

Abstract
We studied the selectivity of a functional model of cytochrome c oxidase's active site that mimics the coordination environment and relative locations of Fea3, CuB, and Tyr244. To control electron flux, we covalently attached this model and analogs lacking copper and phenol onto self-assembled monolayer–coated gold electrodes. When the electron transfer rate was made rate limiting, both copper and phenol were required to enhance selective reduction of oxygen to water. This finding supports the hypothesis that, during steady-state turnover, the primary role of these redox centers is to rapidly provide all the electrons needed to reduce oxygen by four electrons, thus preventing the release of toxic partially reduced oxygen species.
During the final stage of respiration, cytochrome c oxidase (CcO) catalyzes the reduction of O2 to H2O to power its proton pump activity. This four-electron reduction must take place without releasing toxic partially reduced oxygen species (PROS).
Although CcO has been extensively studied in single-turnover experiments, the mechanism by which the enzyme reduces O2 under steady-state conditions, where the electron flux is rate limiting, is poorly understood (1, 2). Specifically, the role of CuB and a posttranslationally modified tyrosine residue (Tyr244) have been frequent subjects of discussion. It has been assumed that, during turnover, CuB and Tyr244 each deliver an electron to bound oxygen in the active site, in addition to the two electrons from the Fe in hemea3. This rapid intramolecular reduction in a hydrophobic site should reduce the lifetimes of potential PROS-releasing intermediates (3). The oxidized active site is then thought to be slowly recharged by ferrous cytochrome c (cyt c) (4, 5) such that O2 only binds when both CuB and Fea3 have been reduced.
This scenario has proved challenging to test in the native enzyme, because of its inherent complexity, and in site-specific mutants, because they have been found to be inactive (6–8). We previously developed functional models of the CcO active site (9–12) and showed that a model that contains two redox sites, an iron heme, and a distal copper, but lacking a phenol to mimic Tyr244 can manifest the selective four-electron catalytic reduction of oxygen at physiological pH and potential. However, our catalysts were adsorbed on graphite electrodes such that electron transfer was very rapid; in contrast to the enzyme, in which electrons arrive slowly from cyt c. We describe the steady-state catalytic reduction of O2 by using synthetic CcO models that also mimic the Tyr244 site and are attached to self-assembled monolayer (SAM) films on gold electrodes in order to control electron flux. This strategy enabled us to examine the influence of each redox center (copper and phenol), while varying the rates of electron delivery and simultaneously measuring the formation of PROS electrochemically. We find that, under conditions where electron delivery limits the turnover rate, the presence of copper and a redox-active phenol to mimic tyrosine sharply reduces the formation of PROS. These model studies support the belief that both CuB and Tyr244 are required to selectively reduce oxygen by four electrons under the physiological condition of rate-limiting electron transfer from cyt c.
Our new structural and functional analog of the CcO active site (catalyst 1a) possesses groups that mimic Fea3, CuB, and Tyr244 (Fig. 1) (13). A myoglobin-like heme was fitted with a proximal imidazole, a distal copper was bound to a trisimidazole ligand about 5 Å above the heme, and a phenol group was covalently attached to one of the copper-ligating imidazoles and directed toward the oxygen binding site on the heme (14). In single-turnover experiments, this fully reduced synthetic model, containing both copper and phenol, activated and reduced oxygen by four electrons in a manner similar to that of the mixed-valence form of the native enzyme. The oxidized product of this reaction contains a ferryl and a phenol radical. Its electron spin resonance (ESR) spectrum is shown in Fig. 2.
Fig. 1.

(A) Crystal structure of the active site of CcO from the bovine heart (13). (B) Model 1a reproduces the key elements of the active site of CcO. (C) Model 2a, in which the phenol is masked as a methyl ether. Model 2a can be treated with dilute acid to yield the iron-only model (2b), which is not shown.
Fig. 2.

ESR spectrum of the product formed by O2 reacting with the fully reduced catalyst 1a.
