

Abstract
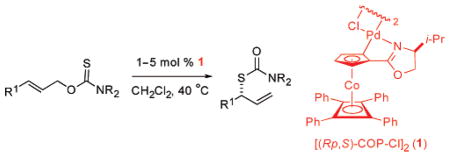
The palladium(II) complex [(Rp,S)-COP-Cl]2 and its enantiomer catalyze the rearrangement of linear prochiral O-allyl carbamothioates under mild conditions to provide branched S-allyl carbamothioates in high yield and high enantiomeric purity.
Catalytic methods for enantioselective construction of C–S bonds are less well developed than those for forming C–C, C–O, and C–N linkages. Among the most powerful methods are enantioselective palladium(0)-catalyzed displacements of allylic ester or carbonate precursors with sulfinate, thiolate, and thiocarboxylate nucleophiles and mechanistically related rearrangements of O-allyl sulfinates and carbamothioates.1–3 However, these methods are only useful for accessing products derived from symmetrically substituted η3-allylpalladium precursors because of low regioselection in the capture of unsymmetrical η3-allylpalladium intermediates by sulfur nucleophiles.2c,3
The formation of allylic sulfur compounds by [3,3]-sigmatropic rearrangements of allylic thiocarbonyl compounds, promoted thermally4 or by metal-catalyzed cyclization-induced rearrangement mechanisms,5,6 typically is not complicated by issues of regioselection. However, to date, catalytic enantioselective variants of such rearrangements have not been reported. Herein, we disclose that the commercially available palladium(II) complex [(Rp,S)-COP-Cl]2 (1)7 and its enantiomer catalyze the rearrangement of linear prochiral O-allyl carbamothioates to provide branched S-allyl carbamothioates in high yield and high enantiomeric purity, products that are readily transformed to the parent allylic thiols.3c,d
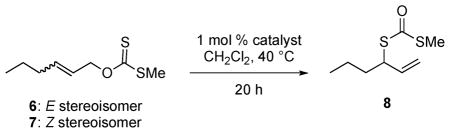
Because of the success of palladium(II) complexes of the COP family for catalyzing enantioselective [3,3]-sigmatropic rearrangements of prochiral allylic imidates to allylic amides,8 we examined the complexes depicted in Figure 1 as catalysts for the rearrangement of (E)- and (Z)-O-2-hexenyl methylxanthates 6 and 7 (Table 1).7,9 Chloride-bridged dimer [(Rp,S)-COP-Cl]2 (1) proved to be the most effective catalyst. With a catalyst loading of 1 mol %, S-methylcarbonodithioate 8 was formed in 66% ee and 90% conversion after 20 h at 40 °C from E precursor 6 (entry 1).10 As expected for a cyclization-induced rearrangement,8d the Z stereoisomer rearranged more slowly (entries 6–10).11 Again, COP complex 1 was the best of the catalysts surveyed in terms of both reaction rate and enantioselection (entry 6).
Figure 1.

COP palladium(II) catalysts.
Table 1.
Performance of Various Palladium(II) COP Catalysts in the Enantioselective Rearrangement of O-2-Hexenyl Methylxanthates 6 and 7
 | ||||
|---|---|---|---|---|
| entry | xanthatea | catalyst | convn. %b | ee %c |
| 1 | 6 | [COP-Cl]2 (1) | 90 | 66 |
| 2 | 6 | COP-acac (4) | 75 | 50 |
| 3 | 6 | [COP-OPiv]2 (3) | 42 | 31 |
| 4 | 6 | [COP-OAc]2 (2) | 45 | 30 |
| 5 | 6 | [COP-NHCOCl3]2 (5) | 35 | 10 |
| 6 | 7 | [COP-Cl]2 (1) | 27 | 50 |
| 7 | 7 | [COP-OPiv]2 (3) | 14 | 49 |
| 8 | 7 | [COP-OAc]2 (2) | 14 | 35 |
| 9 | 7 | COP-acac (4) | 12 | 10 |
| 10 | 7 | [COP-NHCOCl3]2 (5) | 5 | 6 |
Substrate concentration = 0.25 M.
At 20 h, by GC analysis.
Determined by HPLC analysis using a chiral stationary phase.
