overexpressing vector alone or Myc-tTG were immunoblotted with Myc and actin antibodies and were assayed for transamidation activity as read out by the incorporation of BPA into lysate proteins. (F) Cell death assays were performed on the NIH 3T3 stable cell lines maintained in serum-free medium treated with T101, MDC, or 2% CS or left untreated. (G) Anchorage-independent growth assays were performed on the NIH 3T3 stable cell lines. Vector-control fibroblasts were incubated with MDAMB231 cell-derived MV as a positive control. The data shown in B–D, F, and G represent the mean ± SD from at least three independent experiments. (H) Tumor-formation assays were performed in which 5 × 105 MDAMB231 cells mitotically arrested using mitomycin C (Mito-C-MDAMB231) expressing either control siRNA (siCont) or tTG siRNAs (siTG-1 or siTG-2) were injected s.c. alone or combined with 5 × 105 NIH 3T3 fibroblasts into nude mice. As controls, untreated MDAMB231 and NIH 3T3 cells were injected into nude mice. The resulting tumors that formed for each condition were counted, and the results are shown in the table.
These findings raised the question of whether the MV-mediated transfer of activated tTG into recipient fibroblasts might be important for conferring these cells with enhanced survival capability and the characteristics of transformation. To address this question, we took advantage of our earlier findings that tTG is localized on the surfaces of MV so that its cross-linking activity is susceptible to inhibition by the cell-impermeable, irreversible inhibitor T101 (Fig. 3 B, E, and F). By pretreating cancer cell-derived MV with T101 before adding them to fibroblast cultures, we were able to inhibit the cross-linking activity of the MV-associated tTG selectively and irreversibly (Fig. 4B and Fig. S6C). Using this approach, we compared how the survival advantage afforded to NIH 3T3 fibroblasts by MV collected from cancer cells was affected by the inhibition of tTG activity. Fig. 4C and Fig. S7A show that pretreatment of the MV derived from MDAMB231 or U87 cells with T101 severely compromised their ability to protect the recipient fibroblasts from serum deprivation-induced cell death. Importantly, the extent of cell survival achieved by culturing NIH 3T3 cells in medium supplemented with a nominal amount (2%) of calf serum (CS) was unchanged by the addition of T101, indicating that the ability of this small-molecule inhibitor to abolish the protection afforded by the cancer cell-derived MV was not caused by off-target effects that sensitized the fibroblasts to apoptosis. Analogous experiments then were performed in which MDAMB231 cell-derived MV were incubated with serum-starved NIH 3T3 cells in the presence of the cell-permeable tTG inhibitor MDC (Fig. 4C) or MV collected from MDAMB231 cells in which tTG had been knocked down (Fig. S5B) were added to serum-starved NIH 3T3 cells (Fig. S7B). Collectively, the results from these experiments point to a critical role for tTG in mediating the survival advantage imparted to fibroblasts by cancer cell-derived MV.
We then asked whether the transforming abilities of the cancer cell-derived MV were dependent on tTG. As shown previously in Fig. 2E and Fig. S2B, and again in Fig. 4D and Figs. S7C and S8A, incubating normal NIH 3T3 fibroblasts and MCF10A epithelial cells with MV derived from MDAMB231 cells or U87 cells induced their ability to grow (i.e., to form colonies) under anchorage-independent conditions. However, when recipient fibroblasts or epithelial cells were incubated with MV preparations that had been pretreated with T101 (Fig. 4D and Figs. S2B and S8A) or in which tTG had been knocked down (Fig. S7C), the number of colonies that formed was reduced. We then verified that T101 did not generally inhibit cellular transformation by showing that this inhibitor had no influence on the ability of NIH 3T3 cells expressing an activated form of the small GTPase Cdc42 (Cdc42 F28L) to grow under anchorage-independent conditions, even when a fivefold excess of T101 was used (Fig. S8B).