In order to control electron flux to these model compounds, we attached this model and analogs lacking copper and phenol onto mixed SAM–coated gold electrodes (15–19). Electron-transfer rates to redox groups covalently coupled to SAMs can be predictably tuned by varying the length and the degree of conjugation of the SAM (20–22). Although most functional monolayers are made by simply tethering a thiol to the redox molecule of interest, the complexity of our synthetic CcO models and the reactivity of the heme center with free thiols required an alternative method. Recently we demonstrated that azide-terminated mixed SAMs can be modified with acetylene-bearing molecules (23–25) through a Cu(I)-catalyzed azide-alkyne cyclo-addition (26, 27). This very efficient technique yields mixed SAMs that deliver electrons at reproducible rates to covalently attached redox molecules, such as porphyrins (28).
The synthetic CcO models were fitted with a terminal acetylene and then covalently attached to azide-terminated mixed SAMs on electrode surfaces (Fig. 3) (29). The electron delivery pathway is analogous to that in CcO, in which an outer-sphere electron donor (Fea in native CcO and the electrode in our model system) is covalently coupled to the proximal imidazole of an oxygen-binding heme (13).
Fig. 3.

Slow SAM S1 functionalized with model 1a (left) and fast SAM S2 functionalized with model 1a (right).
In order to mimic a situation where electron transfer to the heme is the rate-limiting step during catalysis, we covalently attached these CcO models onto mixed SAMs composed of 1-azidohexadecanethiol and hexadecanethiol (“slow” SAM S1). Such SAMs strongly retard the rate of electron transfer to immobilized redox molecules (28). For comparison, we also attached CcO models to a highly conjugated azide-terminated SAM composed of azidophenylene-ethynylenebenzyl thiol and octanethiol (“fast” SAM S2). SAM S2 is known to facilitate rapid electron transfer from the electrode to immobilized redox molecules (28).
SAMs functionalized with CcO models were characterized by using conventional electrochemical techniques (29). The standard electron-transfer rate constant, k0, between the gold electrode and the iron center of our models was 6 ± 0.1 s−1 on SAM S1. In contrast, k0 on SAM S2 was too fast to measure with conventional electrochemical methods (28) but is at least 1 × 104 s−1. This large variation in k0, controlled by changing the nature of the intervening SAM, allowed us to drastically vary the rate of electron transfer to our CcO models. Catalyst coverage was limited by the azide-terminated thiol to less than 4 × 10−11 moles cm−2, about 5% of the coverage of thiols in the SAMs. At these low coverages, the catalysts do not interact with one another.
In the absence of O2 at pH = 7, the redox potentials of both the heme iron and the distal copper are nearly identical, consistent with the similar potentials of Fea3 and CuB observed in CcO and in our earlier synthetic models (30). In air-saturated aqueous solutions buffered at pH = 7, the reversible couple disappears and is replaced by a large irreversible current caused by O2 reduction (fig. S2). The potential at which catalysis begins is the same on both slow and th fast SAM [0.3 V versus normal hydrogen electrode (NHE)] and is identical to the onset potential observed for O2 reduction by native cyt c/CcO complexes absorbed on gold (31).
Although the SAM does not affect the potential of reduction, it strongly affects the kinetics of catalysis. On SAM S1, the electrochemical process is limited by electron transfer. In contrast, electron delivery to the catalysts immobilized on SAM S2 is sufficiently rapid that the supply of O2 limits the turnover frequency.
In order to measure the selectivity of catalysis under these two rate-limiting regimes (SAM S1 and SAM S2), we used rotating ring-disk voltammetry. There are very few examples reported on the use of rotating ring-disk voltammetry with SAMs (32–34) because of the difficulty of forming reproducible SAMs on such electrodes (18). We have developed a procedure to form gold disk electrodes with the required surface properties to promote high-quality SAM formation (29). A catalyst-modified SAM-coated gold disk electrode is encircled by a PROS-detecting Pt ring electrode. The two-electrode assembly is rotated, and the gold disk is set to a potential at which oxygen reduction takes place. PROS produced during O2 reduction are swept from the disk to the ring, where they are electrochemically detected. For an ideal four-electron electrocatalyst, no PROS would be detected. We measured the percentage of electrons released as PROS for catalysts 1a, 2a, and 2b immobilized on SAM S1 and SAM S2 (Fig. 4).
Fig. 4.