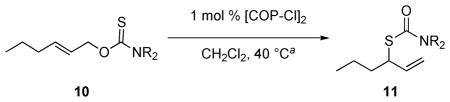
To further optimize enantioselection, the nature of the thiocarbonyl substituent was varied. A series of (E)-O-2-hexenyl carbamothioates were prepared from (E)-2-hexenol (9),12,13 and their rearrangement in the presence of 1 mol % of [(Rp,S)-COP-Cl]2 at 40 °C in CH2Cl2 was examined (Table 2). Although no trend in reaction rate was apparent, enantioselectivity was highest in the rearrangements of O-carbamothioates containing the smallest nitrogen substituents: dimethylamino (10a) and 1-azetidinyl (10c) (entries 1 and 3). Subjection of 10a and 10c to the same reaction conditions in the absence of catalyst resulted in recovery of starting material, establishing that the thermal rearrangement of these substrates was negligible under these conditions.14
Table 2.
[COP-Cl]2-Catalyzed Rearrangements of (E)-O-2-Hexenyl Carbamothioates 10
 | |||||
|---|---|---|---|---|---|
| entry | R2 | catalyst abs. conf. | product |
||
| compd | yield %b | ee %c (abs. conf.)d | |||
| 1 | Me2 (10a) | Sp,R | 11a | 72 | 82e (R) |
| 2 | Et2 (10b) | Sp,R | 11b | 78 | 59f (R) |
| 3 | 1-azetinyl (10c) | Sp,R | 11c | 98 | 83 (R) |
| 4 | 1-pyrrolidinyl (10d) | Rp,S | 11d | 100 | 57 (S) |
| 5 | 1-piperidinyl (10e) | Rp,S | 11e | 71 | 76f (S) |
Substrate concentration = 0.2–0.5 M.
Yield of product after purification by column chromatography.
Determined by HPLC or SFC analysis using a chiral stationary phase.
See the Supporting Information for determination of absolute configuration.
Determined by GC analysis using a chiral stationary phase.
The corresponding S-benzothioates prepared from 11b and 11e were analyzed (see the Supporting Information).
Having determined that O-allyl carbamothioates having dimethylamino or 1-azetinyl substituents rearranged with higher enantioselectivity, we turned our attention to developing an optimized general procedure for the [COP-Cl]2-catalyzed rearrangement of O-allyl methyl- and 1-azetinyl-carbamothioates. As expected, increasing the catalyst loading from 1 to 5 mol % significantly reduced reaction times. However, the higher catalyst loadings complicated the purification of the transposed allyl S-carbamothioates, with traces of COP complexes contaminating the product. Simply adding ethylenediamine (0.5 equiv) to the crude reaction solution at the conclusion of the reaction15 allowed pure products to be isolated reproducibly in high yields.16,17
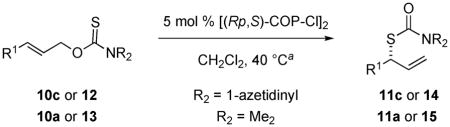
Using this optimized procedure, the catalytic asymmetric rearrangement of various O-allyl dimethyl- and 1-azetidinylcarbamothioates was surveyed (Table 3). The starting 1-azetidinecarbamothioates 10c and 12 were prepared in good overall yields (54–97%) from (E)-allylic alcohol precursors by reaction of O-methylxanthate intermediates with azetidine hydrochloride and triethylamine at room temperature.18 O-Allyl dimethylcarbamothioates 10a and 13 were prepared in one step and high yields by the reaction of dimethyl-thiocarbamoyl chloride with the appropriate allylic alcohol.18 Yields of the branched S-allyl carbamothioate products were generally excellent (85–99%). Two exceptions were products 14e and 14f that contain hydroxyl and Boc-protected aniline substituents, which were formed in lower yields (55–77% yield) (entries 8–10). Products containing linear or branched hydrocarbon substituents (entries 1–3), or homoallylic TBDMS- and TIPS-protected alcohol substituents (entries 3–6), were obtained in high enantiomeric purities (80–88% ee). Enantioselectivity was somewhat reduced in rearrangements of substrates containing unprotected allylic alcohol or a keto substituent at C5 (entries 8 and 11). Carrying out the rearrangement reported in entry 1 with the [(Sp,R)-COP-Cl]2 (ent-1) provided ent-11c in 83% ee and 98% yield. Although the reaction time was longer (67 h), the rearrangement of 10c (entry 1) with ent-1 could be readily carried out at room temperature, giving ent-11c in 82% ee and 96% yield. The catalyst loading could be reduced to 1 mol %, although the reaction had to be run for a longer time. For example, carrying out the rearrangement reported in entry 1 for 47 h at 40 °C at 1 mol % catalyst loading provided 11c in 76% ee and 95% yield. Absolute configurations of 11a–e were determined by chemical correlation with (R)-S-hex-1-yn-3-yl benzothioate, which was prepared by Mitsonobu reaction of (S)-hex-1-yn-3-ol with S-benzothiotic acid.18,19 The absolute configuration of other S-allyl carbamothioates products was assigned by analogy.
