

Abstract
Indenoisoquinoline topoisomerase I (Top1) inhibitors are a novel class of anticancer agents. Modifications of the indenoisoquinoline A, B and D rings have been extensively studied in order to optimize Top1 inhibitory activity and cytotoxicity. To improve understanding of the forces that stabilize drug-Top1-DNA ternary complexes, the five-membered cyclopentadienone C-ring of the indenoisoquinoline system was replaced by six-membered nitrogen heterocyclic rings, resulting in dibenzo[c,h][1,6]naphthyridines that were synthesized by a novel route and tested for Top1 inhibition. This resulted in several compounds that have unique DNA cleavage site selectivities and potent antitumor activities in a number of cancer cell lines.
Introduction
Cells of all living organisms possess topoisomerases to resolve the topological problems associated with DNA supercoiling during various cellular processes (e.g. replication, transcription, repair).1 There are two major families of topoisomerases: type I and type II. Topoisomerase type I (Top1) relaxes both positively and negatively supercoiled DNA via reversible single-strand nicks.2 Top1 forms a covalent link with the 3′-oxygen atom of DNA.3 The free 5′-end is then allowed to rotate about the intact strand, thus relieving tension. Once supercoils are removed, the broken DNA strand is religated and the Top1 released.4 The importance of Top1 for DNA replication and cell division has made it an attractive drug target for anticancer therapy.5, 6 Camptothecin (CPT, 1)5, a natural product isolated from the Chinese tree Camptotheca acuminata, was the first small molecule to be identified as a Top1 inhibitor (Figure 1). Later, topotecan and irinotecan, two clinically relevant analogues of 1, were developed, emphasizing the significance of Top1 as a drug target. The indenoisoquinoline NSC 314622 (2) was originally isolated as a by-product in the total synthesis of nitidine chloride 3, another naturally occurring Top1 inhibitor.7, 8 Interest in indenoisoquinolines was fueled by the fact that despite their similarity to 1 in cytotoxic profile and ability to inhibit Top1, they lack the metabolically unstable lactone ring present in camptothecins.9, 10 Molecules like 1–3 convert Top1 into a poison by stabilizing the covalent Top1-DNA cleavage complex and preventing the religation step, thereby enhancing the formation of persistent DNA breaks that eventually result in cell death.11
Figure 1.

Representative Top1 inhibitors.
Further lead optimization and assessment of synthetic indenoisoquinoline analogues led to the discovery of a series of potent Top1 inhibitors.12–14 To date, the lead optimization of indenoisoquinolines has mainly concentrated on varying the substituents on the A- and D-rings.15, 16 Also, a great deal of effort has been dedicated to finding the optimal replacement for the methyl group on the lactam B-ring of 2.17–19 In particular, the addition of an aminopropyl side chain to the lactam nitrogen was found to be particularly advantageous for improving Top1 inhibitory activity of the indenoisoquinoline class of compounds. This resulted in the identification of potent Top1 inhibitor MJ-III-65 (4),13 as well as MJ-II-38 (5), the latter of which was used for cocrystallization with Top1-DNA cleavage complex to establish the molecular mechanism of Top1 inhibition by indenoisoquinolines.20 As a part of an ongoing study of indenoisoquinoline structure -activity relationships (SAR), the exploration of the role and importance of the C-ring's size, geometry and constitution has been undertaken. For that purpose, the five-membered ring of the indenone fragment of the indenoisoquinoline was expanded to a six-membered pyridone ring. The designed dibenzonaphthyridinediones contain the isoquinolinone moiety of the indenoisoquinolines. The position of the added nitrogen atom was chosen to resemble the polycyclic core of the previously studied synthetic Top1 inhibitor topovale (6).21 The tetracyclic system of the dibenzonaphthyridinediones closely resembles systems 3 and 6. The overall character and position of the substituents on the dibenzonaphthyridinediones were chosen to be related to those of previously published indenoisoquinolines for ease of comparison.
Chemistry
Only two protocols for the preparation of dibenzo[c,h][1,6]naphthyridine-6,11(5H,12H)-diones have been reported so far.22, 23 Unfortunately, neither of the published syntheses is suitable for the preparation of the desired compounds. Therefore, a novel synthetic pathway was established in order to prepare several naphthyridinediones with various chains attached to the isoquinolinone lactam. The synthesis of N-methyl derivative 14 is outlined in Scheme 1. The N-protected o-aminobenzaldehyde 7, prepared in two steps from commercially available o-aminobenzylalcohol,24 was converted to imine 8 by treatment with a methanolic solution of methylamine. The less soluble cis-10 was isolated by filtration of the reaction mixture derived from Schiff base 8 and 4,5-dimethoxyhomophthalic anhydride (9).25 The trans isomer 10 was obtained by evaporation of the filtrate and recrystallization of the crude product. The lower yield of cis-10 relative to its trans analogue can be attributed to the presence of the bulky carbamate in the ortho position. This also explains the nearly complete isomerization within 24 hours of cis to trans configuration that was observed in an NMR sample of cis-10 dissolved in dimethyl-d6 sulfoxide (DMSO-d6). The mixture of both cis- and trans-10 was esterified to produce the more stable trans ester 11. The chemical shifts and coupling constants of H-3 and H-4 were used to establish the cis/trans relationships for acid 10 and ester 11.7 In the case of cis-10, H-3 and H-4 appear as doublets with coupling constants of 6.6 Hz, whereas in the 1H NMR spectrum of trans-10 they appear as singlets, consistent with pseudodiaxial substituents at C-3 and C-4 in trans-10, and a pseudoaxial phenyl substituent and pseudoequatorial carboxylic acid in cis-10. In both diastereomers, the C-3 phenyl substituent is pseudoaxial due to a steric A-strain interaction with the N-methyl group.26–28 The 1H NMR assignments of the relative configurations of Schiff base-homophthalic anhydride condensation products were previously corroborated through their use in the syntheses of a variety of natural products, including nitidine chloride, chelidonine, corynoline, and corydalic acid methyl ester.7, 29–31 In addition, the stereochemical assignment of cis and trans isomers based on the coupling constants of the vicinal methine protons have been more recently supported through the crystallographic studies of condensation products of homophthalic anhydride with imines.32, 33 The enolate formed after treatment of 11 with sodium hexamethyldisilazide (NaHMDS) was quenched with phenylselenyl chloride. Oxidation of the selenide with hydrogen peroxide resulted in dehydrogenated product 12. A similar strategy was previously devised for conversion of trans-isoquinolonic acids and esters into indenoisoquinolines.28 To our surprise, the saponification of ester 12 in the presence of lithium hydroxide, followed by acidification with acetic acid, provided the desired product 14 rather than the carboxylic acid 13.
Scheme 1.
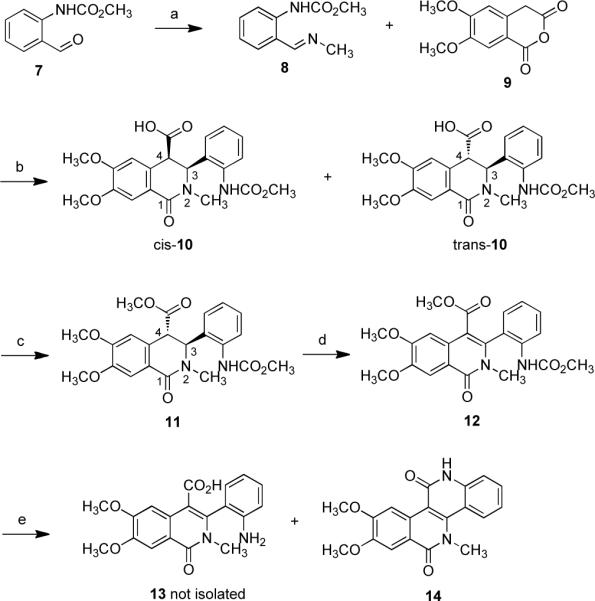
Synthesis of Dibenzo[c,h][1,6]naphthyridinedione 14a
aReagents and conditions: (a) CH3NH2, MgSO4, CH3OH, room temperature, 6 h (99%); (b) CHCl3, room temperature, 2 h (cis-10 35%, trans-10 60%); (c) SOCl2, CH3OH, 0 °C, 6 h (96%); (d) (1) NaHMDS, THF, then PhSeCl, −78 °C to room temperature, 6 h, (2) H2O2, CH3CO2H, 0 °C to room temperature, 6 h (62%); (e) KOH, water-ethylene glycol (1:1), reflux, 4 d (83%).
In order to prepare a variety of analogues similar to previously prepared libraries of indenoisoquinolines for comparison purposes, a method that allows introduction of hydroxy- and aminoalkyl chains in place of the lactam methyl group of the original lead compound 2 was required. The allyl chain was envisioned as a stable enough potential precursor for further modifications and functionalizations once the polycyclic core of the dibenzonaphthyridinedione is complete. Synthesis of the allyl analogue of 14 was accomplished in a similar fashion to that described above (Scheme 2). Imine 15 was isolated from the reaction of aldehyde 7 with allyl amine. The following condensation of 15 with anhydride 9 yieled cis-isoquinolonic acid 16. The esterification of cis-16 was accompanied by only partial isomerization of the cis isomer and resulted in a cis/trans mixture 17. Similar to the case of isoquinolonic acid 10, the coupling constant between H-3 and H-4 (J3–4 = 5.9 Hz) in the 1H NMR spectrum was used to assign the cis stereochemistry of 16. Dehydrogenation followed by saponification and cyclization yielded naphthyridinedione 19.
Scheme 2.
