

Abstract
Background
Aspirin and antagonists of platelet ADP P2Y12 receptors are often co-prescribed for protection against thrombotic events. However, blockade of platelet P2Y12 receptors can inhibit thromboxane A2 (TXA2) dependent pathways of platelet activation independently of aspirin.
Objectives
To assess in vitro whether aspirin adds additional anti-aggregatory effects to strong P2Y12 receptor blockade.
Method
Using platelet-rich plasma from healthy volunteers, determinations were made in 96-well plates of platelet aggregation, TXA2 production and ADP/ATP release caused by ADP, arachidonic acid, collagen, epinephrine, TRAP-6 amide and U46619 (six concentrations of each) in the presence of prasugrel active metabolite (PAM; 0.1-10 μmol L−1), aspirin (30 μmol L−1), PAM+aspirin or vehicle.
Results
PAM concentration-dependently inhibited aggregation; e.g. aggregations to all concentrations of ADP and U46619 were inhibited ≥95% by PAM >3 μmol L−1. In further tests of PAM (3 μmol L−1), aspirin (30 μmol L−1) and PAM+aspirin, aspirin generally failed to produce more inhibition than PAM or additional inhibition to that caused by PAM. The anti-aggregatory effects of PAM were associated with reductions in the platelet releases of both TXA2 and ATP+ADP. Similar effects were found when using either citrate or lepirudin as anti-coagulants, and when conducting traditional light transmission aggregometry at low stirring speeds.
Conclusions
P2Y12 receptors are critical to the generation of irreversible aggregation through the TXA2-dependent pathway. As a result, strong P2Y12 receptor blockade alone causes inhibition of platelet aggregation that is little enhanced by aspirin. The clinical relevance of these observations remains to be determined.
Keywords: P2Y12 receptors, TP receptors, thrombosis, thromboxane, prasugrel
Introduction
Aspirin and antagonists of platelet ADP P2Y12 receptors, such as clopidogrel and prasugrel, are often prescribed together as secondary protection against thrombotic vascular events [1, 2, 3]. This “dual anti-platelet therapy” approach in part reflects the assumption that aspirin and P2Y12 antagonists will independently inhibit the thromboxane A2 (TXA2) -dependent and P2Y12 receptor-dependent pathways of platelet activation and thus have additive effects on inhibition of platelet function [4, 5, 6, 7]. However, antagonism of platelet P2Y12 receptors can inhibit platelet activation and aggregation mediated by TXA2 pathways, both by reducing platelet production of TXA2 and by inhibiting responses following TP receptor activation [8, 9, 10, 11]. To explore these interactions further we conducted a study to examine in vitro the individual and combined effects of aspirin and P2Y12 receptor antagonism, employing prasugrel’s active metabolite (PAM), R-138727 [5], as a pharmacological tool. A particular aim of our study was to assess if aspirin adds additional anti-aggregatory effects in the face of strong P2Y12 receptor antagonism.
Methods
Blood Collection
Blood was collected from healthy volunteers (St Thomas’s Hospital Research Ethics Committee, reference 07 / Q0702 / 24), who had abstained from NSAID consumption for the preceding 14 days, by venepuncture into tri-sodium citrate (3.2%, 1:9 v/v; Sigma, Poole, Dorset, U.K.) or lepirudin (Refludan™; 25 μg mL−1). Platelet-rich plasma (PRP) was obtained by centrifugation at 175 × g for 15 min at 25°C. Platelet-poor plasma (PPP) was obtained by centrifugation of PRP at 15000 × g for 2 min. Platelet counts were made to confirm normal platelet number, 2-4×108 cells mL−1, but samples were not adjusted. All experiments were completed within 2h of blood collection.
96-Well Plate Light Transmission Aggregometry
To assess the aggregation of platelets in 96-well plates, a modified light transmission method was used [12]. Briefly, 100 μL samples of PRP were pre-incubated for 30 min at 37°C with PAM (0.1-10 μmol L1), and/or aspirin (30 μmol L1), or vehicle (0.1% DMSO) before being placed into the individual wells of a 96-well microtitre plate (Nunc, Lutterworth, Leicestershire, U.K.) containing 10 μL of vehicle or agonist: adenosine diphosphate (ADP; 0.1-30 μmol L−1; LabMedics, Salford, Manchester, U.K.), arachidonic acid (AA; 0.03-1 mmol L−1; Sigma, Poole, U.K.), Horm collagen (0.1-30 μg mL−1; Nycomed, Linz, Austria), epinephrine (0.001-100 μmol L−1; LabMedics), the PAR-1 activation peptide TRAP6 amide (SFLLRNamide; 0.1-30 μmol L−1; Bachem, Bubendorf, Switzerland), and the stable TXA2-mimetic U46619 (0.1-30 μmol L−1; Cayman Chemical Company, Ann Arbor, MI, U.S.A.). These agonists were chosen as together they represent the range of platelet agonists most widely used in in vitro platelet testing [13]. The plate was then placed into a 96-well plate reader (Tecan Sunrise) at 37°C, and absorbance was measured at 595 nm every 15 s for 16 minutes with vigorous shaking between readings. Percentage aggregation was calculated with reference to the absorbance of PPP as a surrogate for 100% aggregation. Graphs shown are for aggregation responses at 16 min.
Light Transmission Aggregometry
Briefly, 225 μL samples of PRP were pre-incubated for 30 min at 37°C with PAM (3 μmol L1), and/or aspirin (30 μmol L1), or vehicle (0.1% DMSO) before being placed into individual cuvettes and incubated for 90 s at 37°C without or with stirring (100, 300 or 1200 rpm) in a PAP-8E aggregometer (Alpha Laboratories, Eastleigh, UK). 25 μl Horm collagen (3 μg mL−1; final) was then added and aggregation responses followed for 5 min without or with stirring (100, 300, 1200rpm). The percentage aggregation was calculated with reference to the absorbance of PPP as a surrogate for 100% aggregation. Graphs shown are for aggregation responses at 5 min.
