

Abstract
Metastasis causes most deaths from cancer yet mechanistic understanding and therapeutic options remain limited. Overexpression of the phosphatase PRL-3 is associated with metastasis of colon cancer. Here we show here that PRL-3 is a direct target of signaling by transforming growth factor β (TGFβ), which is broadly implicated in progression and metastasis. We found that suppression of PRL-3 expression by TGFβ was mediated by Smad-dependent inhibition of PRL-3 transcription at the level of promoter activity. PRL-3 activation stimulated PI3K/AKT signaling which caused resistance to stress-induced apoptosis. PRL-3 overexpression promoted metastatic colonization in an orthotopic mouse model of colon cancer, whereas PRK-3 knockdown reduced metastatic potential. Altered metastatic phenotypes were not derivative of primary tumor development or local invasion but could be attributed to PRL-3-mediated cell survival. Our findings suggest that inhibiting PRL-3 expression might be an important mechanism through which TGFβ suppresses metastasis in colon cancer. Additionally, our findings suggest that loss of TGFβ signaling, which occurs commonly during colon cancer progression, is sufficient to activate a PRL-3-mediated cell survival pathway that can selectively promote metastasis. Therefore, a major implication of our findings is that PRL-3 antagonists may offer significant value for anti-metastatic therapy in patients with colon cancer.
INTRODUCTION
Transforming growth factor β (TGFβ) plays an important role in tumorigenesis and metastasis. Upon ligand binding, TGFβ type II receptor (RII) recruits and activates TGFβ type I receptor (RI), which then activates Smad2 and Smad3. Activated Smad2 and Smad3 form complexes with Smad4 and translocate to the nucleus, where they regulate gene expression (1). We and others have demonstrated that TGFβ suppresses tumor initiation in a variety of cancers including colon cancer, and that loss of TGFβ signaling leads to malignancy (2-5). However, the role of TGFβ signaling in metastasis has been controversial. Although many studies have shown that TGFβ promotes metastasis (6), others have demonstrated that TGFβ suppresses metastasis (7;8). Recently, studies of human tumor samples indicate that loss or reduction of TGFβ signaling in human colorectal tumors is associated with development of metastasis (9;10).
Our previous studies indicate that abrogation of TGFβ signaling enables increased survival under stress in colon cancer cells (11). In addition, we have shown that loss of TGFβ signaling is associated with increased metastasis, whereas enhanced TGFβ signaling suppresses metastasis in an orthotopic model of colon cancer (Simms et al., unpublished data). These results suggest that endogenous TGFβ increases stress-induced apoptosis to prevent metastatic progression. In contrast, abrogation of TGFβ signaling leads to activation of oncogenic signals that promote survival and protect tumor cells from stress-induced apoptosis, thereby increasing their metastatic potential. Identification of oncogenes that are suppressed by TGFβ signaling would shed light on mechanisms of TGFβ tumor suppressor function and provide new opportunities of molecular targeting for novel cancer treatment.
Phosphatase of Regenerating Liver 3 (PRL-3), a metastasis-associated protein, belongs to the PRL family of protein tyrosine phosphatases, which includes two other members, PRL-1 and PRL-2 (12). While PRL-1 and PRL-2 are expressed in most tissues, PRL-3 expression is restricted to heart and skeletal muscle (13). Up-regulation of PRL-3 expression correlates with colon cancer progression: undetectable in normal colon epithelial, intermediate levels in 25-45% advanced primary tumors and significantly elevated levels in > 90% metastases regardless of metastatic sites (14;15). Importantly, PRL-3 expression in primary colorectal tumors has prognostic significance in predicting the development of liver and/or lung metastases as well as shortened patient survival (16-19). Therefore, PRL-3 shows promise as a biomarker for advanced malignancy and as a prognostic indicator for poor survival in colorectal cancer. Experimental manipulation of PRL-3 expression in several cell models alters cancer-associated phenotypes including proliferation, migration, invasion, tumorigenesis and metastasis (16;20;21). These studies indicate that up-regulation of PRL-3 expression has a causative role in tumor progression rather than merely being a consequence of these processes, which raises the possibility of PRL-3 being a potential therapeutic target for colon cancer treatment. However, little is known of the regulation of PRL-3 expression. Increased gene copy number is partially responsible for up-regulation of PRL-3 expression in colon cancer (14;15). In this study, we demonstrate that expression of PRL-3 is suppressed by TGFβ signaling in colon cancer cells. Smad3, but not Smad2, is essential for this suppression. Therefore, loss of TGFβ signaling, which occurs in 30-50% of colon cancer (22;23), is likely a mechanism of PRL-3 up-regulation in colon cancer. In addition, our studies show that PRL-3 activates AKT and maintains AKT activation under growth factor deprivation stress (GFDS). Ectopic expression of PRL-3 increases cell survival under GFDS and promotes metastasis in vivo, whereas knockdown of PRL-3 expression sensitizes colon cancer cells to stress-induced apoptosis and reduces metastasis in an orthotopic model.
MATERIALS AND METHODS
Cell Culture and Reagents
The human colon carcinoma FET, GEO and CBS cell lines were described previously (24). They were maintained at 37°C in a humidified incubator with 5% CO2 and cultured in SM medium (McCoy’s 5A serum-free medium, Sigma) supplemented with 10ng/ml epidermal growth factor, 20ug/ml insulin, and 4ug/ml transferrin. When cells were under growth factor and nutrient deprivation stress, they were cultured in SM medium in the absence of growth factor or serum supplements.
An PRL-3 specific monoclonal antibody (clone 318) (25) was used to detect endogenous PRL-3 protein. TGFβ-receptor type I kinase inhibitor SB525334, PI3K inhibitor LY294002 and recombinant human TGFβ1 were purchased from Sigma (St. Louis, MO), Calbiochem (La Jolla, CA) and R&D Systems (Minneapolis, MN) respectively. DNA fragmentation ELISA kits were from Roche Applied Science (Indianapolis, IN).
Reporter plasmids and luciferase assays
A 2033 bp fragment of human Prl-3 promoter (nucleotides 111546 to 113579 of homo sapiens chromosome 8, GenBank AC100803) was amplified by PCR from a BAC clone (ID# 3091L24, Invitrogen, Carlsbad, CA), and cloned into pGL 4.20 luciferase-reporter plasmid (Promega) to generate Prl-3-F5. Site-directed mutagenesis was performed to introduce quadruple point mutations into SBE-like binding sites (GTCTGCTA to CATAGCTA and GTCTGCCC to CATAGCCC) to generate Prl-3-F5-mutE1 and Prl-3-F5-mutE4 respectively. Prl-3-F5-mut E1/4 was generated by mutating both SBE-like binding sites simultaneously. Luciferase activity was measured with Luciferase Assay System (Promega, Madison, WI) and values were normalized by β-galactosidase activity. P values were calculated using Student’s t-test.