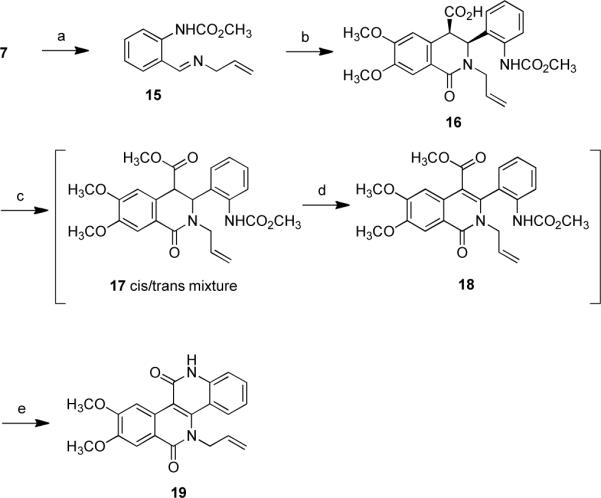
Synthesis of Allyl Derivative 19a
aReagents and conditions: (a) H2C=CHCH2NH2, MgSO4, CHCl3, room temperature, overnight (99%); (b) 9, CHCl3, room temperature, 18 h (26%); (c) SOCl2, CH3OH, 0 °C to room temperature, 6 h; (d) (1) NaHMDS, THF, then PhSeCl, −78 °C to room temperature, overnight, (2) H2O2, CH3CO2H, 0 °C to room temperature, overnight; (e) KOH, water/ethylene glycol (1:3), reflux, 12 h (43% over three steps).
The allyl group proved to be stable enough to allow completion of the synthesis of 19, but unfortunately, further derivatization of the allyl group was difficult to perform, mostly due to the extremely low solubility of the dibenzonaphthyridine 19. This called for further adjustments of the synthetic strategy.
Syntheses of hydroxypropyl- and two different aminopropyldibenzonaphthyridinediones are shown in Scheme 3. Starting from the O-TBDMS-protected Schiff base 20, the hydroxypropyl analogue 28 was prepared. Similarly, bromide 21 became the launching point toward aminopropyl derivatives 29 and 30. Condensation of 20 and 21 with anhydride 9 produced isoquinolonic acids 22 and 23 (Scheme 3). The coupling constants of 5.9 and 6.0 Hz between vicinal methine protons were observed in the 1H NMR spectra of 22 and 23, respectively, indicating cis stereochemistry of the isolated acids. Introduction of bromide and TBDMS-protected alcohol functionalities made it difficult to use the NaHMDS/phenylselenyl chloride/hydrogen peroxide sequence of steps and reagents for the dehydrogenation of the dihydroisoquinolone moieties of 22 and 23. It was shown previously that dehydrogenation of the dihydroisoquinoline fragment could be performed easily on cis-substrates whereas trans-analogues remain unresponsive to the treatment with oxidants like DDQ, CAN, and SeO2.7, 8 This is due to the fact that in the cis diastereomers, the C-4 proton is pseudoaxial, resulting in overlap of the C–H bond with the neighboring π -system of the aromatic ring, thus facilitating deprotonation.28 It is clear that for the strategy to succeed, the retention of cis stereochemistry is required during esterification of 22 and 23. The near-complete retention of cis configuration was achieved by means of esterification with trimethylsilyldiazomethane at low temperatures. The presence of small amounts of the trans isomers of 24 and 25 was confirmed by NMR spectroscopy of the crude products. In order to avoid further loss or isomerization of the cis-24 and cis-25 during column chromatography, the unpurified mixtures of cis- and trans-esters were used in the dehydrogenation step with DDQ. NMR analysis of the products revealed the presence of unchanged trans-24 and trans-25, along with the desired compounds 26 and 27. Treatment of 26 with potassium hydroxide at high temperature, followed by acidification with acetic acid, yielded the cyclized and unprotected hydroxypropyldibenzonaphthyridine 28. Reaction of 27 with morpholine or imidazole in hot DMF provided 29 or 30, respectively. In all cases, the final dibenzonaphthyridines were isolated as less-soluble solids and the more soluble by-products remained in solution.
Scheme 3.

Synthesis of Hydroxy- and Aminopropyl Derivatives 28–30a
aReagents and conditions: (a) RNH2, MgSO4, CHCl3, room temperature, overnight; (b) 9, CHCl3, room temperature, 18 h; (c) TMSCHN2, CH3OH/THF (1:3), −10 to 0 °C, 30–45 min; (d) DDQ (2.2 equiv), 1,4-dioxane, reflux, 3–4 h; (e) KOH, water/ethylene glycol (1:3), reflux for 24 h for 28, or morpholine, DMF, reflux 18 h for 29, or imidazole, DMF, reflux 18–20 h for 30.
The core polycyclic system of the dibenzo[c,h][1,6]naphthyridines has been synthesized before. In the present work, the naphthyridinediones system has been manipulated to provide a variety of substitution patterns. Naphthyridinediones 14 and 29 were reacted with phosphorus(V)oxychloride to yield chloronaphthyridinones 31 and 32 (Scheme 4).
Scheme 4.
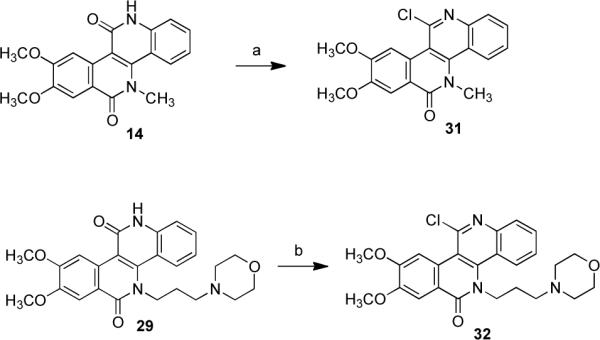
Synthesis of Chloronaphthyridinonesa
aReagents and conditions: (a) POCl3, DMF, room temperature to 70 °C, 7 h (61%); (b) POCl3, PCl5, room temperature, 1 h, reflux, 1.5 h (66%).
Heating a solution of 14 in POCl3 for 6 hours yielded dichloride 33 as the major product (Scheme 5). Both chlorines were subsequently substituted with methoxy groups by the reaction with sodium methoxide providing tetramethoxydibenzonaphthyridine 34.
Scheme 5.

Synthesis of Naphthyridinesa
aReagents and conditions: (a) POCl3, PCl5, reflux, 6 h (96%); (b) CH3ONa, CH3OH, reflux, 10 h (71%).
Biological Results and Discussion
All of the target compounds were tested for induction of DNA damage in Top1-mediated DNA cleavage assays. For this purpose the 32P 3′-end labeled 161 DNA fragment was incubated with Top1 and four 10-fold dilutions starting from 0.1 μM of a tested compound. The DNA fragments were separated on the denaturing gel. Based on the visual inspection of the number and intensities of the bands corresponding to those fragments the Top1 inhibitory activities were assigned. The results of this assay are designated relative to the Top1 inhibitory activity of compounds 1 and 2, and expressed in semiquantitative fashion: 0, no detectable activity; +, weak activity; ++, similar activity to compound 2: +++, greater activity than 2: ++++, equipotent to 1. Naphthyridinediones 14, 19, and 28 expressed no detectable activity as Top1 inhibitors. Introduction of an imidazole or a morpholine at the end of the propyl chain in 29 and 30 made the naphthyridinediones Top1 inhibitors, but only at the lower + level. The loss of Top1 inhibitory activity with expansion of the five-membered indenone ring of the indenoisoquinoline might result from the decreased overall solubility, especially for 14, 19, and 28, rather than differences in intercalation between DNA base pairs at the Top1 cleavage site. With 29 and 30, incorporation of amines increased water solubility and affinity toward DNA due to attraction of their positively charged protonated amines with the phosphates of the DNA backbone. The comparison of the naphthyridinedione 30 to indenoisoquinoline 3534 revealed that despite having similar substituents on the isoquinolinone moiety, the activity of the naphthyridinedione is significantly lower (Figure 2).
Figure 2.

The relative Top1 inhibitory potencies of the compounds are presented as follows: 0: no detectable activity; +: weak activity; ++: similar activity as compound 2; +++ and ++++: greater activity than compound 2; ++++: similar activity as 1 μM 1.
Interestingly, after conversion of the quinolinone fragment to a chloroquinoline, the observed Top1 inhibitory activity increased from 0 for 14 to +++ for 31, and from + for 29 to ++ for 32. The solubilities of 31 and 32 in solvents like dimethyl sulfoxide, methanol and chloroform also increased substantially relatively to their precursors 14 and 29. Further aromatization of the isoquinolinone moiety led to complete loss of activity for dichloride 33 and tetramethoxynaphthyridine 34 despite similarity of their polycyclic systems to nitidine chloride (3). In the case of the indenoisoquinolines, Top1 inhibitory activity could generally be improved by replacement of an N-methyl group by a chain of 2–4 carbon atoms with a polar group attached at its end.17–19 It is particularly noteworthy that this general trend was not observed with the chlorodibenzonaphthyridines 31 and 32. In fact, introduction of the morpholinopropyl fragment in 32 in place of the N-methyl group in 31 led to somewhat weaker Top1 inhibitory activity.
The Top1-mediated DNA fragmentation patterns produced by camptothecin, indenoisoquinolines 2, 4, and synthesized compounds 19 and 28–34, are presented in Figure 3. Inspection of the cleavage band intensities for compound 31 shows that unlike indenoisoquinolines, the cleavages at base pairs 44 and 68 are weak. However, the band for cleavage at the base pair 97 is more intense. This shows that the DNA cleavage site selectivity of chloronaphthyridinone 31 bears greater similarity to camptothecins than to indenoisoquinolines. In the case of compound 32, it is difficult to come to a similar conclusion as only bands for cleavages at the 97 and 119 sites are intense enough to be significant. The behavior of 32 is different from the camptothecins, the indenoisoquinolines, and the chlorodibenzonaphthyridinone 31. Therefore, the dibenzonaphthyridines offer the opportunity to target the genome differently from either the camptothecins or the indenoisoquinolines.
Figure 3.