Total ATP and ADP release
To determine the ATP and ATP+ADP release from the platelets, a modified luminometric method was used [14, 15]. Briefly, 100 μL of PRP was placed in each well of a white clear bottomed 96-well plate (Nunc) containing 10 μl Horm collagen (0.1-30 μg mL−1), TRAP6 amide (0.1-30 μmol L−1), or U46619 (0.1-30 μmol L−1) and placed in the plate reader as for platelet aggregation above. After five minutes the plate was removed from the plate reader. To measure ATP release CHRONO-LUME reagent (1:5 v/v, LabMedics) was then added to individual wells; while to measure ATP+ADP release 50 μl of substrate containing phosphocreatine (1.5 mmol L−1, Sigma), creatine phosphokinase (3000 U mL−1, Sigma), and CHRONO-LUME reagent (1:5 v/v) was added to wells. The plate was shaken at 350 rpm for two minutes at 37°C and then luminescence determined on a 1450 Microbeta Trilux (Perkin Elmer, Waltham, MA, USA). The luminescence of a 20 nmol L−1 ATP standard (LabMedics) in PPP was used to calculate the concentrations of ATP and ATP+ADP released.
Thromboxane B2 ELISA
At the end of the PRP aggregation monitoring (i.e. 16min for 96-well plate aggregometry and 5 min for traditional light transmission aggregometry), cyclooxygenase activity was halted by the addition of 1 mmol L−1 diclofenac (Sigma), the samples were centrifuged at 1300 g for 10 minutes at 5°C, and the supernatants removed and frozen. TXB2 levels in the supernatant, as a measure of TXA2 formation, were determined using selective ELISA (Assay Designs, Cambridge Bioscience, Cambridge, U.K.). In some experiments, to determine the direct effects of aspirin and PAM upon platelet cyclooxygenase activity, 100 μL samples of PRP were pre-incubated for 30 min at 37°C with PAM (0.1-10 μmol L−1), aspirin (30 μmol L−1), or vehicle (0.1% DMSO) before being placed into the individual wells of a 96-well microtitre plate (Nunc) and then rapidly frozen, to lyse the platelets. Plates were then rapidly thawed and incubated (37°C) for 16min with shaking in the presence of arachidonic acid (1 mM). At the end of this period the 96-well plates were processed for the assessment of TXA2 production in the same way as for 96-well plates containing intact platelets.
Statistical Analysis
All statistical analyses were conducted using GraphPad Prism v5 (GraphPad Software Inc, CA, USA). Agonist concentration response curves were plotted and analysed according to the four parameter logistic equation:
Concentration-response curves were compared by two way ANOVA and Bonferroni post-tests with p<0.05 being taken as significant. Other analyses are stated in text as appropriate.
Results
Influence of PAM on platelet aggregation induced by ADP, collagen or U46619 by 96-well plate aggregometry using citrated PRP
PAM caused concentration-dependent inhibitions of aggregation induced by ADP (0.1-30 μmol L−1; Figure 1A), such that aggregation to all ADP concentrations were inhibited ≥95% by PAM at concentrations greater than 3 μmol L−1. Aggregation induced by low concentrations of collagen, <3 μg mL−1, were inhibited more strongly by PAM than those induced by high concentrations (Figure 1B). Notably, PAM also produced insurmountable blockade of aggregation induced by U46619 (0.1-30 μmol L−1; Figure 1C) with very similar IC50 values to those for ADP: e.g. PAM log IC50 (± s.e.m.) values against 1 μmol L−1 of ADP and U46619 were −6.6±0.5 and −6.6±0.3, respectively; the log IC50 values against 10 μmol L−1 of ADP and U46619 were −6.2±0.1 and −5.8±0.1, respectively (Figures 1A and 1C).
Figure 1.
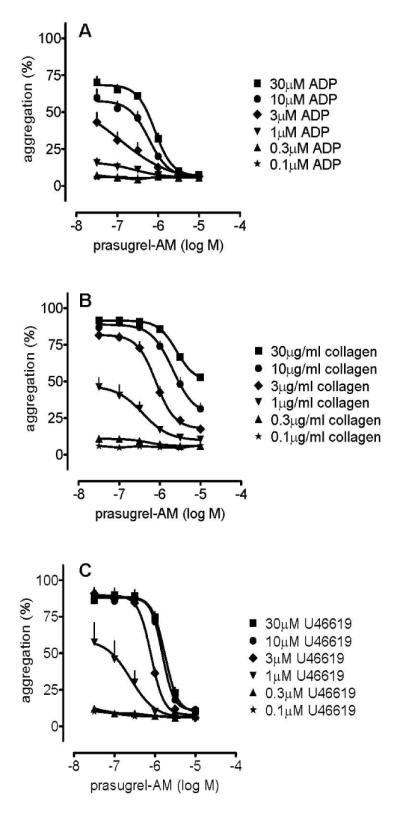
Platelet aggregations induced by ADP (0.1-30 μmol L−1; panel A), collagen (0.1-30 μg mL−1; panel B), or U46619 (0.1-30 μmol L−1; panel C) were inhibited in a concentration-dependent manner by PAM (p<0.05 for all; two way ANOVA). Curves for 0.1 μmol L−1 and 0.3 μmol L−1 ADP are superimposed upon one another (panel A), as are curves for 0.1 μmol L−1 and 0.3 μmol L−1 U46619 (panel B). Data shown are mean ± s.e.m. of responses measured by 96-well plate aggregometry in citrated PRP prepared from 6 different individuals.