Electrophoresis mobility shift assays (EMSA)
Double-stranded oligonucleotides were labeled with [γ-32P] ATP using T4 polynucleotide kinase (Promega). Binding reactions containing GST-Smad3 (SignalChem) or His-Smad4 (Abcam) recombinant proteins and labeled probes were performed for 30 min at room temperature. Protein-DNA complexes were separated on 5% polyacrylamide gels. The sequences of the probes used are in Supplemental Materials.
Chromatin immunoprecipitation (CHIP) assays
CHIP assays were performed with the CHIP assay kit (Upstate Inc.) according to the manufacturer’s protocol. FET cells were treated with 4 ng/ml of TGFβ1 for 90 min. Cell pellets were resuspended in lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris–HCl, pH 8.1, with protease inhibitor cocktail from Sigma-Aldrich). Genomic DNA fragments were purified with a MinElute PCR purification kit (Qiagen). The primers used to amplify E1, E4 and negative control in Supplemental Materials.
In Vivo Orthotopic Model and Immunohistochemistry
Orthotopic implantation was performed as previously described (26). Briefly, exponentially growing GFP-labeled cells (5 × 106 cells) were inoculated subcutaneously into BALB/c nude mice. Once xenografts were established, they were excised and minced into 1 mm3 pieces. Two of these pieces were then sub-serosally implanted into cecum of other BALB/c nude mice. Sixty days post-implantation, animals were euthanized. Organs were explanted and imaged. Tissues were then processed and embedded in paraffin. Slides were cut for hematoxylin and eosin (H&E) and terminal nucleotidyl transferase–mediated nick end labeling (TUNEL) assays (Apotag, Oncor, Gaithersburg, MD). The apoptosis was determined quantitatively by counting the number of positively stained apoptotic bodies per field at 20x magnification. Six animals were analyzed for each type of cells. Three histologically similar fields were randomly selected from each slide for analysis. P values were calculated using Student’s t-test.
RESULTS
TGFβ signaling inhibits expression of PRL-3 in colon cancer cells
Up-regulation of PRL-3 expression is associated with tumor progression and metastasis of colon cancer (14;15). To determine whether TGFβ signaling regulates expression of PRL-3, FET cells were used in our studies. FET cells, derived from an early stage colon carcinoma, are sensitive to TGFβ-mediated growth inhibition and apoptosis (3;11). Our results showed that expression of PRL-3 was suppressed by TGFβ in a concentration-and time-dependent manner in FET cells (Fig. 1A, left and middle panels, Figs. S2A & S2B). SB525334, a potent inhibitor of TGFβ RI kinase, was used to confirm the effect of TGFβ. SB525334 is functional in FET cells as reflected by inhibition of phosphorylation of Smad2 induced by TGFβ treatment (Fig. S1). Treatment of FET cells with SB525334 reversed the inhibitory effect of TGFβ on expression of PRL-3 (Fig. 1A, right panel). In addition, inhibition of TGFβ signaling by transfection of a dominant negative mutant of TGFβ RII (DNRII) increased the expression of PRL-3 in FET cells, whereas activation of TGFβ signaling by transfection of a constitutively active mutant of TGFβ RI T204D (RI/T204D) decreased the expression of PRL-3 (Fig. 1B, left panel, Fig. S2C). Similar results were obtained in other colon cancer cells (FETα, CBS and GEO) that retain responsiveness to TGFβ signaling. Abrogation of TGFβ signaling by ectopic expression of DNRII led to up-regulation of PRL-3 expression, whereas enhancement of TGFβ signaling by overexpression of TGFβ RI or RII resulted in down-regulation of PRL-3 expression as compared to control cells (Fig. 1B, right panel, Fig. S2C).
Figure 1. TGFβ signaling inhibits expression of PRL-3 in colon cancer cells.
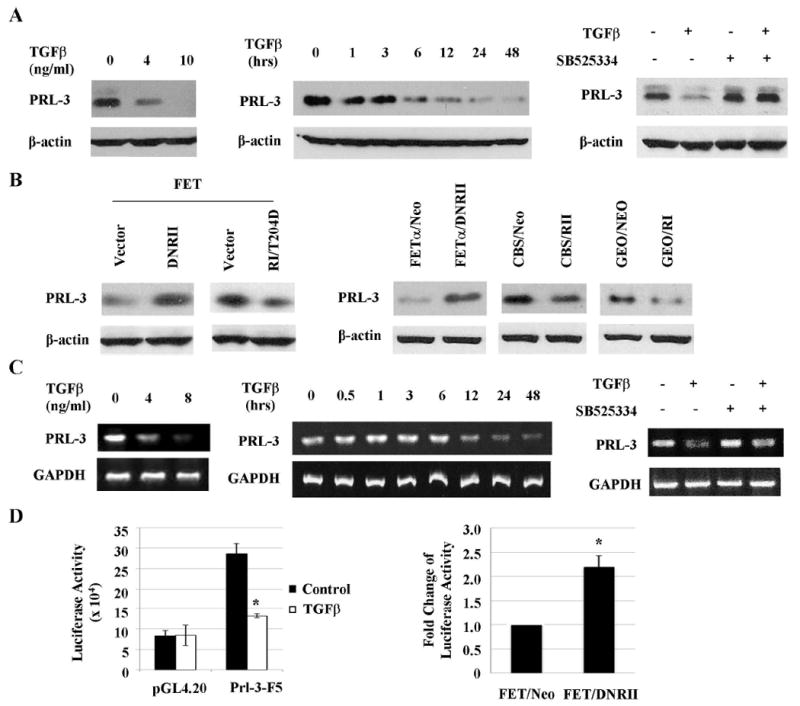
A& C, PRL-3 protein (A) and mRNA (C) expression in FET cells treated with TGFβ for 48 or 24 hrs (left panel), 4 ng/ml TGFβ1 for the indicated time periods (middle panel) or 4 ng/ml TGFβ with or without 200 nM SB525334 for 48 or 24 hrs (right panel). Quantitation of PRL-3 from western blots and RT-PCR analyses is presented in Supplemental Figures S2A, S2B, S3A and S3B. B, PRL-3 expression in FET cells transfected with a DNRII or a constitutively active RI (RI/T204D, left panel) and in FETα, CBS and GEO cells transfected with DNRII, RII or RI respectively (right panel). Quantitation of PRL-3 from western blots analyses is presented in Supplemental Figure S2C. D, PRL-3 Promoter activity in FET cells treated with 4 ng/ml of TGFβ1 for 24 hr (left panel) and in FET Neo and FET DNRII cells (right panel). Firefly luciferase values were normalized to β-galactosidase activity. The data are presented as the mean ± SD of triplicate experiments. *P < 0.03.