A. Lane 1: DNA alone. Lane 2: Top1 alone. Lane 3: Top1 + 1 (1 μM). Lane 4: Top1 + 2 (100 μM). Lane 5: Top1 + 4 (1 μM). Lanes 6–21 (for compounds 19, 28–30): Top1 + indicated compound at 0.1 μM, 1 μM, 10 μM, 100 μM. B. Lane 1:DNA alone. Lane 2: Top1 alone. Lane 3: Top1 + 1 (1 μM). Lane 4: Top1 + 4 (1 μM). Lanes 5–20 (for compounds 31 – 34): Top1 + indicated compound at 0.1 μM, 1 μM, 10 μM, 100 μM. Numbers on right and arrows show the cleavage site positions. The activity of the compounds to produce Top1-mediated DNA cleavage was expressed semiquantitatively as follows: +: weak activity; ++ and +++: moderate activity; ++++: similar activity as 1 μM 1.
Despite the fact that a number of structures of different classes of Top1 inhibitors within drug-Top1-DNA ternary complex have been obtained by X-ray crystallography,20 the binding orientations of 3 and dibenzonaphthyridines like 6 have not been determined. Therefore, hypothetical binding models of naphthyridine-Top1-DNA ternary complexes were constructed in order to better understand the reasons underlying the change of activity in transitioning from indenoisoquinolines to naphthyridinedione and to chloronaphthyridinones. Binding models were constructed by means of docking and molecular mechanics tools. The geometry optimized structures of dibenzonaphthyridines 14 and 31, and indenoisoquinoline 5 were docked using GOLD35 in place of the ligand of the reported crystal structure of the 5-Top1-DNA ternary complex (PDB ID: 1SC720), and the geometries of the docked structures were optimized using the MMFF94s force field in Sybyl 8.1.36 The docking protocol was capable of reproducing the original position of 5 within ternary complex with the 1.15 Å root mean square deviation (RMSD) of the heavy atoms of the indenoisoquinoline polycyclic core. The main goal of this molecular modeling was to calculate the preferred binding orientation of the dibenzonaphthyridine polycyclic core. Both 14 and 31 are oriented with the longer axis of the ligand oriented along the longer axis of the base pair. In the case of 14, the isoquinolinone moiety faces the minor groove of the DNA and the distance of 2.6 Å between isoquinolinone oxygen and a nitrogen of the Arg364 side chain is consistent with a hydrogen bond (Figure 4). The most favorable orientation calculated for 31 is turned almost 180° around the axis perpendicular to the plane of the molecule relative to 14. The chloroquinoline moiety of 31 faces the minor groove with a 3.2 Å distance between quinoline nitrogen and Arg364. Both molecules appear slightly bent out of plane. This bend results in one the benzene rings of 14 being closer to the thymine of the AT base pair, possibly causing disfavorable steric interactions. On the contrary, the bend of 31 nearly perfectly mirrors the out-of-plane distortion of the AT base pair, which may contribute to the better fit and higher Top1 inhibitory activity of this type of inhibitor.
Figure 4.

A. The hypothetical binding mode of 14 within ternary complex. B. The hypothetical binding mode of 31 within ternary complex.
Also, according to calculated binding orientation of these dibenzonaphthyridines, the methoxy groups of 14 would be located on the side of the intact DNA strand causing some additional unfavorable steric interaction with the DNA backbone (Figure 4). On the other hand, the methoxy groups of 31 would be placed toward the less restricted scissile strand. This might explain the difference in activity between 14 and 31. Further similarities between chlorodibenzonaphthyridinones and 1 are evident from comparison of the experimentally determined binding orientation for 1 and the calculated binding mode of 31, which indicates that in both cases the quinoline fragment is located on the side of the intact DNA strand with the quinoline nitrogen facing Arg364 in the minor groove (Figure 5). In the case of the hypothetically determined binding mode of 31, the quinoline nitrogen is placed somewhat closer to the Arg364 compared to 1, forcing the polycyclic core of the dibenzonaphthyridinone to move deeper between flanking base pairs within the ternary complex.
Figure 5.

Comparison of the hypothetical binding orientation of 31 (grey) and the crystallographically obtained orientation of 1 (green). Left: view from the side of the AT base pair (not shown). Right: view from the side of the intact DNA strand.
In order to determine the antipoliferative activity, each compound was tested against 55 different human cancer cell lines in the National Cancer Institute screen.37, 38 The cells were incubated with the tested compounds at 100, 10, 1, 0.1 and 0.01 μM concentrations for 48 hours before treatment with sulforhodamine B dye. Optical densities were recorded, and their ratios relative to that of the control were plotted as percentage growth against the log10 of the tested compound concentrations. The concentration that corresponds to 50% growth inhibition (GI50) is calculated by interpolation between the points located above and below the 50% percentage growth. The chlorodibenzonaphthyridinone 32 bearing a morpholinopropyl chain on the lactam nitrogen was cytotoxic against human cancer cells at low micromolar concentrations. The cytotoxicities of 32 in selected cell lines are presented in Table 1, along with the mean-graph midpoint value of 5.6 μM. Cytotoxicity data recorded for the parent indenoisoquinoline 2 were added to the Table 1 for comparison purposes. In general, cytotoxicity GI50 values were in the low micromolar range, although 32 was particularly cytotoxic in the leukemia K-562 and RPMI-8226 cell lines, the ovarian IGROV1 cancer cell line, the renal UO-31 cancer cell line, and the breast MCF7 cancer cell line.
Table 1.
Antiproliferative Activity of Dibenzonaphthyridine 32 and Indenoisoquinoline 2.
| Cancer cell line | 32 GI50 (μM)a | 2 GI50 (μM)a |
|---|---|---|
| Leukemia | ||
| K-562 | 0.2 | 6.0 |
| RPMI-8226 | 0.1 | >100 |
| Lung | ||
| HOP-62 | 4.7 | 2.8 |
| Colon | ||
| HCT-116 | 4.16 | 11.5 |
| COLO 205 | 0.33 | >100 |
| CNS | ||
| SF-539 | 12.3 | 1.7 |
| Melanoma | ||
| UACC-62 | 6.8 | 0.6 |
| Ovarian | ||
| OVCAR-3 | 6.3 | 22.4 |
| IGROV1 | 0.03 | 0.01 |
| Renal | ||
| SN12C | 13.2 | 25.7 |
| UO-31 | 0.02 | 1.7 |
| Prostate | ||
| DU-145 | 10.5 | 4.8 |
| Breast | ||
| MCF7 | 0.4 | 1.9 |
|
| ||
| MGMb | 4.3 | 8.5 |
The cytotoxicity GI50 values listed are the concentrations corresponding to 50% growth inhibition, and are the result of single determinations.
Mean-graph midpoint for growth inhibition of all human cancer cell lines successfully tested.
In conclusion, a novel method for preparation of dibenzo[c,h][1,6]naphthyridinediones and chlorodibenzo[c,h][1,6]naphthyridinones has been developed. Two different pathways were successfully explored. The synthesis allowed introduction of a variety of chemically sensitive functional groups into the naphthyridinedione structure. The new chloronaphthyridinones represent promising lead compounds with Top1 inhibitory activities and low micromolar to submicromolar cytotoxicity GI50 values. Further replacement of the chloride of the quinoline moiety can serve as a route for expansion of this class of Top1 inhibitors. The tetramethoxy dibenzonaphthyridinedione 34 serves as an example of such modification. All target compounds were evaluated in a Top1-mediated DNA cleavage assay, and unique patterns of DNA cleavage were observed for 31 and 32 relative to the indenoisoquinolines, camptothecins, and each other. The hypothetical binding modes of dibenzonaphthyridinediones and chloronaphthyridinones were constructed by means molecular docking and molecular mechanics energy minimization. Molecular modeling indicates that lack of activity among naphthyridinediones could be attributed to a number of unfavorable steric interactions between the ligand and the cleavage complex. The calculated binding orientation of naphthyridinediones like 14 suggests that the conformationally flexible methoxy groups would face the intact DNA strand within ternary complex resulting in steric repulsion from the phosphodiester backbone and flanking base pairs. Replacement of the quinolinone moiety of 14 with chloroquinoline in 31 drastically changed the calculated binding mode of the tetracyclic core and eliminated such unfavorable interactions, which may explain the greater Top1 inhibitory activity of 31 vs. 14. The lactam substituent effects on Top1 inhibitory activity observed for 31 and 32 are different from those generally seen with the indenoisoquinolines, in which aminoalkyl substituents confer greater potency vs. a methyl substituent.
Experimental Section
General
Melting points were determined using capillary tubes with a Mel-Temp apparatus and are uncorrected. The proton nuclear magnetic resonance (1H and 13C NMR) spectra were recorded using an ARX300 300 MHz and DRX500 500MHz Bruker NMR spectrometers. IR spectra were recorded using a Perkin-Elmer 1600 series FTIR spectrometer. Purity of all tested compounds was ≥95%, as established by combustion analysis. Combustion microanalyses were performed at the Purdue University Microanalysis Laboratory and the reported values were within 0.4% of the calculated values. Analytical thin-layer chromatography was carried out on Baker-flex silica gel IB2-F plates and compounds were visualized with short wavelength UV light. Silica gel flash chromatography was performed using 230–400 mesh silica gel.