Individual and combined effects of PAM and aspirin on platelet aggregation in response to arachidonic acid, ADP, collagen, epinephrine, TRAP6 or U46619 by 96-well plate aggregometry using citrated PRP
Across the range of platelet agonists used, aspirin (30 μmol L−1) did not produce either more inhibition than PAM (3 μmol L−1) or additional inhibition to that caused by PAM alone, except weakly for collagen at the two highest concentrations tested (two ANOVA; Figure 2).
Figure 2.

Platelet aggregation induced by arachidonic acid (0.03-1 mmol L−1; panel A), ADP (0.1-30 μmol L−1; panel B), collagen (0.1-30μg mL−1; panel C), epinephrine (0.001-100 μmol L−1; panel D), TRAP6 (0.1-30 μmol L−1; panel E) and U46619 (0.1-30 μmol L−1; panel F) in the presence of aspirin (30 μmol L−1) and/or PAM (3 μmol L−1). Data shown are mean ± s.e.m. of responses measured by 96-well plate aggregometry in citrated PRP measured prepared from 4 different individuals. * shows p<0.05 difference from vehicle by 2-way ANOVA plus Bonferroni post test; † shows p<0.05 difference between PAM and PAM+aspirin. Symbols at end of lines signify difference in set; symbols at individual points signify particular differences.
Individual and combined effects of PAM and aspirin on platelet TXA2 production in response to arachidonic acid, collagen, or epinephrine using citrated PRP by 96-well plate aggregometry
In control experiments, we measured the production of TXA2 to all platelet agonists (data not shown). From these studies, we selected arachidonic acid, collagen and epinephrine as the most reliable agonists to use in further investigations of the effects of PAM and aspirin. Aspirin (30 μmol L−1) alone caused complete inhibition of the production of TXA2 in response to arachidonic acid (Figure 3A), collagen (Figure 3B) and epinephrine (Figure 3C). Whilst PAM (3 μmol L−1) also reduced TXA2 production to the same three platelet activators, for arachidonic acid and collagen it was to a lesser extent than that found for aspirin. Addition of aspirin on top of PAM significantly (two way ANOVA; p<0.01) inhibited any remaining TXA2 formation in the case of arachidonic acid and collagen, while for epinephrine aspirin added no significant extra effect to that of PAM. In broken platelet preparations, the formation of TXA2 following incubation with arachidonic acid (control: 435±17 ng/ml TXB2; mean±s.e.m., n=24) was markedly inhibited by aspirin (10, 30 and 100 μmol L−1 caused inhibitions of 46±11%, 79±4% and 90±3%, respectively; mean±s.e.m., n=4 for each; p<0.05) whereas PAM was without effect (1, 3 and 10μmol L−1 caused inhibitions of 2±8%, 16±5% and 4±11%, respectively; mean±s.e.m., n=4 for each; n.s.).
Figure 3.
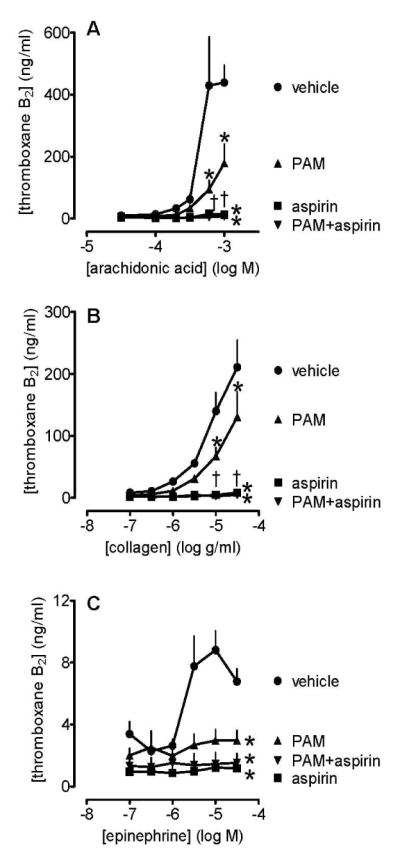
Platelet production of TXA2 induced by arachidonic acid (0.03-1 mmol L−1; panel A), collagen (0.1-30μg mL−1; panel B), and epinephrine (0.1-30 μmol L−1; panel C) in the presence of PAM (3 μmol L−1), and/or aspirin (30 μmol L−1), or vehicle (control, 0.1% DMSO). Data shown are mean ± s.e.m. of responses collected from 96-well plate aggregometry in citrated PRP prepared from 4 different individuals. * shows p<0.05 difference from vehicle by 2-way ANOVA plus Bonferroni post test; † shows p<0.05 difference between PAM and PAM+aspirin. Symbols at end of lines signify difference in set; symbols at individual points signify particular differences.
Individual and combined effects of PAM and aspirin on the release of ATP and ATP+ADP from platelets stimulated with collagen, TRAP6 or U46619 by 96-well plate aggregometry using citrated PRP
In control experiments, we measured the release of ATP and ATP+ADP to all platelet agonists (data not shown). From these studies we selected collagen, TRAP-6 and U46619 as the most reliable agonists to use in further investigations of the effects of PAM and aspirin. Platelet release of ATP (data not shown) and ATP+ADP stimulated by collagen was similarly inhibited by PAM (3 μmol L−1) and aspirin (30 μmol L−1), with no significant additive effect (two way ANOVA; Figure 4A). Release of adenine nucleotides in response to TRAP6 was inhibited by PAM but not by aspirin, with no additive effect (two way ANOVA; Figure 4B). Release of adenine nucleotides in response to U46619 was >90% inhibited by PAM but unaffected by aspirin; there was no additive effect of aspirin to that of PAM (two way ANOVA; Figure 4C).
Figure 4.