We next determined whether TGFβ signaling regulated expression of PRL-3 at the transcriptional level. RT-PCR analyses showed that TGFβ repressed the expression of PRL-3 mRNA in a concentration- and time-dependent manner in FET cells (Fig.1C, left and middle panels, Fig. S3) and that the RI kinase inhibitor SB525334 reversed the inhibitory effect of TGFβ (Fig. 1C, right panel). Of note, levels of PRL-3 mRNA did not decrease until 12 hours after TGFβ treatment (Fig. 1C, middle panel). However, levels of PRL-3 protein decreased significantly as soon as 6 hours after addition of TGFβ (Fig. 1A, middle panel), suggesting that, in addition to regulating PRL-3 transcription, TGFβ may also affect expression of PRL-3 at a post-translational level. Further studies will be necessary to address this possibility and the underlined mechanisms.
To determine how TGFβ signaling might regulate the promoter activity of PRL-3, a reporter plasmid of PRL-3 promoter (Prl-3-F5) was transfected into FET cells followed by treatment of TGFβ. Luciferase assays showed that TGFβ inhibited the activity of this PRL-3 promoter by approximately 55% (Fig. 1D, left panel). The moderate reduction of PRL-3 promoter activity by exogenous TGFβ suggests the existence of strong endogenous TGFβ activity in FET cells. To demonstrate that endogenous TGFβ suppresses the promoter activity of PRL-3, a dominant negative mutant of TGFβ RII was transfected into FET cells (FET DNRII) to inactivate autocrine TGFβ signaling. There was a 2.2-fold increase of PRL-3 promoter activity in FET DNRII cells as compared to FET Neo control cells (Fig. 1D, right panel). Taken together, these results indicate that TGFβ signaling down-regulates expression of PRL-by inhibiting its promoter activity.
Regulation of PRL-3 expression by TGFβ is Smad3-dependent and Smad2-independent
TGFβ signaling is mediated predominantly through Smads. However, Smad-independent TGFβ signaling has also been reported in different cell types (27;28). To determine whether TGFβ-mediated inhibition of PRL-3 expression is Smad-dependent or Smad-independent, Smad2 and Smad3 were knocked down individually or simultaneously in FET cells by shRNAs specific for Smad2 or Smad3. Expression of Smad2 and/or Smad3 was reduced efficiently and specifically in Smad2, Smad3 or Smad2/Smad3 knockdown cells whereas Smad4 expression was not affected (Fig. 2A). Western blot (Fig. 2B, Fig. S4A), RT-PCR (Fig. 2C, Fig. S4B) and promoter analyses (Fig. 2D) indicated that inhibition of PRL-3 promoter activity, mRNA expression and protein expression by TGFβ were partially reversed in Smad3 or Smad2/Smad3 knockdown cells whereas knockdown of Smad2 alone did not affect TGFβ-mediated inhibition. These results indicate that TGFβ suppresses expression of PRL-3 in a Smad3-dependent and Smad2-independent manner. The partial reversal of TGFβ-mediated suppression in Smad3 or Smad2/Smad3 knockdown cells may be attributed to incomplete silencing of Smad3 expression in those cells or the existence of a Smad3-independent mechanism.
Figure 2. Down-regulation of PRL-3 by TGFβ is Smad3-dependent and Smad2-independent.
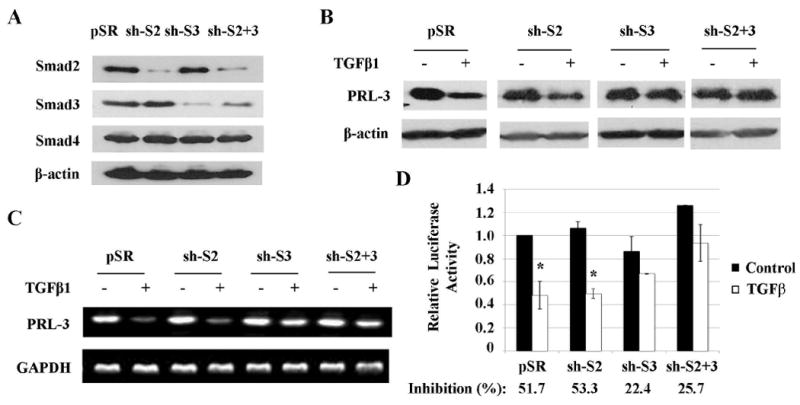
A, Expression of Smad proteins in FET cells with empty vector (pSR), Smad2 shRNA (sh-S2), Smad3 shRNA (sh-S3) or both shRNAs (sh-S2+S3). B&C, PRL-3 protein (B) and mRNA (C) expression in above cells treated with 4 ng/ml of TGFβ. Quantitation of PRL-3 from western blots and RT-PCR analyses is presented in Supplemental Figure S4. D, Relative PRL-3 promoter activity in above cells treated with TGFβ. Inhibition of promoter activity by TGFβ was calculated as percentage of suppression relative to the control. The data are presented as the mean ± SD of triplicate experiments. *P < 0.02.
PRL-3 is a direct target of TGFβ/Smad signaling
Two Smad binding element (SBE)-like sites, E1 and E4, were identified in PRL-3 promoter (Fig. 3A). Smad3 and Smad4 recombinant proteins were tested by an EMSA assay to determine whether they bind to E1 and/or E4. The results showed that GST-Smad3 bound to wild type E1 and E4 oligonucleotides in a concentration-dependent manner (Fig. 3B, left panel). Similarly, His-Smad4 showed strong binding to wild type E1 and E4 (Fig. 3B, right panel). However, both proteins failed to bind to E1 or E4 oligonucleotides carrying mutations in critical nucleotides of SBE consensus sequence (E1mut or E4mut), thus demonstrating the specificity of these binding sites (Fig. 3B). The binding of Smad3 or Smad4 to E1 and E4 was further confirmed in vivo by chromatin immunoprecipitation (CHIP) assays, which revealed that TGFβ increased the binding of Smad3 or Smad4 to E1 and E4 of PRL-3 promoter in FET cells (Fig. 3C). Furthermore, mutations in E1, E4 or both regions of PRL-3 promoter reversed the inhibitory effect of TGFβ on PRL-3 promoter activity (Fig. 3D). These results demonstrate that Smad3/4 directly binds to E1 and E4 of PRL-3 promoter to inhibit its activity.