Methyl 2-[(Methylimino)methyl]phenylcarbamate (8)
A solution of methylamine in methanol (2 N, 3.6 mL, 7.2 mmol) and magnesium sulfate (3 g, 25 mmol) were added to 7 (1 g, 5.6 mmol), and mixture was stirred for 6 h. Then precipitate was filtered off and washed with chloroform (3 × 20 mL). The combined filtrate was concentrated on a rotary evaporator to afford pale-yellow oil (1.08 g, 99%). IR (film) 1733, 1640 cm−1; 1H NMR (500 MHz, CDCl3) δ 12.18 (s, 1 H), 8.41 (d, J = 8.4 Hz, 1 H), 8.28 (d, J = 1.0 Hz, 1 H), 7.41−7.33 (m, 1 H), 7.30−7.24 (m, 1 H), 7.08−6.99 (m, 1 H), 3.77 (s, 3 H), 3.50 (d, J = 1.3 Hz, 3 H); 13C NMR (126 MHz, CDCl3) δ 165.57, 154.76, 140.29, 132.99, 131.32, 121.52, 120.46, 118.05, 52.12, 47.96; EIHRMS m/z M+ calcd. for C10H12N2O2, 192.0899; found, 192.0897.
cis-3-[2-(Methoxycarbonylamino)phenyl]-4-carboxy-3,4-dihydro-6,7-dimethoxy-2-methyl-1(2H)-isoquinolone (10)
4,5-Dimethoxyhomophthalic anhydride (9, 1.16 g, 5.2 mmol) was added to a solution of 8 (1 g, 5.2 mmol) in chloroform (10 mL) and the mixture was stirred for 2 h at room temperature. The precipitate was filtered, washed with chloroform (2 × 10 mL) and dried to yield acid cis 10 (750 mg, 35%): mp 212–213 °C (dec). IR (KBr) 1737, 1626, 1599, 1578 cm−1; 1H NMR (300 MHz, methanol-d4) δ 7.80 (s, 1 H), 7.51 (s, 1 H), 7.29 (m, 2 H), 7.11 (t, J = 7.0 Hz, 1 H), 6.83 (m, 2 H), 5.33 (d, J = 6.60 Hz, 1 H), 4.45 (d, J = 6.60 Hz, 1 H), 3.80 (s, 3 H), 3.74 (s, 3 H), 3.65 (s, 3 H), 2.80 (s, 3 H); ESIMS m/z (rel intensity) 851 (35), 459 (13), 436 (100), 415 (6), 215 (15); ESIHRMS m/z MNa+ calcd. for C21H22N2O7, 437.1325; found, 437.1333.
The combined filtrates were evaporated to dryness and the remaining solid was recrystallized from benzene to provide trans 10 (1.3 g, 60%): mp 148–150 °C (dec). IR (KBr) 1730, 1716, 1640 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 12.91 (s, 1 H), 9.18 (s, 1 H), 7.46 (s, 1 H), 7.36 (s, 1 H), 7.29 (d, J = 7.7 Hz, 1 H), 7.23 (t, J = 7.5 Hz, 1 H), 7.05 (t, J = 7.5 Hz, 1 H), 6.71 (s, 1 H), 6.63 (d, J = 7.8 Hz, 1 H), 5.58 (s, 1 H), 3.81 (s, 3 H), 3.80 (s, 1 H), 3.71 (s, 3 H), 3.69 (s, 3 H), 2.88 (s, 3 H); 13C NMR (126 MHz, DMSO-d6) δ 172.38, 163.02, 155.44, 151.52, 148.23, 135.30, 134.59, 128.34, 128.18, 126.55, 126.25, 125.40, 121.11, 112.12, 109.24, 58.18, 55.63, 55.45, 51.98, 47.78, 33.82.
trans-3-[2-(Methoxycarbonylamino)phenyl]-3,4-dihydro-6,7-dimethoxy-4-methoxycarbonyl-2-methyl-1(2H)-isoquinolone (11)
Thionyl chloride (10 mL) was added slowly to a suspension of cis 10 (0.75 g, 1.8 mmol) and trans 10 (1.35 g, 3.3 mmol) in methanol (100 mL) at 0 °C. The resulting mixture was stirred for 6 h at 0 °C. After completion of the reaction (TLC), the reaction mixture was poured slowly into a mixture of ice (200 g) and a saturated solution of sodium bicarbonate (100 mL). The resulting mixture was extracted with chloroform (3 × 150 mL), and the combined extracts were dried with sodium sulfate, filtered though a thin pad of silica gel, and evaporated to dryness to obtain 11 as amorphous glassy solid (2.1 g, 96%). IR (film) 1733, 1637 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.67 (s, 1 H), 7.52 (d, J = 7.7 Hz, 1 H), 7.25 (t, J = 7.0 Hz, 1 H), 7.03 (t, J = 7.5 Hz, 1 H), 6.83 (d, J = 7.8 Hz, 1 H), 6.73 (s, 1 H), 6.54 (s, 1 H), 5.45 (s, 1 H), 3.95 (s, 3 H), 3.89 (s, 1 H), 3.82 (s, 3 H), 3.72 (s, 3 H), 3.63 (s, 1 H), 3.03 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 172.06, 164.17, 155.49, 152.15, 149.18, 134.39, 132.53, 129.10, 126.67, 126.53, 125.93, 125.14, 121.60, 111.86, 110.17, 59.32, 56.29, 56.20, 53.27, 53.06, 48.66, 34.57; ESIMS m/z (rel intensity) 429 (MH+, 100), 451 (56); ESIHRMS m/z MH+ calcd. for C22H24N2O7, 429,1662; found, 429.1665.
Methyl 6,7-Dimethoxy-3-(2-(methoxycarbonylamino)phenyl)-2-methyl-1-oxo-1,2-dihydroisoquinoline-4-carboxylate (12)
NaHMDS (1 M solution in THF-heptanes, 1.8 mL, 1.8 mmol) was slowly added to a solution of 11 (320 mg, 0.75 mmol) in dry THF (20 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 1 h, and then a solution of phenylselenyl chloride (216 g, 1.13 mmol) in dry THF (5 mL) was added dropwise and the mixture was stirred at −78 °C for 2 h. The reaction mixture was allowed to warm to room temperature and stirred at this temperature for 3 h. The reaction mixture was quenched by slow addition of 1N HCl (5 mL) at 0 °C, diluted with water (50 mL) and extracted with chloroform (4 × 50 mL). The combined extracts were washed with water and brine, dried with sodium sulfate, and evaporated under reduced pressure. The residue was dissolved in THF (10 mL). Acetic acid (1 mL) and hydrogen peroxide (30%, 5 mL) were added sequentially at 0 °C. The reaction mixture was warmed to room temperature and stirred for 6 h. Saturated aqueous sodium bicarbonate (5 mL) was added to the mixture at 0 °C. Chloroform (3 × 10 mL) was used to extract the product. The combined extracts were washed with water and brine, dried with sodium sulfate, and evaporated to afford the product (198 mg, 62%): mp 165–170 °C. IR (film) 1730, 1718, 1638 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.05 (d, J = 6.2 Hz, 1 H), 7.86 (s, 1 H), 7.49 (t, J = 7.9 Hz, 1 H), 7.22−7.16 (m, 2 H), 7.16−7.11 (m, 1 H), 6.53 (s, 1 H), 4.04 (s, 3 H), 3.99 (s, 3 H), 3.72 (s, 3 H), 3.45 (s, 3 H), 3.24 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 167.39, 162.03, 154.22, 154.07, 150.14, 139.65, 136.41, 130.97, 129.29, 128.59, 124.49, 119.35, 112.69, 108.14, 104.80, 56.52, 56.37, 52.75, 52.26, 33.18; ESIMS m/z (rel intensity) 449 (18), 427 (MH+, 100), 395 (78); ESIHRMS m/z MH+ calcd. for C22H22N2O7, 427,1505; found, 427.1507.
8,9-Dimethoxy-5-methyldibenzo[c,h][1,6]naphthyridine-6,11(5H,12H)-dione (14)
The methyl ester 12 (160 mg, 0.38 mmol) was added to a stirred solution of KOH (1.04 g, 18.5 mmol) in water–ethylene glycol mixture (15:15 mL), and the resulting mixture was heated to reflux for 4 d. After the mixture was cooled to room temperature, it was diluted with water (20 mL) and acidified with acetic acid (2.5 mL) and extracted with chloroform (3 × 100 mL), the combined extracts were washed with water (50 mL), brine (50 mL), dried with sodium sulfate, and evaporated to obtain white powder (105 mg, 83%): mp >350 °C. IR (KBr) 1710, 1670, 1651 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 11.86 (s, 1 H), 9.41 (s, 1 H), 8.18 (d, J = 8.30 Hz, 1 H), 7.72 (s, 1 H), 7.55 (t, J = 7.20 Hz, 1 H), 7.44 (d, J = 8.30 Hz, 1 H), 3.92 (s, 3 H), 3.91 (s, 3 H), 3.86 (s, 3 H); positive ESIMS m/z (rel intensity) 374 (60), 337 (MH+, 19), 318 (100); negative ESIMS m/z (rel intensity) 335 ([M − H+], 100). Anal. Calcd for C19H16N2O4: C, 67.85; H, 4.79; N, 8.33. Found: C, 67.52; H, 4.56; N, 7.97.
Methyl 2-[(Allylimino)methyl]phenylcarbamate (15)
Methyl 2-formylphenylcarbamate (7, 1 g, 5.6 mmol) and magnesium sulfate (3 g, 25 mmol) were added to a solution of allylamine (1.5 g, 26 mmol) in chloroform (10 mL) and mixture was stirred overnight. Then mixture was filtered and the residue was washed with chloroform (2 × 10 mL). The combined filtrates were washed with water (3 × 10 mL) brine (10 mL), dried with sodium sulfate, and concentrated on a rotary evaporator to afford 15 as yellow oil (1.2 g, 99%). IR (film) 1732, 1635 cm−1; 1H NMR (500 MHz, CDCl3) δ 12.25 (s, 1 H), 8.43 (d, J = 8.4 Hz, 1 H), 8.28 (s, 1 H), 7.40−7.34 (m, 1 H), 7.27 (dd, J = 7.7, 1.4 Hz, 1 H), 7.02 (td, J = 7.5, 0.9 Hz, 1 H), 6.05 (ddt, J = 17.1, 10.5, 5.3 Hz, 1 H), 5.25 (ddd, J = 17.2, 3.3, 1.6 Hz, 1 H), 5.20−5.13 (m, 1 H), 4.26−4.17 (m, 2 H), 3.76 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 164.94, 154.73, 140.38, 135.38, 133.25, 131.54, 121.49, 120.32, 118.05, 115.99, 62.88, 52.10; ESIMS m/z (rel intensity) 219 (MH+, 100); EIHRMS m/z M+ calcd. for C12H14N2O2, 218.1055; found, 218.1053.