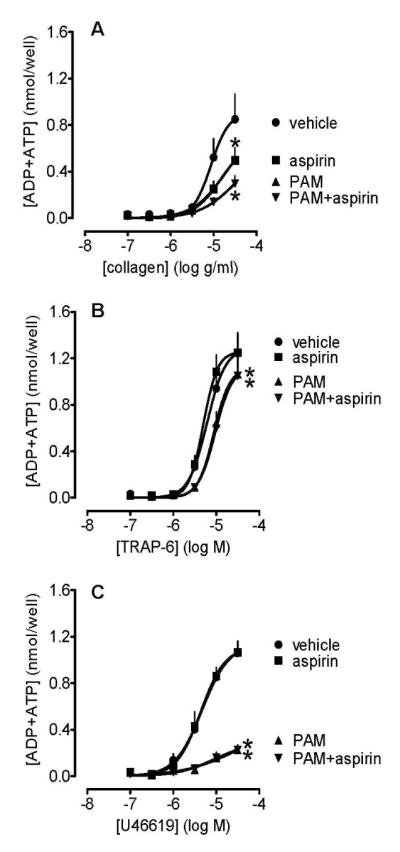
The release of ATP+ADP from platelets stimulated with collagen (0.1-30 μg mL−1; panel A), TRAP6 (0.1-30 μmol L−1; panel B) and U46619 (0.1-30 μmol L−1; panel C) in the presence of PAM (3 μmol L−1), and/or aspirin (30 μmol L−1), or vehicle (control, 0.1% DMSO). Data shown are mean ± s.e.m. of responses measured by 96-well plate aggregometry in citrated PRP prepared from 5 different individuals. * shows p<0.05 difference from vehicle by 2-way ANOVA plus Bonferroni post test. Symbols at end of lines signify difference in set; symbols at individual points signify particular differences.
Individual and combined effects of PAM and aspirin on platelet aggregation in response to arachidonic acid, ADP, collagen, epinephrine, TRAP6 or U46619 by 96-well plate aggregometry using lepirudinized PRP
In lepirudinized PRP, PAM (3 μmol L−1) and aspirin (30 μmol L−1) produced the same pattern of inhibitory effects upon platelet aggregation induced by the range of platelet agonists as was found in citrated PRP. In particular, aspirin did not produce either more inhibition than PAM or additional inhibition to that caused by PAM alone (Figure 5).
Figure 5.

Platelet aggregation induced by arachidonic acid (0.03-1 mmol L−1; panel A), ADP (0.1-30 μmol L−1; panel B), collagen (0.1-30μg mL−1; panel C), epinephrine (0.001-100 μmol L−1; panel D), TRAP6 (0.1-30 μmol L−1; panel E) and U46619 (0.1-30 μmol L−1; panel F) in the presence of aspirin (30 μmol L−1) and/or PAM (3 μmol L−1). Data shown are mean ± s.e.m. of responses measured by 96-well plate aggregometry in lepirudinized PRP prepared from 4 different individuals. * shows p<0.05 difference from vehicle by 2-way ANOVA plus Bonferroni post test. Symbols at end of lines signify difference in set; symbols at individual points signify particular differences.
Individual and combined effects of PAM and aspirin on platelet TXA2 production in response to arachidonic acid or collagen using lepirudinized PRP by 96-well plate aggregometry
The pattern of TXA2 production in lepirudinized PRP was similar to that in citrated PRP, although the amounts produced were approximately halved (Figure 6). Aspirin (30 μmol L−1) alone caused complete inhibition of the production of TXA2 in response to arachidonic acid and collagen, while PAM caused partial inhibitions (Figure 6). Addition of aspirin on top of PAM inhibited any remaining TXA2 formation. Our assays were not sufficiently sensitive to provide robust data regarding the production of TXA2 in response to epinephrine in lepirudinized PRP and the effects of PAM and aspirin (data not shown).
Figure 6.
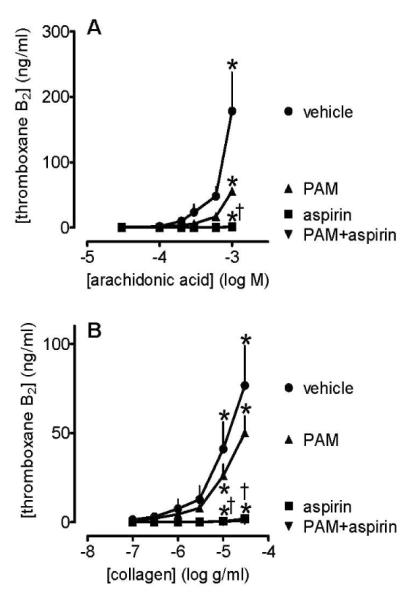
Platelet production of TXA2 induced by arachidonic acid (0.03-1 mmol L−1; panel A) and collagen (0.1-30 μg mL−1; panel B) in the presence of PAM (3 μmol L−1), and/or aspirin (30 μmol L−1), or vehicle (control, 0.1% DMSO). Data shown are mean ± s.e.m. of responses collected from 96-well plate aggregometry in lepirudinized PRP prepared from 4 different individuals. * shows p<0.05 difference from vehicle by 2-way ANOVA plus Bonferroni post test; † shows p<0.05 difference between PAM and PAM+aspirin. Symbols at end of lines signify difference in set; symbols at individual points signify particular differences.