Figure 3. PRL-3 is a direct target of TGFβ/Smad signaling.
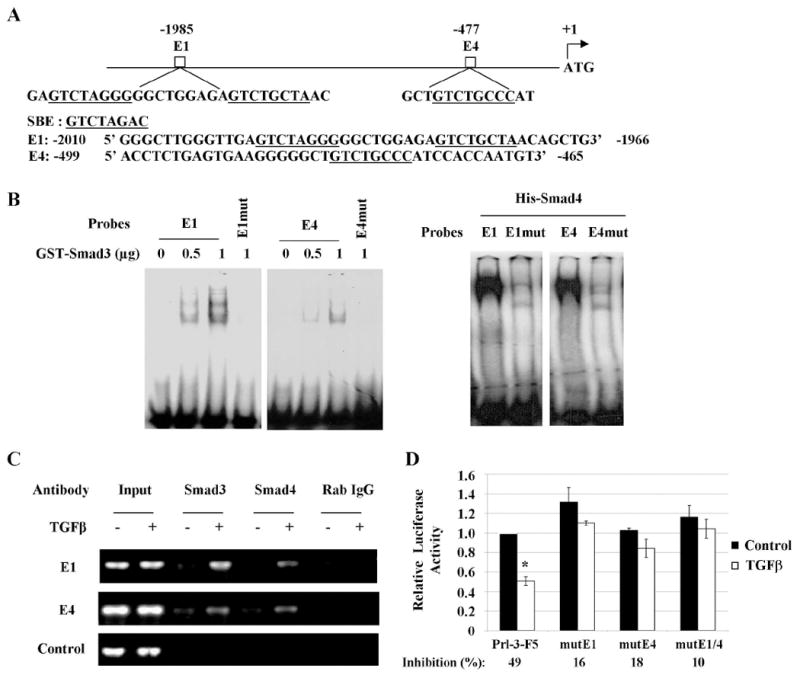
A, Schematic representation of two potential Smad binding sites, E1 and E4, in the PRL-3 promoter. SBE or SBE-like sites are underlined. B, EMSA assays were performed with oligonucleotides containing wild type or mutant E1 or E4 and recombinant proteins GST-Smad3 or His-Smad4. C, CHIP assays were performed as described in Materials and Methods. Rabbit (Rab) IgG was used as a negative control. Input represents 1% of total chromatin used in the assay. D, Relative activity of PRL-3 promoter or promoters containing mutations in E1 and/or E4. Inhibition of promoter activity by TGFβ was calculated as percentage of suppression relative to the control. The data are presented as the mean ± SD of triplicate experiments. *P < 0.02.
PRL-3 promotes cell survival under GFDS by activating the PI3K/AKT pathway
We have identified PRL-3 as a transcriptional target of TGFβ signaling. Because TGFβ has been previously shown to mediate apoptosis under GFDS in colon cancer cells (11), we next determined the role of PRL-3 in cell survival. Wild type PRL-3 and a catalytically inactive PRL-3 mutant (C104S) were stably transduced into FET or GEO cells. Wild type or mutant PRL-3 was abundantly expressed in both cell lines as compared to empty vector-transduced control cells (Fig. 4A, left panel). Under GFDS, ectopic expression of wild type PRL-3 in both FET and GEO cells considerably decreased apoptosis (reflected by reduced levels of caspase 3 cleavage) as compared to control cells whereas mutant PRL-3 showed little effect (Fig. 4A, right panel, Figs. S5A & S5B). In addition, wild type but not mutant PRL-3 protected FET cells from TGFβ-induced apoptosis (Fig. 4A, right panel, Fig. S5A). These observations were further confirmed by DNA fragmentation ELISA assays (Fig. 4B). Taken together, these results indicate that TGFβ suppresses cell survival in part by down-regulation of PRL-3 expression. To determine whether PRL-3 promotes cell survival under other stress conditions besides GFDS, FET cells were exposed to UVC treatment. After UV radiation, wild type PRL-3-expressing cells (PRL-3WT) displayed much lower activation of caspase 3 than vector- or mutant PRL-3-expressing cells (PRL-3C104S; Fig. S6), indicating that PRL-3 also protects FET cells from UV-induced apoptosis.
Figure 4. PRL-3 promotes cell survival under GFDS through activating the PI3K/AKT pathway.
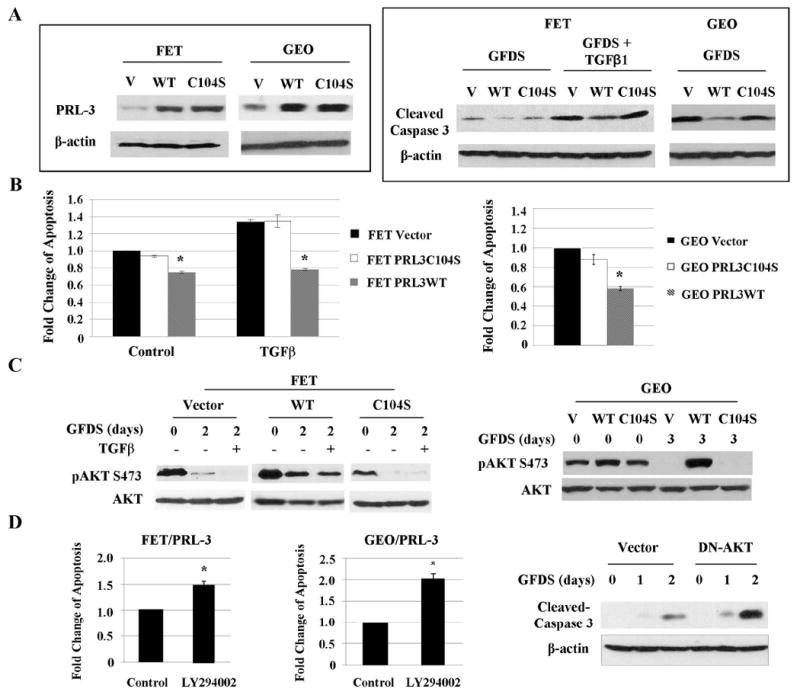
Wild type or catalytic inactive mutant of PRL-3 was stably transduced into FET and GEO cells. A, PRL-3 expression in above-mentioned cells (left panel) and levels of cleaved caspase 3 in FET cells under GFDS with TGFβ for 2 days and in GEO cells under GFDS for 3 days (right panel). Quantitation of cleaved caspase 3 from western blots analyses is presented in Supplemental Figures S5A and S5B. B, DNA fragmentation assays in FET and GEO cells subjected to GFDS as described above. The data are presented as the mean ± SD of triplicate experiments. *P < 0.02. C, AKT expression and phosphorylation in FET cells under GFDS with TGFβ for 2 days (left panel) and GEO cells under GFDS for 3 days (right panel). Quantitation of AKT phosphorylation from western blots analyses is presented in Supplemental Figures S7A and S7B. D, DNA fragmentation assays in FET/PRL-3 and GEO/PRL-3 cells treated with LY294002 (25 nM) for 2 or 3 days respectively. The data are presented as the mean ± SD of triplicate experiments. *P < 0.01 (left and middle panels). Levels of cleaved caspase 3 were determined in FET/PRL-3 cells transfected with a DN-AKT (right panel). Quantitation of cleaved caspase 3 from western blots analyses is presented in Supplemental Figure S5C.