These findings prompted us to consider whether cancer cell-derived MV might function similarly in vivo and promote tumor growth by causing normal cells in the tumor microenvironment to acquire the ability to form a tumor. To investigate this idea, we took advantage of the fact that exposing MDAMB231 cells to the mitotic-arresting agent mitomycin C before injecting them into nude mice inhibited their ability to form tumors under conditions in which their control counterparts (untreated MDAMB231 cells) were quite effective at inducing tumor formation (Fig. 4H). However, when the mitomycin C-treated MDAMB231 cells were coinjected with an equal number of normal (nontransformed) NIH 3T3 fibroblasts into mice, four of six mice formed tumors, suggesting that the MV shed by the mitotically arrested cancer cells were capable of causing the neighboring NIH 3T3 fibroblasts to become transformed, inducing tumor growth. We then went on to show that knocking down tTG expression in the mitotically arrested MDAMB231 cells blocked the ability of the coinjected NIH 3T3 fibroblasts to form tumors in mice. Thus, these results are consistent with the idea that cancer cells can generate MV in vivo and that their ability to cause normal cells in the tumor microenvironment to promote tumor formation is dependent on tTG.
These findings demonstrate that the MV-mediated transfer of tTG into recipient cells is necessary for the ability of MDAMB231 cell- and U87 cell-derived MV to transform fibroblasts. However, is tTG alone sufficient to confer survival and transforming capabilities to the recipient cells? In fact, we found that although NIH 3T3 fibroblasts stably overexpressing Myc-tagged tTG (Fig. 4E) were indeed resistant to serum deprivation-induced apoptosis, an effect that was ablated by treating the cells with MDC (Fig. 4F), they were unable to form colonies in soft agar, unlike vector control-expressing fibroblasts incubated with MV derived from MDAMB231 cells (Fig. 4G). This result shows that although overexpression/overactivation of tTG in normal cells is not sufficient to induce their transformation fully (i.e., NIH 3T3 cells ectopically expressing tTG do not acquire the ability to form colonies when grown under anchorage-independent conditions), it does confer upon normal cells some characteristics of the transformed state, allowing them to grow in a monolayer under low-serum conditions and to become less sensitive to serum deprivation-induced cell death. However, these findings also indicate that for cancer cell-derived MV to enable recipient cells to exhibit one of the major hallmarks of cellular transformation, namely anchorage-independent growth, another protein must be transferred along with tTG. The cytoskeletal component fibronectin (FN) was a particularly attractive candidate, because it is a known binding partner of tTG (16, 17, 19) and was identified in our proteomics screen of MDAMB231 cell- and U87 cell-derived MV (Fig. S3). We confirmed by immunoblot analysis that FN was expressed in the MV collected from each of the cancer cell lines (Fig. 3A). We then assessed the potential role of the MV-associated FN in conferring upon fibroblasts the ability to exhibit anchorage-independent growth by using the arginine-glycine-aspartic acid (RGD) peptide as a means to interfere with the ability of FN to bind to and activate integrins on the surface of the recipient fibroblasts (20, 21). Anchorage-independent growth assays performed on fibroblasts cotreated with MV derived from MDAMB231 or U87 cells and either the RGD peptide or the control arginine-glycine-glutamic acid (RGE) peptide showed that the RGD peptide, like T101, blocked the MV-triggered induction of cellular transformation, whereas the control peptide did not (Fig. 4D and Fig. S8A).
Because both tTG and FN are important for the ability of cancer cell-derived MV to transform recipient cells, might they work together to elicit this cellular outcome? To address this question we first examined whether tTG interacts with FN in MV. Fig. 5A shows that FN coimmunoprecipitates with tTG from MDAMB231 WCL, as previously reported (16, 17, 19), as well as with tTG from lysates of MV shed by these cells. In addition to binding the monomeric form of FN, tTG associated with a larger form of FN with an apparent molecular mass of ~440 kDa that likely represented cross-linked FN dimers and was detectable only in the MV lysate. Pretreating intact MV collected from MDAMB231 cells or U87 cells with the tTG inhibitor T101 before lysing the MV and subjecting the extracts to immunoblot analysis did not affect the ability of tTG to be coimmunoprecipitated with monomeric FN from the MV lysates (Fig. S9). However, pretreating the MV with the tTG inhibitor resulted in a marked reduction in the amount of the ~440-kDa FN species detected in the MV lysate samples (Fig. 5B), suggesting that the higher molecular mass form of FN in the cancer cell-derived MV is generated through the ability of tTG to interact with and cross-link FN.