Individual and combined effects of PAM and aspirin on platelet aggregation in response to collagen in traditional LTA using citrated PRP
When conducting LTA at different stir speeds (100, 300, and 1200 rpm), we noted no significant differences in the maximal responses to collagen (3 μg mL−1; Figure 7A) although the AUC was less at 100 rpm than at 300 rpm or 1200 rpm due to the slower development of the response (Figure 7B). PAM (3 μmol L−1) inhibited the collagen-induced aggregations with a very notable sensitivity to stirring speed, such that at 100 rpm PAM inhibited the maximum aggregation by 70±7% whereas at 1200rpm it inhibited by only 15±4% (n=4; Figure 7C); the associated values for AUC were 67±8% and 15±4% (n=4; Figure 7D). Aspirin produced much greater additive effects at 1200 rpm than at 100 rpm, such that there were no significant differences between the responses to collagen in the presence of PAM+aspirin at any speed (100, 300 and 1200 rpm; Figures 7E and 7F).
Figure 7.

Platelet aggregation induced by collagen (3 μg mL−1) as measured by traditional light transmission aggregometry at stir speeds of 100, 300 or 1200 rpm using citrated PRP. Responses are shown as % maximum aggregation (panels A, C and E), and AUC of aggregation (panels B, D and F), as calculated over a 5 min aggregation period. Control responses shown in panels A and B; responses in the presence of PAM (3 μmol L−1) in panels C and D; responses in the presence of PAM (3 μmol L−1) + aspirin (30 μmol L−1) in panels E and F. Data shown are mean ± s.e.m. of responses in PRP prepared from 4 different individuals; * p<0.05, one-way ANOVA.
Individual and combined effects of PAM and aspirin on platelet TXA2 production in response to collagen in traditional LTA using citrated PRP
There was a clear association between stir speed of PRP and the production of TXA2 in response to collagen (Figure 8). Aspirin (30 μmol L−1) either alone or in combination with PAM (3 μmol L−1) inhibited TXA2 production by more than 98% in all cases (n=32). PAM alone inhibited TXA2 production at 100 rpm by 19±11%, at 300rpm by 36±5%, and at 1200rpm by 30±6% (n=4 for each).
Figure 8.
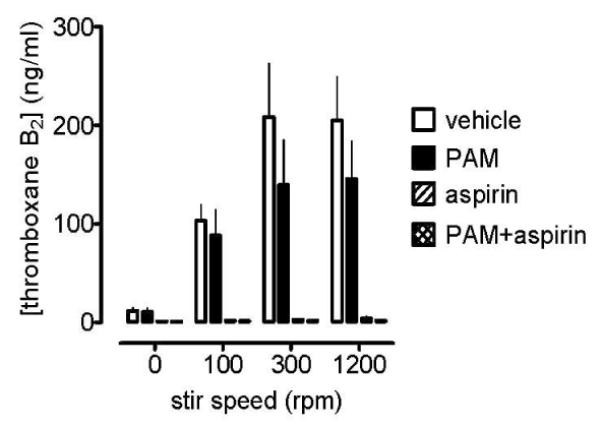
Platelet production of TXA2 induced by collagen (3μg mL−1) in the presence of PAM (3 μmol L−1), and/or aspirin (30 μmol L−1), or vehicle (control, 0.1% DMSO). Data shown are mean ± s.e.m. of samples collected from traditional light transmission aggregometry in citrated PRP prepared from 4 different individuals.
Discussion
In experiments using multiple platelet agonists across wide concentration ranges, we found that aspirin, at a concentration that completely inhibited TXA2 formation, neither produced more inhibition of platelet aggregation than PAM nor substantially increased inhibition of platelet aggregation when added to PAM, as assessed by either 96-well plate aggregometry or traditional light transmission aggregometry at low speed. Furthermore, our data suggest that the failure of aspirin to produce additional anti-aggregatory effects in the presence of strong P2Y12 blockade reflects blockade by PAM of TXA2 pathways of platelet activation due to (i) a reduction in the formation of TXA2 and (ii) an inhibition of TXA2-driven aggregation mediated by TP receptor stimulation. These observations indicate, at least in vitro, that under certain conditions the platelet inhibition resulting from combined aspirin and P2Y12 receptor blockade is not appreciably greater than that produced by high levels of P2Y12 receptor blockade alone.
In the studies presented here, we initially characterised the effects of PAM upon platelet aggregation induced by ADP, to confirm the ability of our 96-well plate aggregation assay to characterise concentration-dependent P2Y12 receptor blockade [5, 16, 17, 18, 19, 20]. Then, consistent with the roles of platelet-derived ADP in collagen-induced aggregation, we demonstrated PAM to be inhibitory with more activity against low than high concentrations of collagen [5, 18]. We next showed that PAM blocked platelet aggregations induced by the TP receptor agonist, U46619, at concentrations very similar to those that blocked responses to ADP. This is consistent with reports that the second, irreversible wave of platelet aggregation following TP receptor activation is dependent upon platelet-derived ADP acting upon platelet P2Y12 receptors [8, 9, 16]. We also found that aggregation in response to ADP was associated with increased production of thromboxane A2 (e.g. TXB2 levels in presence of ADP 10 μM: control, 9.9±1.9 ng/ml; aspirin, 2.6±0.8 ng/ml; p<0.05, two-way ANOVA plus Dunnett’s test, n=8 for each: full curves not shown), even though we found only a weak anti-aggregatory effect of aspirin (significant at ADP 3μM; p<0.05; 2-way ANOVA; Figure 2B). This weak inhibition was not seen in experiments using lepirudinized PRP, which may well be explained by the enhancement of TXA2-dependent responses in the presence of low extracellular calcium (as discussed below) [21, 22, 23, 24, 25]. From these experiments we selected concentrations of PAM (3 μmol L1) and aspirin (30 μ mol L1) for further study, consistent with the effective peak plasma concentrations of these drugs in clinical use [5, 10, 11, 12]. These studies showed clearly that, with the exception of a weak additive effect for aggregation induced by collagen, there were no circumstances in which aspirin produced either more inhibition of platelet aggregation than PAM or additional inhibition of platelet aggregation when added to PAM. PAM also blocked platelet aggregation induced by arachidonic acid, a response which is well characterised as being TXA2 dependent and widely used as an ex vivo test for the effects of aspirin [26]. We also found that PAM caused reductions in the production of TXA2 following stimulation of platelets with arachidonic acid, collagen or epinephrine, but that this effect was lost in broken platelets confirming that PAM, unlike aspirin, is not a direct inhibitor of cyclooxygenase. These observations are in agreement with our previous report that treatment with standard dose of clopidogrel for one week reduces the ex vivo production of TXA2 by platelets [10], and demonstrates that this production is partially dependent upon activation