Because PI3K/AKT is a major survival pathway in colon cancer cells, we next determined the effect of PRL-3 expression on AKT activation. Phosphorylation of AKT at Thr308 and Ser473 leads to activation of its kinase activity (29). As shown in Figure 4C, FET and GEO cells expressing wild type PRL-3 displayed higher levels of AKT phosphorylation at Ser473 than those expressing vector or mutant PRL-3 under normal culture conditions. However, phosphorylation of AKT at Thr308 remained unchanged in all cell types (data not shown). In addition, AKT Ser473 phosphorylation was maintained at a high level in FET and GEO cells expressing wild type PRL-3 under GFDS, whereas it decayed rapidly in vector- or mutant PRL-3- expressing cells (Fig. 4C, Figs. S7A & S7B). Moreover, wild type but not mutant PRL-3 antagonized TGFβ-induced decrease of AKT phosphorylation in FET cells under GFDS (Fig. 4C, left panel). These results indicate that PRL-3 promotes AKT signaling under stress in colon cancer cells, suggesting that PRL-3 may increase cell survival via activation of the PI3K/AKT pathway.
Functionally, targeting PI3K/AKT with a potent PI3K inhibitor, LY294002, reduced survival of FET and GEO cells expressing wild type PRL-3 (designated FET/PRL-3 and GEO/PRL-3 respectively) under GFDS (Fig. 4D, left and middle panels). This effect correlated with reduced phosphorylation of AKT (data not shown). In addition, transfection of a dominant negative mutant of AKT (AKT/K179D) into FET/PRL-3 cells increased caspase 3 activation under GFDS (Fig. 4D, right panel, Fig. S5C), consistent with the results obtained from LY294002 treatment.
Knockdown of PRL-3 sensitizes colon cancer cells to GFDS-induced apoptosis
To further define the role of PRL-3 in cell survival, we used an RNA interference approach to knock down expression of PRL-3. Stable transfection of a PRL-3 shRNA vector into FET and GEO cells led to a significant reduction of PRL-3 expression in these cells (Fig. 5A). As a result, there was increased apoptosis in PRL-3 knockdown cells (shPRL3) relative to control cells (pSR) under GFDS (Fig. 5B, Fig. S5D). These results were confirmed by DNA fragmentation assays (Fig. 5C). These data demonstrate that down-regulation of PRL-3 expression sensitizes colon cancer cells to GFDS-induced apoptosis. In addition, knockdown of PRL-3 expression accelerated decay of AKT phosphorylation under GFDS (Fig. 5B, Figs. S7C & S7D), providing further evidence that PRL-3 regulates AKT signaling.
Figure 5. Knockdown of PRL-3 expression inhibits AKT activation and sensitizes colon cancer cells to GFDS-induced apoptosis.
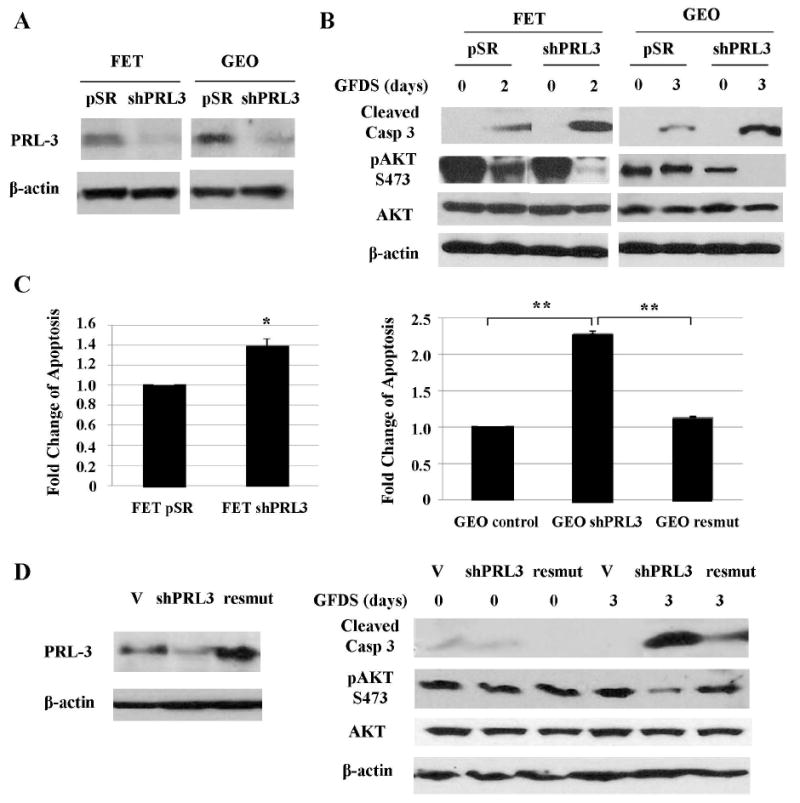
A, PRL-3 expression was significantly reduced in PRL-3 shRNA-expressing cells (shPRL3). B & C, Cells were under GFDS for 2 or 3 days. AKT expression, phosphorylation and caspase 3 cleavage were determined (B). Quantitation of cleaved caspase 3 and AKT phosphorylation from western blots analyses is presented in Supplemental Figures S5D, S7C and S7D. DNA fragmentation assays were performed. The data are presented as the mean ± SD of triplicate experiments. *P < 0.04; **P < 0.001 (C). D, Transfection of a PRL-3 cDNA harboring silent mutations (resmut) into GEO shPRL3 cells restored PRL-3 expression (left panel). Cells were under GFDS for 3 days. AKT expression, phosphorylation and caspase 3 cleavage were determined (right panel).