cis-2-Allyl-6,7-dimethoxy-3-(2-(methoxycarbonylamino)phenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (16)
4,5-Dimethoxyhomophthalic anhydride (9, 444 mg, 2 mmol) was added to a solution of 15 (700 mg, 2 mmol) in chloroform (5 mL) and the mixture was stirred at room temperature for 18 h. The precipitate was collected and washed with chloroform (2 × 20 mL) to obtain a white solid (300 mg, 26%): mp 219–220 °C (dec). IR (KBr) 1746, 1618 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 12.80 (s, 1 H), 8.79 (s, 1 H), 7.52 (s, 1 H), 7.37 (d, J = 7.9 Hz, 1 H), 7.30−7.20 (m, 1 H), 7.12−7.01 (m, 2 H), 6.87 (s, 1 H), 5.72−5.50 (m, 1 H), 5.39 (d, J = 5.9 Hz, 1 H), 4.99 (dd, J = 10.3, 1.3 Hz, 1 H), 4.91 (dd, J = 17.2, 1.5 Hz, 1 H), 4.46−4.36 (m, 2 H), 3.83 (s, 3 H), 3.77 (s, 3 H), 3.63 (s, 3 H), 3.29 (dd, J = 15.4, 6.6 Hz, 1 H); 13C NMR (126 MHz, DMSO-d6) δ 171.54, 164.02, 155.37, 151.91, 148.35, 136.46, 133.58, 131.43, 128.66, 128.30, 128.26, 125.43, 121.80, 117.18, 110.54, 110.25, 55.95, 55.84, 54.54, 52.18, 48.37, 47.13; ESIMS m/z (rel intensity) 479 (MK+, 100), 441 (MH+, 26); ESIHRMS m/z MH+ calcd. for C23H24N2O7, 441.1662; found, 441.1657.
5-Allyl-8,9-dimethoxydibenzo[c,h][1,6]naphthyridine-6,11(5H,12H)-dione (19)
Thionyl chloride (10 mL) was added slowly to a suspension of 16 (990 mg, 2.24 mmol) in methanol (50 mL) at 0 °C. The resulting mixture was stirred for 6 h at room temperature. After completion of the reaction, the mixture was poured slowly into ice (200 g) and neutralized with a saturated aqueous sodium bicarbonate solution (100 mL). The organic products were extracted with chloroform (3 × 50 mL), and the combined extracts were dried with sodium sulfate and concentrated. The solid residue (17, cis/trans mixture) was redissolved in dry THF (30 mL). NaHMDS (1 M solution in THF-heptanes, 5 mL, 5 mmol) was slowly added to the resulting solution. The reaction mixture was stirred at −78 °C for 2 h and then a solution of phenylselenyl chloride (728 mg, 3.1 mmol) in dry THF (10 mL) was added dropwise and the mixture was stirred at −78 °C for additional 2 h. The reaction mixture was allowed to warm to room temperature and stirred at this temperature overnight. The reaction mixture was quenched by slow addition of 1N HCl (55 mL) at 0 °C, diluted with water (50 mL) and extracted with chloroform (4 × 50 mL). The combined extracts were washed with water and brine, dried with sodium sulfate, and evaporated under reduced pressure. The residue was dissolved in THF (50 mL). Acetic acid (5 mL) and hydrogen peroxide (30%, 30 mL) were added sequentially at 0 °C. The reaction mixture was warmed to room temperature and stirred overnight. Saturated sodium bicarbonate (15 mL) was added to the mixture at 0 °C. The precipitate was collected by filtration, washed with water and hot methanol, providing 18 as white solid mass (118 mg, 15%). The combined filtrates were extracted with chloroform (3 × 10 mL). The combined extracts were washed with water and brine, dried with sodium sulfate, and evaporated to dryness. The residue was added to a mixture of potassium hydroxide (560 mg, 10 mmol), water (10 mL), and ethylene glycol (30 mL). The resulting solution was heated to reflux for 12 h. After cooling down to ambient temperature, acetic acid was used to neutralize the solution. The precipitate was collected by filtration and washed with water and hot methanol providing the rest of 19 (215 mg, 28%, total yield 343 mg, 43%): mp 298–300 °C. IR (KBr) 1691, 1602, 1500 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 11.86 (s, 1 H), 9.38 (s, 1 H), 8.14 (d, J = 8.4 Hz, 1 H), 7.69 (s, 1 H), 7.53 (t, J = 7.6 Hz, 1 H), 7.43 (d, J = 8.2 Hz, 1 H), 7.18 (t, J = 7.7 Hz, 1 H), 6.33−6.14 (m, 1 H), 5.31 (d, J = 10.7 Hz, 1 H), 5.17 (d, J = 17.5 Hz, 1 H), 4.87 (s, 2 H), 3.91 (s, 3 H), 3.90 (s, 3 H); 13C NMR (126 MHz, DMSO-d6) δ 162.69, 161.09, 153.31, 149.53, 144.72, 137.67, 135.41, 130.73, 129.05, 126.25, 121.17, 118.98, 116.65, 116.25, 113.17, 108.21, 107.71, 107.41, 55.93, 55.79, 53.72; ESIMS m/z (rel intensity) 475 (MNa+, 100), 453 (MH+, 18). Anal. Calcd for C21H18N2O4: C, 69.60; H, 5.01; N, 7.73. Found: C, 69.22; H, 4.88; N, 7.64.
Methyl 2-[(3-Bromopropylimino)methyl]phenylcarbamate (21)
Methyl 2-formylphenylcarbamate (7, 2.5 g, 14 mmol) and magnesium sulfate (5 g, 42 mmol) were added to a solution of 3-bromopropylamine hydrochloride (3.68 g, 16.8 mmol) and triethylamine in chloroform (30 mL) and mixture was stirred for 24 h. The mixture was filtered and the residue was washed with chloroform (2 × 30 mL). The combined filtrates were washed with water (2 × 30 mL), brine (30 mL), dried with sodium sulfate, and concentrated to afford 21 as a yellow oil (3.95 g, 94.5%). IR (film) 1730, 1638 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.38 (d, J = 8.4 Hz, 1 H), 8.30 (s, 1 H), 7.35 (t, J = 7.7 Hz, 2 H), 7.26 (d, J = 7.7 Hz, 1 H), 7.00 (t, J = 7.4 Hz, 1 H), 3.74 (s, 3 H), 3.69 (t, J = 6.3 Hz, 2 H), 3.50 (t, J = 6.6 Hz, 2 H), 2.20 (pent, J = 6.3 Hz, 2 H); 13C NMR (75 MHz, CDCl3) δ 164.74, 154.30, 140.07, 133.00, 131.32, 121.25, 119.79, 117.69, 58.17, 52.87, 33.16, 31.15; ESIMS m/z (rel intensity) 299/301 (MH+, 100/99); EIHRMS m/z M+ calcd. for C12H15BrN2O2, 298.0317; found, 298.0320.
cis-2-[3-(tert-Butyldimethylsilyloxy)propyl]-6,7-dimethoxy-3-(2-(methoxycarbonylamino)phenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (22)
Methyl 2-formylphenylcarbamate (7, 1.79 g, 10 mmol) and magnesium sulfate (3 g, 25 mmol) were added to a solution of 3-(tert-butyldimethylsilyloxy)propan-1-amine (1.89 g, 10 mmol) in chloroform (10 mL) and the mixture was stirred overnight. Then mixture was filtered and the residue was washed with chloroform (2×10 mL). The combined filtrate was concentrated on a rotary evaporator to afford crude 20 as yellow oil (3.37 g, 96%) that was used without additional purification. 1H NMR (300 MHz, CDCl3) δ 8.38 (d, J = 8.4 Hz, 1 H), 8.30 (s, 1 H), 7.35 (t, J = 7.7 Hz, 2 H), 7.26 (d, J = 7.7 Hz, 1 H), 7.00 (t, J = 7.4 Hz, 1 H), 3.71−3.69 (m, 5 H), 3.60 (m, 2 H), 1.83 (pent, J = 6.3 Hz, 2 H), 0.84 (s, 9 H), −0.01 (s, 6 H); ESIMS m/z (rel intensity) 299/301 (MH+, 100/99). Anhydride 9 (444 mg, 2 mmol) was added to a solution of 20 (700 mg, 2 mmol) in chloroform (5 mL) and the mixture was stirred at room temperature for 18 h. The precipitate was collected and washed with chloroform (2 × 20 mL) to obtain a white solid (300 mg, 26%): mp 252–258 °C (dec). IR (KBr) 3357, 1733 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.91 (s, 1 H), 7.50 (s, 1 H), 7.41 (d, J = 8.3 Hz, 1 H), 7.28−7.19 (m, 1 H), 7.01 (d, J = 6.2 Hz, 2 H), 6.86 (s, 1 H), 5.46 (d, J = 5.9 Hz, 1 H), 4.39 (d, J = 5.9 Hz, 1 H), 3.85−3.72 (m, 7 H), 3.64 (s, 3 H), 3.52−3.39 (m, 2 H), 2.78 (ddd, J = 13.7, 9.1, 5.0 Hz, 1 H), 1.54 (ddd, J = 21.7, 13.6, 7.4 Hz, 2 H), 0.78 (s, 9 H), −0.07 (s, 6 H); 13C NMR (126 MHz, DMSO-d6) δ 171.17, 163.66, 155.01, 151.40, 147.92, 136.25, 128.22, 127.86, 124.82, 121.78, 110.13, 109.91, 60.88, 55.58, 55.49, 54.52, 51.81, 48.07, 42.37, 30.23, 25.79, 17.88, −5.42, −5.45; ESIMS m/z (rel intensity) 573 (MH+, 64), 441 (100); ESIHRMS m/z MH+ calcd. for C29H40N2O8Si, 573.2632; found, 573.2636.