of P2Y12 receptors, consistent with previous reports [25]. Finally, we also found that PAM had generally greater effects upon ADP and ATP release than aspirin, and for aspirin to have only weak, or no, additive effects to PAM. We are aware that the effects we report here from studies employing 96-well plate aggregometry differ in some important regards to those previously reported using traditional light transmission aggregometry. In particular, studies using hirudin as an anti-coagulant and/or employing stirred platelet preparations have shown PAM, and other P2Y12 receptor blockers, to have relatively weak effects against aggregation induced by collagen with a much more important additive effect of aspirin [19, 20]. One reason for the difference in results could accord with the understanding that analysis of platelet aggregation in the presence of citrate may over emphasise the role of TXA2. This phenomenon occurs because the production of TXA2 in response to a range of platelet agonists becomes enhanced when the concentration of extracellular calcium is decreased, as can be seen in washed platelet preparations using calcium-free buffers and similarly in citrated PRP [21, 22, 23, 24, 25]. However, in studies using lepirudinized PRP, we found the same pattern of results as in citrated PRP, although with an approximate 50% reduction in the associated production of TXA2. Indeed, the response to collagen actually became less sensitive to the effects of aspirin, with a proportionately greater difference with regard to the effects of PAM. As the differences between our results and those previously published were not explainable by differences in the anti-coagulant we considered they must be explained by the methods of mixing in 96-well plate versus traditional light transmission aggregometry. In particular, we noted that PAM was considerably more active against collagen-induced aggregation in 96-well plates than previously reported in traditional light transmission aggregometry [19, 20]. To explore the reasons for this, we repeated our studies of aggregation responses to collagen using a traditional (PAP-8E) light aggregometer, and paid particular attention to the speed of the stirrer bar, since we hypothesised that the shaking provided by the 96-well plate reader would be better mimicked at low stirrer bar speeds. We found that the effects of PAM and aspirin against collagen-induced aggregation at 1200 rpm were as reported previously, i.e. that PAM was only poorly effective with a marked additional effect of aspirin [19, 20]. However, as we reduced the speed we found that the effects of PAM become relatively larger, such that at 100 rpm it inhibited collagen-induced aggregation by around 70%. Interestingly, despite these marked differences in sensitivity to PAM, aggregatory responses to collagen were little different between 100 rpm and 1200 rpm. These data show that the effects of PAM against collagen-induced aggregation are strongly influenced by physical conditions and offer a good explanation for differences between our results and those reported previously.
Of course in vitro systems are of value if they assist in understanding how systems work in vivo, and so consideration must be given to determining if aggregometry conducted in 96-well plates has any usefulness in this regard. One way to address this issue is to assess whether the mechanistic in vitro data we present here accords with mechanistic in vivo data; i.e. does aspirin fail to add to the anti-thrombotic effects of strong P2Y12 receptor blockade in vivo. A number of studies suggest that our mechanistic in vitro data is indeed in accord. For example, thrombotic occlusion of injured canine coronary arteries can be abolished by strong P2Y12 receptor blockade without the need for aspirin [27, 28], and the same observation has been made for platelet deposition in a porcine denuded arterial shunt [29]. Furthermore, in wild type mice, FeCl3 damage of mesenteric arteries has been reported to result in occlusion within about 15 min, whereas in P2Y12 mice the same was only seen in 1 out of 9 animals [30]. This result led the authors to conclude that ‘more aggressive strategies of P2Y12 antagonism will be anti-thrombotic without the requirement of aspirin co-therapy’. Of course this does not deny that aspirin can add anti-thrombotic effects when P2Y12 receptor blockers are used at doses producing lower levels of P2Y12 receptor blockade, even a potent P2Y12 blocker such as prasugrel [31, 32].
In conclusion, we show that P2Y12 receptors underpin key components of the TXA2 pathway of platelet activation and appear to be critical to the generation of irreversible aggregation following TP receptor activation by TXA2, especially at lower speeds of platelet mixing which could accord with low shear areas of thrombus formation in vivo [33]. As a result strong P2Y12 receptor blockade alone produces inhibition of platelet aggregation to a broad range of platelet agonists that is not further enhanced by addition of aspirin, an observation that is consistent with data from some pre-clinical in vivo models. The clinical relevance of these observations remains to be determined in appropriately designed clinical investigations. However, in a highly speculative manner, one could postulate that addition of aspirin to individuals receiving strong P2Y12 receptor blockers has the potential to produce side effects secondary to inhibition of cyclooxygenase at non-platelet sites, as has recently become apparent for non-steroidal anti-inflammatory drugs [34, 35, 36, 37].
Acknowledgements
This research was supported by European Community FP6 funding (“Eicosanox”; LSHM-CT-2004-0050333), the British Heart Foundation, the National Heart and Lung Institute Foundation and the Wellcome Trust. This publication reflects only the authors’ views. The European Community is not liable for any use that may be made of information herein. This work forms part of the research themes contributing to the translational research portfolio of Barts and the London Cardiovascular Biomedical Research Unit which is supported and funded by the National Institute of Health Research. Prasugrel’s active metabolite, R-138727, was kindly supplied by Daiichi-Sankyo Co. Ltd, Tokyo, Japan.