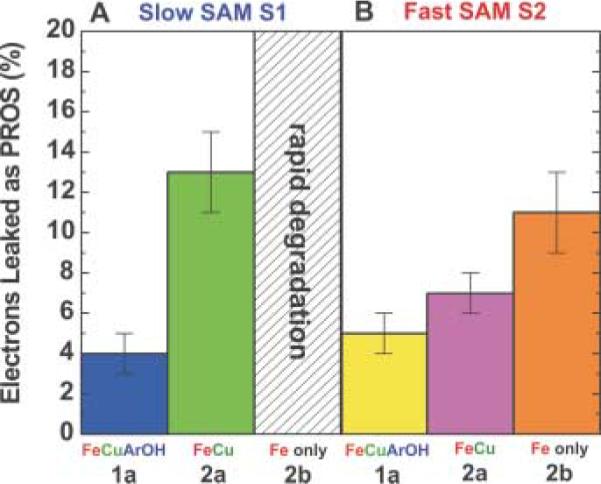
Percentage of electrons released as PROS for catalysts 1a, 2a, and 2b on either (A) SAM S1 or (B) SAM S2. Data taken at 0.1 V versus NHE in air-saturated electrolyte buffered at pH = 7. The catalyst coverage on either SAM was 4 × 10−11 moles cm−2. Ring-disk assembly was rotated at 300 revolutions per minute. Error bars indicate standard deviation from the mean.
When attached to SAM S2, the incomplete CcO model 2b, which lacks both copper and a redox-active phenol, is moderately selective at reducing O2 to H2O; 89 ± 2% of the electrons are used to form H2O. Addition of copper (catalyst 2a) decreases the amount of peroxide detected at the ring by over 30%. Copper is known to increase the oxygen-binding affinity of both synthetic CcO models and CcO itself and to improve the selectivity of CcO models absorbed on edge-plane graphite (6, 30, 35). Interestingly, the copper- and phenol-containing catalyst 1a on SAM S2 shows only a slightly improved selectivity over catalyst 2a, in which the phenol is masked. We believe this limited influence of the redox-active phenol results from the ability of the electrode to deliver an additional electron rapidly through SAM S2. This behavior is reminiscent of fully reduced CcO during single turnover experiments; Fea can rapidly (~30 μs) provide an electron to a partially reduced oxygen intermediate, and a tyrosine radical is not observed (2). This result demonstrates that during turnover Tyr244 is not required when a fourth electron can be rapidly delivered from outside the active site.
During in vivo operation, CcO is never in the fully reduced state because electron delivery by cyt c is far slower than the rate at which internal electrons are delivered to oxygen bound at the active site (36). Thus, studies of CcO models immobilized on SAM S1, where electron transfer limits turnover, have greater physiological relevance. Compared with SAM S2, on SAM S1 models lacking either copper or phenol show a marked decrease in selectivity. For example, the iron-only model 2b on SAM S1 rapidly degrades and is likely consumed by substantial PROS formation. This is unlike the case with similar iron-only porphyrins directly absorbed on edge-plane graphite (30) but is similar to our previous work using lipid films to force slow diffusional delivery of electrons (37). When immobilized on SAM S1, model 2a, which contains both iron and copper but not a redox-active phenol, is more stable than the iron-only model but still shows a marked degree of PROS leakage. This catalyst can store no more than three electrons at the active site before oxygen binding (two from iron and one from copper); therefore, one additional electron would be required from the electrode in order for oxygen to be reduced by four electrons. If the electron transfer rate between electrode and catalyst is slow, partially reduced intermediates must persist before additional electrons are delivered, and in the interim PROS can leak and degrade the catalyst (Fig. 5A).
Fig. 5.

Possible intermediates during the reduction of oxygen to the redox level of water. (A) A monometallic heme, such as catalyst 2b, requires electrons to be delivered externally in order to reduce oxygen by four electrons. If those electrons are delivered slowly, the lifetimes of PROS-releasing intermediates increase. (B) Catalyst 1a equipped with both copper and phenol can bind oxygen and rapidly reduce it to the redox level of water without requiring external electrons. PROS release is minimized.
In contrast, catalyst 1a, equipped with both copper and phenol, is highly selective at reducing oxygen to water on SAM S1 (Fig. 5B). Compared with the catalyst without a redox-active phenol 2a, the complete catalyst 1a releases over threefold less PROS. Remarkably, the selectivity of catalyst 1a is nearly identical whether immobilized on SAM S1 or SAM S2. Thus, 1a mimics one of the most important and fascinating features of CcO: its ability to selectively reduce oxygen by four electrons when the electron-transfer flux is rate limiting.