cis-2-(3-Bromopropyl)-6,7-dimethoxy-3-[2-(methoxycarbonylamino)phenyl]-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (23)
4,5-Dimethoxyhomophthalic anhydride (9, 222 mg, 1 mmol) was added to a solution of 21 (300 mg, 10 mmol) in chloroform (5 mL) and the mixture was stirred at room temperature for 16 h. The precipitate was collected and washed with chloroform (2 × 20 mL) to obtain a white solid (208 mg, 40%): mp 264–266 °C (dec). IR (KBr) 3391, 1726 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 12.81 (s, 1 H), 8.84 (s, 1 H), 7.52 (s, 1 H), 7.40 (d, J = 8.1 Hz, 1 H), 7.30−7.20 (m, 1 H), 7.02 (t, J = 7.3 Hz, 1 H), 6.96 (dd, J = 7.9, 1.3 Hz, 1 H), 6.86 (s, 1 H), 5.50 (d, J = 6.0 Hz, 1 H), 4.51 (d, J = 6.0 Hz, 1 H), 3.91−3.79 (m, 4 H), 3.76 (d, J = 14.9 Hz, 3 H), 3.66 (d, J = 12.4 Hz, 3 H), 3.45−3.37 (m, 2 H), 2.84 (ddd, J = 13.6, 8.2, 5.6 Hz, 1 H), 1.96 (ddt, J = 21.7, 14.2, 7.0 Hz, 2 H); 13C NMR (126 MHz, DMSO-d6) δ 171.03, 163.68, 155.04, 151.52, 147.93, 136.30, 128.36, 127.63, 127.56, 125.03, 121.49, 110.05, 109.99, 55.58, 55.50, 54.67, 51.89, 47.82, 44.09, 32.09, 30.63; ESIMS m/z (rel intensity) 299/301 (MH+, 100/99) 543/545 (MNa+, 68/63), 441 (100).
5-(3-Hydroxypropyl)-8,9-dimethoxydibenzo[c,h][1,6]naphthyridine-6,11(5H,12H)-dione (28)
Trimethylsilyldiazomethane (0.12 mL, 2.0 M in diethyl ether, 0.25 mmol) was added dropwise to a suspension of 22 (110 mg, 0.19 mmol) in methanol (1 mL) and THF (3 mL) at −10 to 0 °C, and the mixture was stirred at −10 °C for 30–45 min after addition. The solvent was removed under reduced pressure at 20–25 °C. The residue (24, cis-trans mixture) was dissolved in anhydrous 1,4-dioxane (5 mL) and DDQ (100 mg, 0.44 mmol) was added to the solution. The reaction mixture was heated at reflux for 3–4 h. 1,4-Dioxane was evaporated under reduced pressure and chloroform (30 mL) was added to the residue. The mixture was washed with sodium bicarbonate (5%, 2 × 10 mL), water (15 mL), dried with sodium sulfate, and filtered through a thin layer of silica gel, eluting with chloroform. The combined filtrates were evaporated under reduced pressure. The amorphous solid containing 26 was added to a stirred solution of KOH (180 mg, 18.5 mmol) in water-ethylene glycol mixture (1 + 3 mL) at room temperature, and the mixture was heated at reflux on oil bath for 24 h. After the mixture was cooled to room temperature, it was diluted with water (5 mL) and acidified with acetic acid (0.5 mL). The white precipitate was collected by filtration to obtain 28 as a white powder (24 mg, 47%): mp 313 °C. IR (KBr) 1650, 1604 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 11.85 (s, 1 H), 9.36 (s, 1 H), 8.02 (d, J = 8.3 Hz, 1 H), 7.71 (s, 1 H), 7.54 (t, J = 7.6 Hz, 1 H), 7.44 (d, J = 8.1 Hz, 1 H), 7.23 (t, J = 7.6 Hz, 1 H), 4.51 (t, J = 6.8 Hz, 2 H), 4.43 (t, J = 4.8 Hz, 1 H), 3.92 (s, 3 H), 3.91 (s, 3 H), 3.22 (dd, J = 10.8, 5.5 Hz, 2 H), 2.05−1.81 (m, 2 H); 13C NMR (126 MHz, DMSO-d6) δ 162.82, 160.88, 152.96, 149.17, 144.41, 137.37, 130.28, 128.54, 126.19, 121.07, 118.95, 115.89, 113.14, 107.83, 107.40, 57.90, 55.63, 55.50, 48.77, 31.35; positive ESIMS m/z (rel intensity) 381 (MH+, 100). Anal. Calcd for C21H20N2O5: C, 66.31; H, 5.30; N, 7.36. Found: C, 66.03; H, 5.16; N, 7.26.
8,9-Dimethoxy-5-(3-morpholinopropyl)dibenzo[c,h][1,6]naphthyridine-6,11(5H,12H)-dione (29)
Trimethylsilyldiazomethane (0.12 mL, 2.0 M in diethyl ether, 0.25 mmol) was added dropwise to a suspension of 23 (100 mg, 0.19 mmol) in methanol (1 mL) and THF (3 mL) at −10 to 0 °C. The mixture was kept in this temperature range for 30 min after addition. The reaction mixture became clear after stirring at 0 °C for 30–45 min. The solvent was removed under reduced pressure at 20–25 °C to yield solid residue 24 that melts at 183–184 °C. Without additional purification the residue was dissolved in anhydrous 1,4-dioxane (5 mL) and DDQ (100 mg, 0.44 mmol) was added to the solution. The reaction mixture was heated at reflux for 3–4 h. 1,4-Dioxane was evaporated under reduced pressure and chloroform (30 mL) was added to the residue. The resulting mixture was washed with sodium bicarbonate (5%, 2 × 10 mL), water (15 mL), dried with sodium sulfate, and filtered through a thin layer of silica gel, eluting with chloroform. The combined filtrates were evaporated under reduced pressure. The residue containing 27 was redissolved in dry DMF (5 mL). Morpholine (200 mg, 2.3 mmol) was added to the solution and the mixture was heated to reflux for 18 h. The mixture was then cooled to room temperature (precipitate started forming prior to cooling). The precipitate was collected by filtration and washed with methanol and diethyl ether on a filter, and the product was dried to yield pure 29 (48 mg, 56%): mp 250–252 °C. IR (KBr) 1649, 1603 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 11.83 (s, 1 H), 9.35 (s, 1 H), 7.94 (d, J = 8.3 Hz, 1 H), 7.71 (d, J = 2.5 Hz, 1 H), 7.62−7.51 (m, 1 H), 7.44 (dd, J = 8.2, 1.1 Hz, 1 H), 7.29−7.14 (m, 1 H), 4.57 (t, J = 6.1 Hz, 2 H), 3.91 (s, 3 H), 3.91 (s, 3 H), 3.13 (s, 4 H), 1.94 (s, 4 H), 1.89−1.82 (m, 2 H), 1.79 (d, J = 5.6 Hz, 2 H); 13C NMR (126 MHz, DMSO-d6) δ 162.66, 160.94, 152.99, 149.12, 145.03, 137.34, 130.16, 128.54, 126.05, 120.89, 118.80, 115.78, 113.64, 107.73, 107.42, 107.09, 65.78, 55.61, 55.50, 53.85, 52.65, 48.80; positive ESIMS m/z (rel intensity) 450 (MH+, 92); negative ESIMS m/z (rel intensity) 448 ([M − H+], 100). Anal. Calcd for C25H27N3O5: C, 66.80; H, 6.05; N, 9.35. Found: C, 66.49; H, 5.73; N, 9.22.
5-(3-(1H-Imidazol-1-yl)propyl)-8,9-dimethoxydibenzo[c,h][1,6]-naphthyridine-6,11(5H,12H)dione (30)
Trimethylsilyldiazomethane (0.12 mL, 2.0 M in diethyl ether, 0.25 mmol) was added dropwise to a suspension of 23 (100 mg, 0.19 mmol) in methanol (1 mL) and THF (3 mL) at −10 to 0 °C. The mixture was kept at this temperature for 30 min after addition. The reaction mixture became clear after stirring at 0 °C for 30–45 min. The solvent was removed under reduced pressure at 20–25 °C to yield solid residue 24 that melts at 183–184 °C. Without additional purification, the residue was dissolved in anhydrous 1,4-dioxane (5 mL) and DDQ (100 mg, 0.44 mmol) was added to the solution. The reaction mixture was heated at reflux for 3–4 h. 1,4-Dioxane was evaporated under reduced pressure and chloroform (30 mL) was added to the residue. The mixture was washed with aqueous sodium bicarbonate (5%, 2 × 10 mL), water (15 mL), dried with sodium sulfate, and filtered through a thin layer of silica gel, eluting with chloroform. The combined filtrates were evaporated under reduced pressure. The residue containing 27 was redissolved in dry DMF (5 mL). Imidazole (160 mg, 2.3 mmol) was added to the solution and the mixture was heated to reflux for 18–20 h. The mixture was then cooled to room temperature (precipitate started forming prior to cooling). The precipitate was collected by filtration and washed with methanol and diethyl ether on a filter, and the product was dried to yield pure 30 (47 mg, 58%): mp 288–290 °C. IR (KBr) 1659, 1636 1601 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 11.86 (s, 1 H), 9.36 (s, 1 H), 7.77 (d, J = 8.3 Hz, 1 H), 7.71 (s, 1 H), 7.56−7.48 (m, 2 H), 7.42 (d, J = 8.2 Hz, 1 H), 7.13 (t, J = 7.7 Hz, 1 H), 7.08 (s, 1 H), 6.81 (s, 1 H), 4.40−4.28 (m, 2 H), 3.99−3.91 (m, 5 H), 3.91 (s, 3 H), 2.40−2.23 (m, 2 H); 13C NMR (126 MHz, DMSO-d6) δ 162.77, 160.78, 153.03, 149.23, 143.94, 137.34, 137.14, 130.29, 128.59, 128.46, 125.56, 121.15, 119.16, 118.82, 115.92, 112.77, 107.86, 107.55, 107.39, 55.63, 55.50, 48.67, 43.54, 29.61; positive ESIMS m/z (rel intensity) 431 (MH+, 100); negative ESIMS m/z (rel intensity) 429 ([M − H+], 100); ESIHRMS m/z MH+ calcd. for C24H22N4O4, 431.1719; found, 431.1120. Anal. Calcd for C24H22N4O4·0.7H2O: C, 65.06; H, 5.32; N, 12.65. Found: C, 64.98; H, 5.40; N, 12.37.