To test the specificity of PRL-3 shRNA, we ectopically expressed a mutant PRL-3 cDNA in GEO shPRL3 cells (designated GEO resmut) to determine whether it could rescue the phenotypes caused by knockdown of PRL-3 expression. The mutant PRL-3 cDNA harbored a silent mutation in the region targeted by the PRL-3 shRNA so that it is resistant to silencing. Figure 5D showed that PRL-3 expression was restored in GEO resmut cells as compared to GEO shPRL3 cells (left panel). As a result, GEO resmut cells displayed a higher level of AKT phosphorylation under GFDS and were more resistant to GFDS induced apoptosis than GEO shPRL3 cells (Fig. 5D, right panel). DNA fragmentation assays confirmed that re-expression of PRL-3 in GEO shPRL-3 cells decreased GFDS-induced apoptosis to the level that is similar to that of GEO control cells (Fig. 5C, right panel). Taken together, these results indicate that PRL-3 activates PI3K/AKT survival signaling and enhances stress resistance in colon cancer cells.
PRL-3 mediates metastasis of colon cancer cells in an orthotopic model
Since cell survival under stress is an important determinant of metastasis (30;31;32), we next used an orthotopic model to determine the effect of PRL-3 expression on metastasis of GEO cells. PRL-3 knockdown cells (shRPL3), PRL-3-expressing cells (PRL-3), or control cells were stably transfected with GFP and characterized in the orthotopic model as described previously (26).
In vivo studies showed that animals implanted with xenografts formed by GEO control, GEO shPRL3 or GEO PRL-3 cells demonstrated 100% primary growth at the site of implantation with clear invasion of the bowel on histological evaluation (Fig. 6A & 6C). In addition, sizes of primary tumors were very similar in animals bearing different types of cells (Fig. 6A). However, compared to control cells, orthotopic implantation of GEO PRL-3 cells gave rise to significantly increased metastatic localization to the liver, whereas implantation of GEO shRPL3 cells in the same model resulted in markedly diminished liver metastasis (Fig. 6B). Moreover, fluorescence imaging of explanted liver showed a remarkable increase in lesion size of liver metastases in the animals implanted with GEO PRL-3 cells relative to control animals (Fig. 6B). The livers of each animal were serially sectioned at 1 mm intervals and the presence or absence of metastatic disease was confirmed by microscopic histological analysis (Fig. 6C). These results indicate that PRL-3 is necessary and sufficient for metastatic colonization in the liver. To determine whether PRL-3-mediated cell survival was associated with metastatic potential in vivo, TUNEL assays were performed. TUNEL staining of primary tumors showed that, as compared to control cells, there were considerably increased numbers of apoptotic cells in the tumors of GEO shPRL3 cells as opposed to decreased numbers of apoptotic cells in those of GEO PRL-3 cells (Figure 6D). These data are fully consistent with cell survival as a key factor in determining metastasis. Taken together, these in vivo results demonstrate an important role for PRL-3 in distant colony formation, indicating a strong potential for treatment of patients with colon cancer metastases by PRL-3 antagonism.
Figure 6. PRL-3 mediates metastasis of colon cancer cells in an orthotopic model.
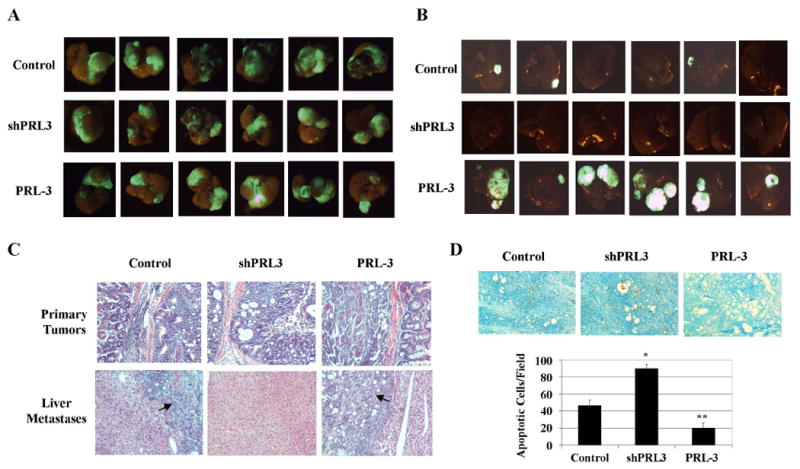
A, GFP images of primary tumors. B, GFP images of liver metastasis. C, H&E stain of primary tumors and liver metastases. Arrows indicate liver metastases. D, TUNEL stain of primary tumors. TUNEL images were captured at 10x magnification. The panels are representatives of multiple fields of tumor sections from at least 6 tumors per group. Numbers of apoptotic cells were determined as described in Materials and Methods. The data are presented as the mean ± SD. *P < 0.001; **P < 0.005.
DISCUSSION
Metastasis has been associated with resistance to stress-induced apoptosis that can be attributed to the aberrant survival capacity of cancer cells (30;31;32). Therefore, inducing cell death has become an important therapeutic strategy to treat metastasis. PRL-3 mediates cell proliferation, migration, invasion, and metastasis in different cell models (16;33-35). However, the role of PRL-3 in cell survival has not been studied. We have demonstrated in this study that, although PRL-3 has little effect on cell proliferation and motility in FET and GEO cells (Figs. S8 & S9), it increases cell survival capacity under stress. When tested in an in vivo orthotopic model that recapitulates the pattern of colorectal cancer metastases in humans, PRL-3 is necessary and sufficient for metastatic colonization. Previously, increased metastasis by PRL-3 has been attributed to its effect on promoting cell motility in experimental metastasis models (16;21). However, PRL-3 has little effect on cell motility in our cell model (Fig. S9). In addition, the orthotopic assays show 100% invasion in primary tumors of control, shPRL-3 and PRL-3 cells (Fig. 6C), despite differences in their metastatic potential. Taken together with the results of TUNEL staining in Figure 6D, our studies indicate that enhanced cell survival conferred by PRL-3 is partially responsible for increased metastasis of colon cancer cells in vivo.
PRL-1 and PRL-2 are two additional members of PRL family (12). Although PRL-1 has been shown to play similar roles as PRL-3 in cell proliferation, migration and invasion (36;37), little is known about the function of PRL-2. Our studies show that knockdown of PRL-1 expression in GEO cells has little effect on cell survival under GFDS (Fig. S10), indicating that PRL-1 is likely not involved in protecting these cells from stress-induced apoptosis. These observations suggest that PRL-1 and PRL-3 have non-redundant functions and that the unique role of PRL-3 in cell survival might account for how its elevated expression correlates with metastatic disease.