Table 3.
Scope of the [(Rp,S)-COP-Cl]2-Catalyzed Rearrangement of (E)-O-Allyl Carbamothioates
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 | R2 | time, hc | product |
||
| compd | yield %d | ee %e | ||||
| 1b | n-Pr | 1-azetinyl | 15 | ent-11c | 98 | 83 |
| 2b | n-Pr | Me2 | 18 | ent-11a | 72 | 82 |
| 3 | CH2CH(CH3)2 | 1-azetinyl | 42 | 14a | 85 | 80 |
| 4 | CH2OTBDMS | 1-azetinyl | 20 | 14b | 85 | 88 |
| 5 | CH2OTBDMS | Me2 | 18 | 15a | 97 | 87f,g |
| 6 | CH2OTIPS | 1-azetinyl | 42 | 14c | 99 | 87f |
| 7 | CH2OTBDPS | 1-azetinyl | 42 | 14d | 86 | 76f |
| 8 | CH2OH | 1-azetinyl | 13 | 14e | 55 | 61 |
| 9 | CH2NPh(Boc) | 1-azetinyl | 44 | 14f | 68 | 71 |
| 10 | CH2NPh(Boc) | Me2 | 43 | 15b | 77 | 81 |
| 11 | (CH2)2COMe | 1-azetinyl | 11 | 14g | 85 | 76 |
Substrate concentration = 0.5 M.
Sp,R enantiomer of [COP–Cl]2 was used.
Time for disappearance of starting material by TLC analysis.
Yield of product after purification by column chromatography.
Determined by SFC analysis using a chiral stationary phase; results from duplicate experiments agreed within ± 1%.
Determined after conversion of the product to alcohol 14e.
Determined by GC analysis using a chiral stationary phase; results from duplicate experiments agreed within ±2%.
In summary, a new catalytic asymmetric method for preparing allylic thiol derivatives has been developed. It is the first catalytic asymmetric method that provides branched allylic thiol derivatives in high regioselectivity from prochiral linear allylic precursors. Attractive features of the method include the ready synthesis of (E)-S-allyl carbamothioates from allylic alcohol precursors, the high yields and good enantioselection observed in their catalytic asymmetric rearrangement with [COP-Cl]2, and the ability to transform the branched allyl S-carbamothioate product to the corresponding enantioenriched branched allylic thiol by reduction with lithium aluminum hydride.3c,d,18
Supplementary Material
Acknowledgments
We thank the NSF (CHE-0200786) and the National Heart, Lung, and Blood Institute (HL-25854) for financial support and the Royal Commission for the Exhibition of 1851 for postdoctoral fellowship support of H.F.S. NMR and mass spectra were determined at UC Irvine using instruments acquired with the assistance of NSF and NIH Shared Instrumentation Programs. We thank Nicole White (UCI) and Professor Neil Garg (UCLA) for assistance with SFC analysis.
Footnotes
Supporting Information Available: Experimental procedures; characterization data for new compounds (1H, 13C, and HMQC NMR spectra); copies of SFC, GC, and HPLC traces used to establish the enantiopurity of (E)-S-allylic carbamothioate products; and details of chemical correlations to establish absolute configuration of products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Friesen RW. Science of Synthesis. Vol. 1. Thieme; Stuttgart: 2001. pp. 201–208. version 3.4. [Google Scholar]
- 2.(a) Hiroi K, Makino K. Chem Lett. 1986:617–620. [Google Scholar]; (b) Hiroi K, Makino K. Chem Pharm Bull. 1988;36:1744–1749. [Google Scholar]; (c) Eichelmann H, Gais H-J. Tetrahedron: Asymmetry. 1995;6:643–646. [Google Scholar]; (d) Trost BM, Krische MJ, Radinov R, Zanoni G. J Am Chem Soc. 1996;118:6297–6298. [Google Scholar]
- 3.For representative examples, see: Trost BM, Organ MG, O’Doherty GA. J Am Chem Soc. 1995;117:9662–9670.Valk JM, Claridge TDW, Brown JM. Tetrahedron: Asymmetry. 1995;6:2597–2610.Böhme A, Gais HJ. Tetrahedron: Asymmetry. 1999;10:2511–2514.Gais HJ, Böhme A. J Org Chem. 2002;67:1153–1161. doi: 10.1021/jo010668b.Lüssem BJ, Gais HJ. J Org Chem. 2004;69:4041–4052. doi: 10.1021/jo049756x.