Fig. 5.

tTG cooperates functionally with FN to mediate the transforming actions of MV on recipient fibroblasts. (A) WCL of MDAMB231 and lysates of the MV shed by these cells were immunoblotted (Input) or were subjected to immunoprecipitation using a tTG antibody (IP: tTG) and then immunoblotted with FN, tTG, and actin antibodies. Note the detection of cross-linked FN in the MV lanes (FN dimer). (B) Intact MV collected from MDAMB231 or U87 cells were treated with T101 or were left untreated before being lysed. The MV extracts then were immunoblotted with FN and tTG antibodies. Note that the cross-linked forms of FN detected in the MV samples (FN dimer) were reduced significantly by T101 treatment. (C) Lysates of fibroblasts that were incubated with or without MV derived from MDAMB231 and U87 cells that had been pretreated or not with T101 were immunoblotted with antibodies against FAK and ERK or with antibodies that specifically recognize the activated forms of these protein kinases. (D) Diagram depicting how MV transform recipient cells. MV containing tTG and fibronectin are generated and released from the surfaces of human cancer cells. The MV then can be taken up by or can directly alter the microenvironment of neighboring normal cells, where the cotransfer of tTG and FN function cooperatively on the recipient cells to induce signaling events that promote cell survival and aberrant cell growth.
The dimerization of FN strongly enhances its ability to bind and activate integrins on the surfaces of cells (22, 23). The ability of tTG to cross-link FN in MV shed by cancer cells suggested that this covalently modified form of FN is capable of potentiating integrin activation. Indeed, we found that preparations of intact MV isolated from the medium of serum-deprived MDAMB231 cells or U87 cells were capable of stimulating signaling activities that are well known to be downstream from activated integrins including focal adhesion kinase (FAK) and ERK (Fig. 5C). Moreover, the activation of these kinases by MV was blocked by using either the tTG inhibitor T101 or the RGD peptide that interferes with integrin signaling, thus further demonstrating the importance of tTG and FN for the signaling functions of MV.
Discussion
The findings described here shed important light on an unconventional and poorly understood mechanism of cell-to-cell communication and how that communication may have significant consequences in human cancer progression. In particular, we have shown that exposing normal recipient cells to bioactive MV that are constitutively shed by certain human cancer cells can cause the recipient cells to acquire a transformed phenotype (Fig. 5D). We have identified the protein cross-linking enzyme tTG and the extracellular matrix protein FN as components of cancer cell-derived MV that functionally cooperate to mediate the transforming activities of the MV. The fact that tTG and FN are aberrantly regulated in several types of human cancers (24–30) raises exciting possibilities regarding the broad roles played by MV in cancer progression.
One aspect of our work that merits further consideration involves the potential effects of MV on tumor growth. A growing body of evidence demonstrates that cancer cells are capable of generating MV in vivo, suggesting that the shedding of MV by cancer cells is not merely an artifact of tissue culture but rather represents a tumor-relevant process. A particularly good example comes from a recent study which showed that brain tumor cell-derived MV could be detected routinely in blood samples taken from human patients afflicted with glioblastoma multiforme (4). Moreover, it also was shown that when MV shed by cultures of several different human primary tumor cells or established cancer cell lines were added back to the same cancer cells, the growth and the survival of these cells were enhanced significantly (4–7). When these findings are considered in the context of a tumor setting, the increased proliferative capacity afforded by sharing MV among cancer cells could be envisioned as a mechanism to augment tumor growth. However, our results now suggest that MV may impact cancer progression in another way, specifically, by conferring upon normal cell lineages that are major constituents of the tumor microenvironment (i.e., fibroblasts and epithelial cells), the transformed characteristics of cancer cells. This effect would be consistent with the idea that the expansion of a tumor mass would not necessarily depend solely on the proliferation of the cancer cells but rather could include as well the aberrant growth in the tumor microenvironment exhibited by stromal cells (including fibroblasts) and normal epithelium that have been exposed to MV shed by cancer cells. If this notion turns out to be true, we must adjust our understanding of cancer progression in vivo to include the potential contribution of MV.