High frequency of Smad4 mutation and inactivation is closely associated with increased metastases and poor prognosis in colon cancer (38;39). Although TGFβ signaling acts as a tumor suppressor in FET and GEO cells that express wild type Smad4 (2;40), Smad4-independent TGFβ signaling has been shown to promote colon cancer metastasis (41). To determine the effects of TGFβ on PRL-3 expression in Smad4-null cells, we treated SW480 cells with TGFβ and found that TGFβ increased PRL-3 mRNA and protein expression (Fig. S11). This indicates that TGFβ-mediated activation of Smad4-independent pathways is involved in the up-regulation of PRL-3, which may cooperate with other pro-oncogenic pathways to promote metastasis. Further studies will be necessary to determine the mechanisms by which Smad4-independent TGFβ signaling regulates PRL-3 expression.
We showed that wild type but not catalytically inactive PRL-3 mediates AKT activation and maintenance under GFDS (Fig. 4C). We do not know yet the mechanisms by which PRL-3 activates AKT signaling. However, a number of kinases including ILK, PKCα and mTORC2 have been shown to directly phosphorylate AKT at S473 (42-44), the activating phosphorylation site most notably regulated by PRL-3. We are currently investigating whether PRL-3 affects their activity in colon cancer cells. In addition, several proteins have been proposed to be the substrates of PRL-3 including ezrin and paxillin (45;46). However, ectopic expression of PRL-3 did not affect phosphorylation of these proteins in our cell model (data not shown).
In summary, we have identified PRL-3 as a direct target of TGFβ signaling. Loss of TGFβ signaling leads to up-regulation of PRL-3 expression, which is likely a mechanism of increased PRL-3 expression in colon cancer. Up-regulation of PRL-3 expression leads to activation of the PI3K/AKT axis, enhanced survival and resistance to stress-induced apoptosis and increased metastatic potential. Our studies suggest that PRL-3 may be a promising target for cancer treatment, especially in patients with defective TGFβ signaling, which occurs in 30-50% of colon cancer patients (22;23). This is highly important considering that TGFβ signaling may actually promote metastasis at a late stage of carcinogenesis (6), which makes restoring TGFβ signaling complicated or impossible as a therapeutic strategy.
Supplementary Material
Acknowledgments
This work was supported by NIH P20RR018759 and R01CA140988-01.
Reference List
- 1.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 2.Wang J, Han W, Zborowska E, et al. Reduced expression of transforming growth factor beta type I receptor contributes to the malignancy of human colon carcinoma cells. J Biol Chem. 1996;271:17366–71. doi: 10.1074/jbc.271.29.17366. [DOI] [PubMed] [Google Scholar]
- 3.Ye SC, Foster JM, Li W, et al. Contextual effects of transforming growth factor beta on the tumorigenicity of human colon carcinoma cells. Cancer Res. 1999;59:4725–31. [PubMed] [Google Scholar]
- 4.Wang J, Sun L, Myeroff L, et al. Demonstration that mutation of the type II transforming growth factor beta receptor inactivates its tumor suppressor activity in replication error-positive colon carcinoma cells. J Biol Chem. 1995;270:22044–9. doi: 10.1074/jbc.270.37.22044. [DOI] [PubMed] [Google Scholar]
- 5.Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–8. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 6.Wakefield LM, Roberts AB. TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002;12:22–9. doi: 10.1016/s0959-437x(01)00259-3. [DOI] [PubMed] [Google Scholar]
- 7.Forrester E, Chytil A, Bierie B, et al. Effect of conditional knockout of the type II TGF-beta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res. 2005;65:2296–302. doi: 10.1158/0008-5472.CAN-04-3272. [DOI] [PubMed] [Google Scholar]
- 8.Yang L, Huang J, Ren X, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veenendaal LM, Kranenburg O, Smakman N, Klomp A, Borel R, I, van Diest PJ. Differential Notch and TGFbeta signaling in primary colorectal tumors and their corresponding metastases. Cell Oncol. 2008;30:1–11. doi: 10.1155/2008/839076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bacman D, Merkel S, Croner R, Papadopoulos T, Brueckl W, Dimmler A. TGF-beta receptor 2 downregulation in tumour-associated stroma worsens prognosis and high-grade tumours show more tumour-associated macrophages and lower TGF-beta1 expression in colon carcinoma: a retrospective study. BMC Cancer. 2007;7:156. doi: 10.1186/1471-2407-7-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Yang L, Yang J, et al. Transforming growth factor beta induces apoptosis through repressing the phosphoinositide 3-kinase/AKT/survivin pathway in colon cancer cells. Cancer Res. 2008;68:3152–60. doi: 10.1158/0008-5472.CAN-07-5348. [DOI] [PubMed] [Google Scholar]
- 12.Bessette DC, Qiu D, Pallen CJ. PRL PTPs: mediators and markers of cancer progression. Cancer Metastasis Rev. 2008;27:231–52. doi: 10.1007/s10555-008-9121-3. [DOI] [PubMed] [Google Scholar]
- 13.Matter WF, Estridge T, Zhang C, et al. Role of PRL-3, a human muscle-specific tyrosine phosphatase, in angiotensin-II signaling. Biochem Biophys Res Commun. 2001;283:1061–8. doi: 10.1006/bbrc.2001.4881. [DOI] [PubMed] [Google Scholar]
- 14.Bardelli A, Saha S, Sager JA, Romans KE, Xin B, Markowitz SD, Lengauer C, Velculescu VE, Kinzler KW, Vogelstein B. PRL-3 expression in metastatic cancers. Clin Cancer Res. 2003:5607–15. [PubMed] [Google Scholar]
- 15.Saha S, Bardelli A, Buckhaults P, et al. A phosphatase associated with metastasis of colorectal cancer. Science. 2001;294:1343–6. doi: 10.1126/science.1065817. [DOI] [PubMed] [Google Scholar]
- 16.Kato H, Semba S, Miskad UA, Seo Y, Kasuga M, Yokozaki H. High expression of PRL-3 promotes cancer cell motility and liver metastasis in human colorectal cancer: a predictive molecular marker of metachronous liver and lung metastases. Clin Cancer Res. 2004;10:7318–28. doi: 10.1158/1078-0432.CCR-04-0485. [DOI] [PubMed] [Google Scholar]