CcO is believed to reduce oxygen by four electrons with >99% selectivity (36). Catalyst 1a on SAM S1 is only about 96% selective for four-electron reduction (38). We suggest two plausible reasons for PROS formation. First, our Fe-Cu models do not require complete reduction of the metal centers in order to bind O2, whereas in CcO an apparent cooperativity between Fea3 and CuB redox centers ensures that only the fully reduced active site binds O2 (1, 36). In the present model, it is possible to form a partially reduced active site (Fe+2/Cu+2) that would bind O2. This process could lead to occasional production of PROS when electron transfer is rate limiting.
A second disparity is that these active-site models are in direct contact with water, whereas CcO is buried in a membrane. Exposure to a water interface should favor hydrolytic autoxidation, thereby increasing PROS formation (39). To test this hypothesis, we treated the SAM films with a hydrophobic surfactant (29). It has been demonstrated that such surfactant films can act as hydrophobic blocking layers and influence the physical properties of redox molecules within the SAM (40, 41). In preliminary studies, we have found that this treatment reduces the percent of electrons contributing to PROS by almost a factor of 3 (29).
Supplementary Material
Acknowledgments
We dedicate this scientific contribution to the memory of the late H. Taube. We thank J. I. Brauman, T. D. P. Stack, C. C. Cummins, R. Boulatov, and P. H. Dinolfo for helpful discussions. N.K.D., R.A.D., and W.E. acknowledge a Stanford graduate fellowship, a Lavoisier fellowship, and a Dreyfus undergraduate fellowship, respectively. This material has its basis in work supported by the NIH under grant GM-17880-35.
Footnotes
Supporting Online Material www.sciencemag.org/cgi/content/full/315/5818/1565/DC1 Materials and Methods Figs. S1 to S4 References
References and Notes
- 1.Brunori M, Giuffre A, Sarti P. J. Inorg. Biochem. 2005;99:324. doi: 10.1016/j.jinorgbio.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 2.Morgan JE, Verkhovsky MI, Palmer G, Wikstrom M. Biochemistry. 2001;40:6882. doi: 10.1021/bi010246w. [DOI] [PubMed] [Google Scholar]
- 3.Proshlyakov DA, et al. Science. 2000;290:1588. doi: 10.1126/science.290.5496.1588. [DOI] [PubMed] [Google Scholar]
- 4.Chazotte B, Hackenbrock CR. J. Biol. Chem. 1989;264:4978. [PubMed] [Google Scholar]
- 5.Gupte SS, Hackenbrock CR. J. Biol. Chem. 1988;263:5248. [PubMed] [Google Scholar]
- 6.Mogi T, Hirano T, Nakamura H, Anraku Y, Orii Y. FEBS Lett. 1995;370:259. doi: 10.1016/0014-5793(95)00852-z. [DOI] [PubMed] [Google Scholar]
- 7.Das TK, Pecoraro C, Tomson FL, Gennis RB, Rousseau DL. Biochemistry. 1998;37:14471. doi: 10.1021/bi981500w. [DOI] [PubMed] [Google Scholar]
- 8.Pfitzner U, et al. J. Bioenerg. Biomembr. 1998;30:89. doi: 10.1023/a:1020515713103. [DOI] [PubMed] [Google Scholar]
- 9.Collman JP, Fu L, Herrmann PC, Zhang X. Science. 1997;275:949. doi: 10.1126/science.275.5302.949. [DOI] [PubMed] [Google Scholar]
- 10.Collman JP, et al. J. Am. Chem. Soc. 1994;116:9783. [Google Scholar]
- 11.Collman JP, et al. J. Am. Chem. Soc. 1999;121:1387. [Google Scholar]
- 12.Collman JP, Sunderland CJ, Boulatov R. Inorg. Chem. 2002;41:2282. doi: 10.1021/ic011191p. [DOI] [PubMed] [Google Scholar]
- 13.Tsukihara T, et al. Science. 1995;269:1069. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 14.Collman JP, Decréau RA, Zhang C. J. Org. Chem. 2004;69:3546. doi: 10.1021/jo0499625. [DOI] [PubMed] [Google Scholar]