However, the ability of the cancer cell-derived MV to induce a transformed phenotype in normal (nontransformed) recipient cell types is not sustained. For MV derived from MDAMB231 breast cancer cells or U87 brain tumor cells to promote the growth of normal cells (i.e., NIH 3T3 fibroblasts and MCF10A mammary epithelial cells) in low serum and to induce their ability to form colonies in soft agar, the recipient cells had to be treated repeatedly with freshly prepared MV throughout the duration of these growth assays. This requirement implies that the proteins and RNA transcripts contained within MV that are involved in promoting their transforming activity have a finite lifespan after being added to normal recipient cells and need to be replenished continuously. In the context of a tumor setting, the chronic shedding of MV by the cancer cells into their microenvironment might provide the continuous supply of MV required by the nearby recipient stroma and normal epithelium to induce and maintain a transformed phenotype.
The MV field is still in its infancy, and many of the advances made in our understanding of MV formation, as well as the identity of their contents and their mechanisms of action (i.e., transforming capability), have been largely limited thus far to tissue culture experiments. However, as our methods for isolating and characterizing cancer cell-derived MV continue to advance, it ultimately will be important to extend these studies into animal models. Toward this end, we currently are focusing our efforts on understanding the signaling events that are coupled to the formation and shedding of MV from the surface of human cancer cells and on determining whether additional proteins (i.e., aside from tTG and FN) contribute to the abilities of cancer cell-derived MV to transform recipient cells.
Materials and Methods
The isolation of MV from cancer cells was carried out essentially as described previously (4, 5). A detailed description of the protocol is provided in SI Materials and Methods. All reagents and additional procedures used in this study, including cell culture, cell growth and survival assays, electron and fluorescent microscopy, immunoprecipitation, immunoblot analysis, flow cytometry, proteomic analyses, transamidation assays, and mouse xenograft experiments are described in SI Materials and Methods.
Supplementary Material
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1017667108/-/DCSupplemental.
References
- 1.Al-Nedawi K, Meehan B, Rak J. Microvesicles: Messengers and mediators of tumor progression. Cell Cycle. 2009;8:2014–2018. doi: 10.4161/cc.8.13.8988. [DOI] [PubMed] [Google Scholar]
- 2.Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006;20:1487–1495. doi: 10.1038/sj.leu.2404296. [DOI] [PubMed] [Google Scholar]
- 4.Skog J, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Nedawi K, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619–624. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- 6.Graner MW, et al. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 2009;23:1541–1557. doi: 10.1096/fj.08-122184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Vizio D, et al. Oncosome formation in prostate cancer: Association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009;69:5601–5609. doi: 10.1158/0008-5472.CAN-08-3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mathivanan S, Simpson RJ. ExoCarta: A compendium of exosomal proteins and RNA. Proteomics. 2009;9:4997–5000. doi: 10.1002/pmic.200900351. [DOI] [PubMed] [Google Scholar]