References
- 1.Saw J, Madsen EH, Chan S, Maurer-Spurej E. The ELAPSE (Evaluation of Long-Term Clopidogrel Antiplatelet and Systemic Anti-Inflammatory Effects) Study. J Am Coll Cardiol. 2008;52:1826–33. doi: 10.1016/j.jacc.2008.08.047. [DOI] [PubMed] [Google Scholar]
- 2.Antithrombotic Trialists’ Collaboration Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehta SR, Yusuf S, Peters RJG, Bertrand ME, Lewis BS, Natarajan MK, Malmberg K, Rupprecht H-J, Zhao F, Chrolavicius S, Copland I, Fox KAA. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet. 2001;358:527–33. doi: 10.1016/s0140-6736(01)05701-4. [DOI] [PubMed] [Google Scholar]
- 4.Bhatt DL. Role of antiplatelet therapy across the spectrum of patients with coronary artery disease. Am J Cardiol. 2009;103:11A–19A. doi: 10.1016/j.amjcard.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 5.Jakubowski JA, Winters KJ, Naganuma H, Wallentin L. Prasugrel: a novel thienopyridine antiplatelet agent. A review of preclinical and clinical studies and the mechanistic basis for its distinct antiplatelet profile. Cardiovasc Drug Rev. 2007;25:357–74. doi: 10.1111/j.1527-3466.2007.00027.x. [DOI] [PubMed] [Google Scholar]
- 6.Angiolillo DJ. Variability in responsiveness to oral antiplatelet therapy. Am J Cardiol. 2009;103:27A–34A. doi: 10.1016/j.amjcard.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 7.Jennings LK. Role of platelets in atherothrombosis. Am J Cardiol. 2009;103:4A–10A. doi: 10.1016/j.amjcard.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 8.Li Z, Zhang G, Le Breton GC, Gao X, Malik AB, Du X. Two waves of platelet secretion induced by thromboxane A2 receptor and a critical role for phosphoinositide 3-kinases. J Biol Chem. 2003;278:30725–31. doi: 10.1074/jbc.M301838200. [DOI] [PubMed] [Google Scholar]
- 9.Paul BZS, Jin J, Kunapuli SP. Molecular mechanism of thromboxane A2-induced platelet aggregation. Essential role for P2Tac and alpha2a receptors. J Biol Chem. 1999;274:29108–14. doi: 10.1074/jbc.274.41.29108. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong PC, Dhanji AR, Tucker AT, Mitchell JA, Warner TD. Reduction of platelet thromboxane A production ex-vivo and in-vivo by clopidogrel therapy. J Thromb Haemost. 2010;8:613–5. doi: 10.1111/j.1538-7836.2009.03714.x. [DOI] [PubMed] [Google Scholar]
- 11.Armstrong PC, Dhanji AR, Truss NJ, Zain NJ, Tucker AT, Mitchell JA, Warner TD. Utility of 96-well plate aggregometry and measurement of thrombi adhesion to determine aspirin and clopidogrel effectiveness. Thromb Haemost. 2009;102:772–8. doi: 10.1160/TH09-04-0215. [DOI] [PubMed] [Google Scholar]
- 12.Armstrong PC, Truss NJ, Ali FY, Dhanji AA, Vojnovic I, Zain ZN, Bishop-Bailey D, Paul-Clark MJ, Tucker AT, Mitchell JA, Warner TD. Aspirin and the in vitro linear relationship between thromboxane A2-mediated platelet aggregation and platelet production of thromboxane A2. J Thromb Haemost. 2008;6:1933–43. doi: 10.1111/j.1538-7836.2008.03133.x. [DOI] [PubMed] [Google Scholar]
- 13.Cattaneo M, Hayward CP, Moffat KA, Pugliano MT, Liu Y, Michelson AD. Results of a worldwide survey on the assessment of platelet function by light transmission aggregometry: a report from the platelet physiology subcommittee of the SSC of the ISTH. J Thromb Haemost. 2009;7:1029. doi: 10.1111/j.1538-7836.2009.03458.x. [DOI] [PubMed] [Google Scholar]
- 14.Sun B, Tandon NN, Yamamoto N, Yoshitake M, Kambayashi J. Luminometric assay of platelet activation in 96-well microplate. Biotechniques. 2001;31:1174–78. doi: 10.2144/01315dd02. [DOI] [PubMed] [Google Scholar]
- 15.Truss NJ, Armstrong PC, Liverani E, Vojnovic I, Warner TD. Heparin but not citrate anticoagulation of blood preserves platelet function for prolonged periods. J Thromb Haemost. 2009;7:1897–905. doi: 10.1111/j.1538-7836.2009.03589.x. [DOI] [PubMed] [Google Scholar]
- 16.Gachet C. Regulation of platelet functions by P2 receptors. Annu Rev Pharmacol Toxicol. 2006;46:277–300. doi: 10.1146/annurev.pharmtox.46.120604.141207. [DOI] [PubMed] [Google Scholar]
- 17.Cattaneo M. ADP receptors: inhibitory strategies for antiplatelet therapy. Drug News Perspect. 2006;19:253–9. doi: 10.1358/dnp.2006.19.5.985936. [DOI] [PubMed] [Google Scholar]
- 18.Gachet C. The platelet P2 receptors as molecular targets for old and new antiplatelet drugs. Pharmacol Ther. 2005;108:180–92. doi: 10.1016/j.pharmthera.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 19.Storey RF, Newby LJ, Heptinstall S. Effects of P2Y1 and P2Y12 receptor antagonists on platelet aggregation induced by different agonists in human whole blood. Platelets. 2001;12:443–7. doi: 10.1080/09537100120085450. [DOI] [PubMed] [Google Scholar]