- 4.For a brief review, see: Kocovsky P, Stary I. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, de Meijere A, editors. II. Wiley; New York: 2002. pp. 2014–2015.
- 5.Overman LE. Angew Chem, Int Ed. 1984;23:579–586. [Google Scholar]
- 6.Hg(II), Pd(II), Pt(II), Rh(I), and Ir(I) complexes have been employed. See inter alia.: Overman LE, Campbell CB, Knoll FM. J Am Chem Soc. 1978;100:4822–4834.Tamaru Y, Kagotani M, Yoshida Z. J Org Chem. 1980;45:5221–5223.Auburn PR, Whelan J, Bosnich B. Organometallics. 1986;5:1533–1537.Auburn PR, Whelan J, Bosnich B. Israel J Chem. 1986;27:250–254.
- 7.Stevens AM, Richards CJ. Organometallics. 1999;18:1346–1348.Anderson CE, Kirsch SF, Overman LE, Richards CJ, Watson MP. Org Synth. 2007;84:148–155.Anderson CE, Overman LE, Richards CJ, Watson MP, White N. Org Synth. 2007;84:139–147.(d) Both enantiomers of [COP-Cl]2 are available from Aldrich Chemical Co. (products # 661791 and 646636).
- 8.(a) Anderson CE, Overman LE. J Am Chem Soc. 2003;125:12412–12413. doi: 10.1021/ja037086r. [DOI] [PubMed] [Google Scholar]; (b) Overman LE, Owen CE, Pavan MM, Richards CJ. Org Lett. 2003;5:1809–1812. doi: 10.1021/ol0271786. [DOI] [PubMed] [Google Scholar]; (c) Kirsch SF, Overman LE, Watson MP. J Org Chem. 2004;69:8101–8104. doi: 10.1021/jo0487092. [DOI] [PubMed] [Google Scholar]; (d) Watson MP, Overman LE, Bergman RG. J Am Chem Soc. 2007;129:5031–5044. doi: 10.1021/ja0676962. [DOI] [PubMed] [Google Scholar]
- 9.The preparation and characterization of COP complexes 3 and 5 will be described in a forthcoming publication.
- 10.The half-life of the thermal background reaction is ~60 h under these conditions, which likely contributed somewhat to lowering enantioselection.
- 11.The thermal background reaction for rearrangement of (Z)-O-2-hexenyl methylxanthate 7 was negligible.
- 12.O-Hexenyl carbamothioate 10a was prepared by reaction of alcohol 9 with commercially available dimethylthiocarbamoyl chloride, whereas substrates 10b–e were prepared by reaction of (E)-O-2-hexenyl methyl xanthate 6 with the corresponding secondary amine. Details can be found in the Supporting Information.
- 13.Barrett AGM, Prokopiou PA, Barton DHR. J Chem Soc, Perkin Trans. 1981;1:1510–1515. [Google Scholar]
- 14.The (E)-O-2-hexenyl carbamothioate containing an NHPh substituent, prepared from the reaction of allylic alcohol 9 with phenyl isothiocyanate, rearranged in low enantioselectivity. The O-benzothioate derivative of 9 rearranged with good enantioselectivity; however, its relative instability led to low yields of the product, S-hex-1-en-3-yl benzothioate.
- 15.Urawa Y, Miyazawa M, Ozeki N, Ogura K. Org Process Res Dev. 2003;7:191–195. [Google Scholar]
- 16.Adding polymer-supported ethylenediamine, followed by filtration, was initially attempted. However, the product still contained ~5% of a palladium impurity.
- 17.Many literature reports address the removal of palladium residues from reaction mixtures. See inter alia: Larsen RD, King AO, Chen CY, Corley EG, Foster BS, Roberts FE, Yang C, Lieberman DR, Reamer RA, Tschaen DM, Verhoeven TR, Reider PJ, Lo YS, Rossano LT, Brookes AS, Meloni D, Moore JR, Arnett JF. J Org Chem. 1994;59:6391–6394.Rosso VW, Lust DA, Bernot PJ, Grosso JA, Modi SP, Rusowicz A, Sedergran TC, Simpson JH, Srivastava SK, Humora MJ, Anderson NG. Org Process Res Dev. 1997;1:311–314.Chen C, Dagneau P, Grabowski EJJ, Oballa R, O’Shea P, Prasit P, Robichaud J, Tillyer R, Wang X. J Org Chem. 2003;68:2633–2638. doi: 10.1021/jo0205614.
- 18.See the Supporting Information for details.
- 19.Corey EJ, Cimprich KA. Tetrahedron Lett. 1992;33:4099–4102. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.