11-Chloro-8,9-dimethoxy-5-methyldibenzo[c,h][1,6]naphthyridin-6(5H)-one (31)
Dry DMF (1 mL) was added slowly to a mixture of 14 (84 mg, 0.25 mmol) and phosphoryl chloride (10 mL, 108 mmol) at 0 °C. The mixture was allowed to warm up to room temperature. The mixture was heated to 70 °C and kept at 65–70 °C for 2 h. After disappearance of starting material (TLC), the mixture was cooled to 0 °C, poured into the ice (50 g), and neutralized with concentrated ammonium hydroxide. The precipitate was collected by filtration and purified by preparative TLC to get an off-white solid (54 mg, 61 %): mp 242–243 °C. IR (KBr) 1650, 1605 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.69 (s, 1 H), 8.20 (d, J = 8.6 Hz, 1 H), 8.01 (d, J = 8.3 Hz, 1 H), 7.94 (s, 1 H), 7.71 (t, J = 7.6 Hz, 1 H), 7.54 (t, J = 7.7 Hz, 1 H), 4.07 (s, 3 H), 4.06 (s, 3 H), 4.03 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 163.12, 152.95, 150.30, 146.61, 145.57, 145.40, 130.36, 128.69, 126.51, 125.75, 124.93, 119.96, 118.71, 111.14, 108.76, 107.97, 56.48, 40.73; positive ESIMS m/z (rel intensity) 355/357 (MH+, 100/33). Anal. Calcd for C19H15ClN2O3·0.5H2O: C, 62.73; H, 4.43; N, 7.70. Found: C, 62.74; H, 4.75; N, 7.38.
11-Chloro-8,9-dimethoxy-5-(3-morpholinopropyl)dibenzo[c,h][1,6]-naphthyridin-6(5H)-one (32)
Precursor 29 (22.5 mg, 0.05 mmol) was mixed with phosphoryl chloride (3 mL, 32 mmol) and phosphorus pentachloride (100 mg, 0.48 mmol) was added slowly at room temperature. Starting material dissolved upon PCl5 addition, forming a clear yellow solution. The reaction mixture was stirred for 1 h and then heated at reflux for 1.5 h. After disappearance of starting material, the reaction mixture was cooled to room temperature and concentrated under reduced pressure. The brown oily residue was dissolved completely in ice-cold water (3 mL) and concentrated ammonium hydroxide solution was added dropwise to neutral pH. The cloudy aqueous solution was extracted with ethyl acetate (3 × 30 mL). The combined extracts were washed with concentrated ammonium hydrochloride solution, dried with sodium sulfate and evaporated to dryness under reduced pressure to yield 32 as an amorphous solid (15 mg, 66%): mp 185–190 °C (dec). IR (KBr) 1651, 1606 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.52 (s, 1 H), 8.17 (d, J = 8.5 Hz, 1 H), 7.95 (d, J = 7.6 Hz, 1 H), 7.82 (t, J = 7.6 Hz, 1 H), 7.77 (s, 1 H), 7.71 (t, J = 7.4 Hz, 1 H), 4.56−4.28 (m, 2 H), 3.97 (s, 3 H), 3.96−3.88 (m, 5 H), 3.76 (t, J = 12.0 Hz, 2 H), 3.42 (d, J = 11.9 Hz, 2 H), 3.18 (s, 2 H), 3.05 (d, J = 11.1 Hz, 2 H), 2.42 (s, 2 H); 13C NMR (126 MHz, CDCl3) δ 161.90, 152.44, 149.88, 145.75, 144.76, 144.57, 130.45, 127.92, 126.50, 125.47, 124.66, 119.34, 117.80, 110.25, 108.06, 107.89, 63.23, 55.91, 55.78, 53.12, 50.89, 48.68, 22.93; positive ESIMS m/z (rel intensity) 468/470 (MH+, 100/32), 381/383 (97/30). Anal. Calcd for C25H28Cl3N3O4·1.2H2O: C, 53.38; H, 5.45; N, 7.47. Found: C, 53.36; H, 5.45; N, 7.10.
6,11-Dichloro-8,9-dimethoxydibenzo[c,h][1,6]naphthyridine (33)
Compound 14 (700 mg, 2.04 mmol) and phosphorus pentachloride (868 mg, 4 mmol) were dissolved in phosphoryl chloride (25 mL, 270 mmol) at room temperature. The resulting solution was stirred for 2 h, and then heated at reflux for 6 h. The reaction mixture was cooled to room temperature, concentrated to about 5–6 mL, and quenched by pouring slowly into ice (50 g). The mixture was neutralized by adding a concentrated solution of ammonium hydroxide. The precipitate was separated by filtration and washed several times with small portions (5 mL) of ice-cold water providing light-gray powder (690 mg, 96%): mp 245–246 °C. IR (KBr) 1655, 1616, 1570, 1559, 1517 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.23 (s, 1 H), 9.03 (d, J = 7.8 Hz, 1 H), 8.06 (d, J = 8.1 Hz, 1 H), 7.82 (t, J = 7.2 Hz, 1 H), 7.79−7.69 (m, 2 H), 4.15 (s, 3 H), 4.10 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 154.73, 153.76, 150.54, 146.72, 146.51, 144.52, 131.02, 129.64, 128.02, 127.96, 124.95, 122.10, 114.65, 106.93, 106.83, 56.67, 56.39; positive ESIMS m/z (rel intensity) 359/360/361 (MH+, 100/22/65). Anal. Calcd for C18H12Cl2N2O2·0.6H2O: C, 58.43; H, 3.60; N, 7.57. Found: C, 58.32; H, 3.43; N, 7.53.
6,8,9,11-Tetramethoxydibenzo[c,h][1,6]naphthyridine (34)
Sodium methoxide (63 mg, 1.17 mmol) and 33 (70 mg, 0.2 mmol) were mixed in methanol (10 mL). The mixture was heated to reflux for 10 h. The reaction mixture was cooled to room temperature and ice-cold water (10 mL) was added. The precipitated white amorphous solid (50 mg, 71%) was collected by filtration: mp 173–175 °C. IR (KBr) 1650, 1610, 1590, 1515 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.80 (d, J = 7.2 Hz, 1 H), 8.57 (s, 1 H), 7.78 (d, J = 7.8 Hz, 1 H), 7.70 (t, J = 7.3 Hz, 1 H), 7.59−7.45 (m, 2 H), 4.24 (s, 3 H), 4.19 (s, 3 H), 3.95 (s, 3 H), 3.91 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 160.78, 159.51, 153.11, 149.48, 146.29, 143.82, 129.96, 129.49, 126.60, 124.42, 124.34, 123.50, 114.22, 107.61, 105.68, 103.90, 55.82, 55.72, 54.24, 53.95; positive ESIMS m/z (rel intensity) 351 (MH+, 100). Anal. Calcd for C20H18N2O4·0.6H2O: C, 66.51; H, 5.36; N, 7.76. Found: C, 66.41; H, 5.08; N, 7.54.
Topoisomerase I-Mediated DNA Cleavage Reactions
Human recombinant Top1 was purified from Baculovirus as described previously.39 The 161 bp fragment from pBluescript SK(−) phagemid DNA (Stratagene, La Jolla, CA) was cleaved with the restriction endonucleases PvuII and HindIII (New England Biolabs, Beverly, MA) supplied in NE buffer 2 (50 μL reactions) for 1 h at 37 °C, and separated by electrophoresis in a 1% agarose gel made in 1 × TBE buffer. The 161 bp fragment was eluted from the gel slice using the QIAEX II kit (QIAGEN Inc., Valencia, CA). Approximately 200 ng of the fragment was 3'-end labeled at the HindIII site by fill-in reaction with [alpha-32P]-dGTP and 0.5 mM dATP, dCTP, and dTTP, in React 2 buffer (50 mM Tris-HCl, pH 8.0, 100 mM MgCl2, 50 mM NaCl) with 0.5 unit of DNA polymerase I (Klenow fragment). Unincorporated 32P-dGTP was removed using mini Quick Spin DNA columns (Roche, Indianapolis, IN), and the eluate containing the 3'-end-labeled 161 bp fragment was collected. Aliquots (approximately 50,000 dpm/reaction) were incubated with topoisomerase I at 22 °C for 30 min in the presence of the tested drug. Reactions were terminated by adding SDS (0.5% final concentration). The samples (10 μL) were mixed with 30 μL of loading buffer (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue, pH 8.0). Aliquots were separated in denaturing gels (16% polyacrylamine, 7 M urea). Gels were dried and visualized by using a Phosphoimager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Molecular Modeling
Structures of 14 and 31 were built and geometry optimized with Sybyl 8.1 using the MMFF94s force field and MMFF94 charges.36 The X-ray crystal structure was obtained from the Protein Data Bank (PDB ID: 1SC7). Hydrogens were added to all atoms and MMFF94 charges were assigned. The positions of hydrogen atoms were optimized with the MMFF94s force field. The original ligand 5 was removed from the structure of the ternary complex,20 and 100 docking runs were performed for both 14 and 31 using the docking genetic algorithm and Goldscore fitness function within GOLD 3.2.35 The best solutions were merged with the Top1-DNA cleavage complex. In order to refine the position of the naphthyridine ligands, geometry optimizations of the ligands within newly obtained ternary complexes were performed by 100 iterations with steepest descent minimization followed by 200 iterations with conjugate gradient using the MMFF94s force field and MMFF94 charges within Sybyl 8.1.
Acknowledgment
This work was made possible by the National Institute of Health (NIH) through support of this work with Research Grant UO1 CA89566.