- 15.Alleman KS, Weber K, Creager SE. J. Phys. Chem. 1996;100:17050. [Google Scholar]
- 16.Zou SZ, Clegg RS, Anson FC. Langmuir. 2002;18:3241. [Google Scholar]
- 17.Offord DA, et al. J. Am. Chem. Soc. 1998;120:4478. [Google Scholar]
- 18.Hutchison JE, Postlethwaite TA, Murray RW. Langmuir. 1993;9:3277. [Google Scholar]
- 19.Zak J, Yuan HP, Ho M, Woo LK, Porter MD. Langmuir. 1993;9:2772. [Google Scholar]
- 20.Chidsey CED. Science. 1991;251:919. doi: 10.1126/science.251.4996.919. [DOI] [PubMed] [Google Scholar]
- 21.Creager S, et al. J. Am. Chem. Soc. 1999;121:1059. [Google Scholar]
- 22.Finklea HO, Hanshew DD. J. Am. Chem. Soc. 1992;114:3173. [Google Scholar]
- 23.Collman JP, Devaraj NK, Eberspacher TPA, Chidsey CED. Langmuir. 2006;22:2457. doi: 10.1021/la052947q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collman JP, Devaraj NK, Chidsey CED. Langmuir. 2004;20:1051. doi: 10.1021/la0362977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JK, Chi YS, Choi IS. Langmuir. 2004;20:3844. doi: 10.1021/la049748b. [DOI] [PubMed] [Google Scholar]
- 26.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 27.Tornoe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 28.Devaraj NK, Decreau RA, Ebina W, Collman JP, Chidsey CED. J. Phys. Chem. B. 2006;110:15955. doi: 10.1021/jp057416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Materials and methods are available as supporting material on Science Online.
- 30.Boulatov R, Collman JP, Shiryaeva IM, Sunderland CJ. J. Am. Chem. Soc. 2002;124:11923. doi: 10.1021/ja026179q. [DOI] [PubMed] [Google Scholar]
- 31.Haas AS, et al. J. Phys. Chem. B. 2001;105:11351. [Google Scholar]
- 32.Cha SK. Bull. Korean Chem. Soc. 2004;25:786. [Google Scholar]
- 33.Degefa TH, Schon P, Bongard D, Walder L. J. Electroanal. Chem. 2004;574:49. [Google Scholar]
- 34.Katz E, Schmidt HL. J. Electroanal. Chem. 1994;368:87. [Google Scholar]
- 35.Sigman JA, Kwok BC, Lu Y. J. Am. Chem. Soc. 2000;122:8192. [Google Scholar]
- 36.Babcock GT, Wikstrom M. Nature. 1992;356:301. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 37.Collman JP, Boulatov R. Angew. Chem. Int. Ed. 2002;41:3487. doi: 10.1002/1521-3773(20020916)41:18<3487::AID-ANIE3487>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 38.The peroxide detected at the ring electrode could arise either from direct release of peroxide during the catalytic cycle or from release and rapid disproportionation of superoxide (42). The hydrolytic autoxidation of heme oxygen complexes in aqueous solution is a well-documented phenomenon (39, 43). This process is known to be catalyzed by protons, and preliminary results indicate that the selectivity of all of our catalysts can be markedly improved by raising the pH (to the range of 8 to 12). Furthermore, our prior work with CcO models on edge-plane graphite provided evidence that superoxide is a substantial source of PROS (30).
- 39.Shikama K. Chem. Rev. 1998;98:1357. doi: 10.1021/cr970042e. [DOI] [PubMed] [Google Scholar]
- 40.Creager SE, Rowe GK. Langmuir. 1993;9:2330. [Google Scholar]
- 41.Sigal GB, Mrksich M, Whitesides GM. Langmuir. 1997;13:2749. [Google Scholar]
- 42.Sawyer DT, Valentine JS. Acc. Chem. Res. 1981;14:393. [Google Scholar]
- 43.Collman JP, Yan YL, Eberspacher T, Xie XJ, Solomon EI. Inorg. Chem. 2005;44:9628. doi: 10.1021/ic0516717. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.