- 17.Mollevi DG, Aytes A, Padulles L, et al. PRL-3 is essentially overexpressed in primary colorectal tumours and associates with tumour aggressiveness. Br J Cancer. 2008;99:1718–25. doi: 10.1038/sj.bjc.6604747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng L, Ning J, Meng L, Shou C. The association of the expression level of protein tyrosine phosphatase PRL-3 protein with liver metastasis and prognosis of patients with colorectal cancer. J Cancer Res Clin Oncol. 2004;130:521–6. doi: 10.1007/s00432-004-0563-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Li ZF, He J, et al. Expression of the human phosphatases of regenerating liver (PRLs) in colonic adenocarcinoma and its correlation with lymph node metastasis. Int J Colorectal Dis. 2007;22:1179–84. doi: 10.1007/s00384-007-0303-1. [DOI] [PubMed] [Google Scholar]
- 20.Guo K, Li J, Tang JP, Koh V, Gan BQ, Zeng Q. Catalytic domain of PRL-3 plays an essential role in tumor metastasis: formation of PRL-3 tumors inside the blood vessels. Cancer Biol Ther. 2004;3:945–51. doi: 10.4161/cbt.3.10.1111. [DOI] [PubMed] [Google Scholar]
- 21.Wu X, Zeng H, Zhang X, et al. Phosphatase of regenerating liver-3 promotes motility and metastasis of mouse melanoma cells. Am J Pathol. 2004;164:2039–54. doi: 10.1016/S0002-9440(10)63763-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salovaara R, Roth S, Loukola A, et al. Frequent loss of SMAD4/DPC4 protein in colorectal cancers. Gut. 2002;51:56–9. doi: 10.1136/gut.51.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grady WM, Myeroff LL, Swinler SE, et al. Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res. 1999;59:320–4. [PubMed] [Google Scholar]
- 24.Boyd D, Florent G, Kim P, Brattain M. Determination of the levels of urokinase and its receptor in human colon carcinoma cell lines. Cancer Res. 1988;48:3112–6. [PubMed] [Google Scholar]
- 25.Li J, Guo K, Koh VW, et al. Generation of PRL-3- and PRL-1-specific monoclonal antibodies as potential diagnostic markers for cancer metastases. Clin Cancer Res. 2005;11:2195–204. doi: 10.1158/1078-0432.CCR-04-1984. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Rajput A, Kan JL, et al. Knockdown of Ron kinase inhibits mutant PI3 kinase and reduces metastasis in human colon carcinoma. J Biol Chem. 2009;284:10912–22. doi: 10.1074/jbc.M809551200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu L, Hebert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002;21:3749–59. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu B, Zhai J, Zhu H, Kyprianou N. Prohibitin regulates TGF-beta induced apoptosis as a downstream effector of Smad-dependent and -independent signaling. Prostate. 2010;70:17–26. doi: 10.1002/pros.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alessi DR, Andjelkovic M, Caudwell B, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–51. [PMC free article] [PubMed] [Google Scholar]
- 30.Inbal B, Cohen O, Polak-Charcon S, et al. DAP kinase links the control of apoptosis to metastasis. Nature. 1997;390:180–4. doi: 10.1038/36599. [DOI] [PubMed] [Google Scholar]
- 31.Mehrotra S, Languino LR, Raskett CM, Mercurio AM, Dohi T, Altieri DC. IAP regulation of metastasis. Cancer Cell. 2010;17:53–64. doi: 10.1016/j.ccr.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–26. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 33.Li Z, Zhan W, Wang Z, et al. Inhibition of PRL-3 gene expression in gastric cancer cell line SGC7901 via microRNA suppressed reduces peritoneal metastasis. Biochem Biophys Res Commun. 2006;348:229–37. doi: 10.1016/j.bbrc.2006.07.043. [DOI] [PubMed] [Google Scholar]
- 34.Liang F, Liang J, Wang WQ, Sun JP, Udho E, Zhang ZY. PRL3 promotes cell invasion and proliferation by down-regulation of Csk leading to Src activation. J Biol Chem. 2007;282:5413–9. doi: 10.1074/jbc.M608940200. [DOI] [PubMed] [Google Scholar]
- 35.Polato F, Codegoni A, Fruscio R, et al. PRL-3 phosphatase is implicated in ovarian cancer growth. Clin Cancer Res. 2005;11:6835–9. doi: 10.1158/1078-0432.CCR-04-2357. [DOI] [PubMed] [Google Scholar]
- 36.Achiwa H, Lazo JS. PRL-1 tyrosine phosphatase regulates c-Src levels, adherence, and invasion in human lung cancer cells. Cancer Res. 2007;67:643–50. doi: 10.1158/0008-5472.CAN-06-2436. [DOI] [PubMed] [Google Scholar]
- 37.Sun JP, Luo Y, Yu X, et al. Phosphatase activity, trimerization, and the C-terminal polybasic region are all required for PRL1-mediated cell growth and migration. J Biol Chem. 2007;282:29043–51. doi: 10.1074/jbc.M703537200. [DOI] [PubMed] [Google Scholar]
- 38.Losi L, Bouzourene H, Benhattar J. Loss of Smad4 expression predicts liver metastasis in human colorectal cancer. Oncol Rep. 2007;17:1095–9. [PubMed] [Google Scholar]
- 39.Miyaki M, Iijima T, Konishi M, et al. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene. 1999;18:3098–103. doi: 10.1038/sj.onc.1202642. [DOI] [PubMed] [Google Scholar]
- 40.Wu SP, Theodorescu D, Kerbel RS, et al. TGF-beta 1 is an autocrine-negative growth regulator of human colon carcinoma FET cells in vivo as revealed by transfection of an antisense expression vector. J Cell Biol. 1992;116:187–96. doi: 10.1083/jcb.116.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang B, Halder SK, Kashikar ND, et al. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology. 2010;138:969–80. doi: 10.1053/j.gastro.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA. 1998;95:11211–6. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Partovian C, Simons M. Regulation of protein kinase B/Akt activity and Ser473 phosphorylation by protein kinase Calpha in endothelial cells. Cell Signal. 2004;16:951–7. doi: 10.1016/j.cellsig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 44.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 45.Forte E, Orsatti L, Talamo F, Barbato G, De Francesco R, Tomei L. Ezrin is a specific and direct target of protein tyrosine phosphatase PRL-3. Biochim Biophys Acta. 2008;1783:334–44. doi: 10.1016/j.bbamcr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 46.Wang H, Quah SY, Dong JM, Manser E, Tang JP, Zeng Q. PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 2007;67:2922–6. doi: 10.1158/0008-5472.CAN-06-3598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.