List of abbreviations
- CAN
ceric ammonium nitrate
- DDQ
2,3-dichloro-5,6-dicyanobenzoquinone
- DMSO-d6
dimethyl-d6 sulfoxide
- MGM
mean-graph midpoint
- NaHMDS
sodium hexamethyldisilazide
- RMSD
root mean square deviation
- TBDMS
tert-butyldimethylsilyl
- Top1
Topoisomerase type I
- SAR
structure-activity relationships
References
- (1).Wang JC. DNA Topoisomerases. Annu. Rev. Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- (2).Koster DA, Croquette V, Dekker C, Shuman S, Dekker NH. Friction and Torque Govern the Relaxation of DNA Supercoils by Eukaryotic Topoisomerase IB. Nature. 2005;434:671–674. doi: 10.1038/nature03395. [DOI] [PubMed] [Google Scholar]
- (3).Stewart L, Redinbo MR, Qiu X, Hol WGJ, Champoux JJ. A Model for the Mechanism of Human Topoisomerase I. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- (4).Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WGJ. Crystal Structures of Human Topoisomerase I in Covalent and Noncovalent Complexes with DNA. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- (5).Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca acuminata. J. Am. Chem. Soc. 1966;88:3888–3890. [Google Scholar]
- (6).Pommier E. Topoisomerase I Inhibitors: Camptothecins and Beyond. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- (7).Cushman M, Cheng L. Total Synthesis of Nitidine Chloride. J. Org. Chem. 1978;43:286–288. [Google Scholar]
- (8).Cushman M, Cheng L. Stereoselective Oxidation by Thionyl Chloride Leading to the Indeno[1,2-c]isoquinoline System. J. Org. Chem. 1978;43:3781–3783. [Google Scholar]
- (9).Kohlhagen G, Paull K, Cushman M, Nagafuji P, Pommier Y. Protein-Linked DNA Strand Breaks Induced by NSC 314622, a Novel Noncamptothecin Topoisomerase I Poison. Mol. Pharmacol. 1998;54:50–58. doi: 10.1124/mol.54.1.50. [DOI] [PubMed] [Google Scholar]
- (10).Pommier Y. Eukaryotic DNA topoisomerase I: Genome gatekeeper and its intruders, camptothecins. Seminars in Oncology. 1996;23:3–10. [PubMed] [Google Scholar]
- (11).Zunino F, Dallavalle S, Laccabue D, Beretta G, Merlini L, Pratesi G. Current Satus and Perspectives in the Development of Camptothecins. Curr. Pharm. Des. 2002;8:2505–2520. doi: 10.2174/1381612023392801. [DOI] [PubMed] [Google Scholar]
- (12).Strumberg D, Pommier Y, Paull K, Jayaraman M, Nagafuji P, Cushman M. Synthesis of Cytotoxic Indenoisoquinoline Topoisomerase I Poisons. J. Med. Chem. 1999;42:446–457. doi: 10.1021/jm9803323. [DOI] [PubMed] [Google Scholar]
- (13).Cushman M, Jayaraman M, Vroman JA, Fukunaga AK, Fox BM, Kohlhagen G, Strumberg D, Pommier Y. Synthesis of New Indeno[1,2-c]isoquinolines: Cytotoxic Non-Camptothecin Topoisomerase I Inhibitors. J. Med. Chem. 2000;43:3688–3698. doi: 10.1021/jm000029d. [DOI] [PubMed] [Google Scholar]
- (14).Pommier Y, Cushman M. The Indenoisoquinoline Noncamptothecin Topoisomerase I Inhibitors: Update and Perspectives. Mol. Cancer Ther. 2009;8:1008–1014. doi: 10.1158/1535-7163.MCT-08-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Morrell A, Placzek M, Parmley S, Grella B, Antony S, Pommier Y, Cushman M. Optimization of the Indenone Ring of Indenoisoquinoline Topoisomerase I Inhibitors. J. Med. Chem. 2007;50:4388–4404. doi: 10.1021/jm070307+. [DOI] [PubMed] [Google Scholar]
- (16).Morrell A, Placzek M, Parmley S, Antony S, Dexheimer TS, Pommier Y, Cushman M. Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors: A Systematic Study and Optimization. J. Med. Chem. 2007;50:4419–4430. doi: 10.1021/jm070361q. [DOI] [PubMed] [Google Scholar]
- (17).Nagarajan M, Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. Design, Synthesis, and Biological Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Featuring Polyamine Side Chains on the Lactam Nitrogen. J. Med. Chem. 2003;46:5712–5724. doi: 10.1021/jm030313f. [DOI] [PubMed] [Google Scholar]
- (18).Nagarajan M, Morrell A, Ioanoviciu A, Antony S, Kohlhagen G, Agama K, Hollingshead M, Pommier Y, Cushman M. Synthesis and Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Substituted with Nitrogen Heterocycles. J. Med. Chem. 2006;49:6283–6289. doi: 10.1021/jm060564z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Morrell A, Placzek MS, Steffen JD, Antony S, Agama K, Pommier Y, Cushman M. Investigation of the Lactam Side Chain Length Necessary for Optimal Indenoisoquinoline Topoisomerase I Inhibition and Cytotoxicity in Human Cancer Cell Cultures. J. Med. Chem. 2007;50:2040–2048. doi: 10.1021/jm0613119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB. Structures of Three Classes of Anticancer Agents bound to the Human Topoisomerase I-DNA Covalent Complex. J. Med. Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- (21).Li TK, Houghton PJ, Desai SD, Daroui P, Liu AA, Hars ES, Ruchelman AL, LaVoie EJ, Liu LF. Characterization of ARC-111 As a Novel Topoisomerase I-Targeting Anticancer Drug. Cancer Res. 2003;63:8400–8407. [PubMed] [Google Scholar]
- (22).Stadbauer W, Kappe T. Synthesis of Indoles and Isoquinolones from Phenylmalonate Heterocycles. Monatsh. Chem. 1984;115:467–475. [Google Scholar]
- (23).Mrkvicka V, Klasek A, Kimmel R, Pevec A, Kosmrlj J. Thermal Reaction of 3aH,5HThiazolo[5,4-c]quinoline-2,4-diones - an Easy Pathway to 4-Amino-1H-quinolin-2-ones and Novel 6H-Thiazolo[3,4-c]quinazoline-3,5-Diones. ARKIVOC (Gainsville, FL, United States) 2008;14:289–302. [Google Scholar]
- (24).Chong PY, Janicki SZ, Petillo PA. Multilevel Selectivity in the Mild and High-Yielding Chlorosilane-Induced Cleavage of Carbamates to Isocyanates. J. Org. Chem. 1998;63:8515–8521. [Google Scholar]
- (25).Potts KT, Robinson R. Synthetical Experiments Related to Indole Alkaloids. J. Chem. Soc. 1955:2675–2686. [Google Scholar]
- (26).Johnson F. Allylic Strain in Six-Membered Rings. Chem. Rev. 1968;68:375–413. [Google Scholar]
- (27).Johnson F. Steric Interference in Allylic and Pseudoallylic Systems. I. Two Stereochemical Theorems. J. Am. Chem. Soc. 1965;87:5492–5493. [Google Scholar]
- (28).Xiao X, Cushman M. A Facile Method to Transform trans-4-Carboxy-3,4-dihydro-3-phenyl-1(2H)-isoquinolones to Indeno[1,2-c]isoquinolines. J. Org. Chem. 2005;70:6496–6498. doi: 10.1021/jo050831t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cushman M, Choong T-C, Valko JT, Koleck M. A Total Synthesis of Chelidonine. J. Org. Chem. 1980;45:5067–5073. [Google Scholar]
- (30).Cushman M, Abbaspour A, Gupta YP. Total Synthesis of (+/−)-14-Epicorynoline, (+/−)-Corynoline, and (+)-6-Oxocorynoline. J. Am. Chem. Soc. 1983;105:2873–2879. [Google Scholar]
- (31).Cushman M, Wong WC. Total Synthesis of (+/−)-Corydalic Acid Methyl-Ester. J. Org. Chem. 1984;49:1278–1280. [Google Scholar]
- (32).Zhang HW, Vogels CM, Wheaton SL, Baerlocher FJ, Decken A, Westcott SA. Synthesis of cis-Isoquinolonic Acids Containing Boronate Esters. Synthesis. 2005:2739–2743. [Google Scholar]
- (33).Vara Y, Bello T, Aldaba E, Arrieta A, Pizarro JL, Arriortua MI, Lopez X, Cossio FP. Trans-Stereoselectivity in the Reaction between Homophthalic Anhydride and Imines. Organic Lett. 2008;10:4759–4762. doi: 10.1021/ol801757r. [DOI] [PubMed] [Google Scholar]
- (34).Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis of Benz[d]indeno[1,2-b]pyran-5,11-diones: Versatile Intermediates for the Design and Synthesis of Topoisomerase I Inhibitors. Bioorg. Med. Chem. Lett. 2006;16:1846–1849. doi: 10.1016/j.bmcl.2006.01.008. [DOI] [PubMed] [Google Scholar]
- (35).Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved Protein-Ligand Docking Using GOLD. Protein Struct. Funct. Genet. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- (36).SYBYL 7.3. Tripos International; St. Louis: [Google Scholar]
- (37).Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- (38).Boyd MR, Paull KD. Some Practical Considerations and Applications of the National Cancer Institute In Vitro Anticancer Drug Discovery Screen. Drug Development Res. 1995;34:91–109. [Google Scholar]
- (39).Pourquier P, Ueng L-M, Fertala J, Wang D, Park H-J, Essigmann JM, Bjornsti M-A, Pommier Y. Induction of Reversible Complexes between Eukaryotic DNA Topoisomerase I and DNA-containing Oxidative Base Damages. 7,8-Dihydro-8-Oxoguanine and 5-Hydroxycytosine. J. Biol. Chem. 1999;274:8516–8523. doi: 10.1074/jbc.274.13.8516. [DOI] [PubMed] [Google Scholar]