- 9.Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci USA. 2009;106:3794–3799. doi: 10.1073/pnas.0804543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li B, et al. EGF potentiated oncogenesis requires a tissue transglutaminase-dependent signaling pathway leading to Src activation. Proc Natl Acad Sci USA. 2010;107:1408–1413. doi: 10.1073/pnas.0907907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaguchi H, Wang HG. Tissue transglutaminase serves as an inhibitor of apoptosis by cross-linking caspase 3 in thapsigargin-treated cells. Mol Cell Biol. 2006;26:569–579. doi: 10.1128/MCB.26.2.569-579.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hwang JY, et al. Clinical and biological significance of tissue transglutaminase in ovarian carcinoma. Cancer Res. 2008;68:5849–5858. doi: 10.1158/0008-5472.CAN-07-6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mann AP, et al. Overexpression of tissue transglutaminase leads to constitutive activation of nuclear factor-kappaB in cancer cells: Delineation of a novel pathway. Cancer Res. 2006;66:8788–8795. doi: 10.1158/0008-5472.CAN-06-1457. [DOI] [PubMed] [Google Scholar]
- 14.Mangala LS, Fok JY, Zorrilla-Calancha IR, Verma A, Mehta K. Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene. 2007;26:2459–2470. doi: 10.1038/sj.onc.1210035. [DOI] [PubMed] [Google Scholar]
- 15.Yuan L, et al. Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene. 2007;26:2563–2573. doi: 10.1038/sj.onc.1210048. [DOI] [PubMed] [Google Scholar]
- 16.Balklava Z, et al. Analysis of tissue transglutaminase function in the migration of Swiss 3T3 fibroblasts: The active-state conformation of the enzyme does not affect cell motility but is important for its secretion. J Biol Chem. 2002;277:16567–16575. doi: 10.1074/jbc.M109836200. [DOI] [PubMed] [Google Scholar]
- 17.Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol. 2000;148:825–838. doi: 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Y, et al. Exo1: A new chemical inhibitor of the exocytic pathway. Proc Natl Acad Sci USA. 2003;100:6469–6474. doi: 10.1073/pnas.0631766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaudry CA, et al. Cell surface localization of tissue transglutaminase is dependent on a fibronectin-binding site in its N-terminal beta-sandwich domain. J Biol Chem. 1999;274:30707–30714. doi: 10.1074/jbc.274.43.30707. [DOI] [PubMed] [Google Scholar]
- 20.Yang W, Lin Q, Guan JL, Cerione RA. Activation of the Cdc42-associated tyrosine kinase-2 (ACK-2) by cell adhesion via integrin beta1. J Biol Chem. 1999;274:8524–8530. doi: 10.1074/jbc.274.13.8524. [DOI] [PubMed] [Google Scholar]
- 21.Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238:491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 22.Wierzbicka-Patynowski I, Schwarzbauer JE. The ins and outs of fibronectin matrix assembly. J Cell Sci. 2003;116:3269–3276. doi: 10.1242/jcs.00670. [DOI] [PubMed] [Google Scholar]
- 23.Leiss M, Beckmann K, Girós A, Costell M, Fässler R. The role of integrin binding sites in fibronectin matrix assembly in vivo. Curr Opin Cell Biol. 2008;20:502–507. doi: 10.1016/j.ceb.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 24.Shao M, et al. Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 2009;69:9192–9201. doi: 10.1158/0008-5472.CAN-09-1257. [DOI] [PubMed] [Google Scholar]
- 25.Hettasch JM, et al. Tissue transglutaminase expression in human breast cancer. Lab Invest. 1996;75:637–645. [PubMed] [Google Scholar]
- 26.Singer CF, et al. Tissue array-based expression of transglutaminase-2 in human breast and ovarian cancer. Clin Exp Metastasis. 2006;23:33–39. doi: 10.1007/s10585-006-9015-0. [DOI] [PubMed] [Google Scholar]
- 27.Williams CM, Engler AJ, Slone RD, Galante LL, Schwarzbauer JE. Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation. Cancer Res. 2008;68:3185–3192. doi: 10.1158/0008-5472.CAN-07-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barkan D, et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008;68:6241–6250. doi: 10.1158/0008-5472.CAN-07-6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 30.Antonyak MA, et al. Augmentation of tissue transglutaminase expression and activation by epidermal growth factor inhibit doxorubicin-induced apoptosis in human breast cancer cells. J Biol Chem. 2004;279:41461–41467. doi: 10.1074/jbc.M404976200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}