- 20.Dawood BB, Wilde J, Watson SP. Reference curves for aggregation and ATP secretion to aid diagnose of platelet-based bleeding disorders: effect of inhibition of ADP and thromboxane A2 pathways. Platelets. 2007;18:329–45. doi: 10.1080/09537100601024111. [DOI] [PubMed] [Google Scholar]
- 21.Cattaneo M, Gachet C, Cazenave JP, Packham MA. Adenosine diphosphate(ADP) does not induce thromboxane A2 generation in human platelets. Blood. 2002;99:3868–9. doi: 10.1182/blood-2002-01-0313. author reply 9-70. [DOI] [PubMed] [Google Scholar]
- 22.Mustard JF, Perry DW, Kinlough-Rathbone RL, Packham MA. Factors responsible for ADP-induced release reaction of human platelets. Am J Physiol. 1975;228:1757–65. doi: 10.1152/ajplegacy.1975.228.6.1757. [DOI] [PubMed] [Google Scholar]
- 23.Packham MA, Bryant NL, Guccione MA, Kinlough-Rathbone RL, Mustard JF. Effect of the concentration of Ca2+ in the suspending medium on the responses of human and rabbit platelets to aggregating agents. Thromb Haemost. 1989;62:968–76. [PubMed] [Google Scholar]
- 24.Falcon CR, Cattaneo M, Ghidoni A, Mannucci PM. The in vitro production of thromboxane B2 by platelets of diabetic patients is normal at physiological concentrations of ionized calcium. Thromb Haemost. 1993;70:389–92. [PubMed] [Google Scholar]
- 25.Jin J, Quinton TM, Zhang J, Rittenhouse SE, Kunapuli SP. Adenosine diphosphate (ADP)-induced thromboxane A(2) generation in human platelets requires coordinated signaling through integrin alpha(IIb)beta(3) and ADP receptors. Blood. 2002;99:193–8. doi: 10.1182/blood.v99.1.193. [DOI] [PubMed] [Google Scholar]
- 26.Born G, Patrono C. Antiplatelet drugs. Br J Pharmacol. 2006;147(Suppl 1):S241–51. doi: 10.1038/sj.bjp.0706401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao SK, Ober JC, McNatt J, Benedict CR, Rosolowsky M, Anderson HV, Cui K, Maffrand JP, Campbell WB, Buja LM. ADP plays an important role in mediating platelet aggregation and cyclic flow variations in vivo in stenosed and endothelium-injured canine coronary arteries. Circ Res. 1992;70:39–48. doi: 10.1161/01.res.70.1.39. [DOI] [PubMed] [Google Scholar]
- 28.Wang YX, Vincelette J, da Cunha V, Martin-McNulty B, Mallari C, Fitch RM, Alexander S, Islam I, Buckman BO, Yuan S, Post JM, Subramanyam B, Vergona R, Sullivan ME, Dole WP, Morser J, Bryant J. A novel P2Y12 adenosine diphosphate receptor antagonist that inhibits platelet aggregation and thrombus formation in rat and dog models. Thromb Haemost. 2007;97:847–55. [PubMed] [Google Scholar]
- 29.Vilahur G, Segales E, Salas E, Badimon L. Effects of a novel platelet nitric oxide donor (LA816), aspirin, clopidogrel, and combined therapy in inhibiting flow- and lesion-dependent thrombosis in the porcine ex vivo model. Circulation. 2004;110:1686–93. doi: 10.1161/01.CIR.0000142296.19558.99. [DOI] [PubMed] [Google Scholar]
- 30.Andre P, Delaney SM, LaRocca T, Vincent D, DeGuzman F, Jurek M, Koller B, Phillips DR, Conley PB. P2Y12 regulates platelet adhesion/activation, thrombus growth, and thrombus stability in injured arteries. J Clin Invest. 2003;112:398–406. doi: 10.1172/JCI17864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niitsu Y, Jakubowski JA, Sugidachi A, Asai F. Pharmacology of CS-747 (prasugrel, LY640315), a novel, potent antiplatelet agent with in vivo P2Y12 receptor antagonist activity. Semin Thromb Hemost. 2005;31:184–94. doi: 10.1055/s-2005-869524. [DOI] [PubMed] [Google Scholar]
- 32.Sugidachi A, Asai F, Ogawa T, Inoue T, Koike H. The in vivo pharmacological profile of CS-747, a novel antiplatelet agent with platelet ADP receptor antagonist properties. Br J Pharmacol. 2000;129:1439–46. doi: 10.1038/sj.bjp.0703237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nesbitt WS, Westein E, Tovar-Lopez FJ, Tolouei E, Mitchell A, Fu J, Carberry J, Fouras A, Jackson SP. A shear gradient-dependent platelet aggregation mechanism drives thrombus formation. Nat Med. 2009;15:665–73. doi: 10.1038/nm.1955. [DOI] [PubMed] [Google Scholar]
- 34.Warner TD, Mitchell JA. COX-2 selectivity alone does not define the cardiovascular risks associated with non-steroidal anti-inflammatory drugs. Lancet. 2008;371:270–3. doi: 10.1016/S0140-6736(08)60137-3. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell JA, Warner TD. COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nature Rev Drug Discov. 2006;5:75–86. doi: 10.1038/nrd1929. [DOI] [PubMed] [Google Scholar]
- 36.White WB. Cardiovascular effects of the cyclooxygenase inhibitors. Hypertension. 2007;49:408–18. doi: 10.1161/01.HYP.0000258106.74139.25. [DOI] [PubMed] [Google Scholar]
- 37.Kearney PM, Baigent C, Godwin J, Hallss H, Emberson JR, Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332:1302–8. doi: 10.1136/bmj.332.7553.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]