

Abstract
O-GlcNAc transferase (OGT) is an essential mammalian enzyme that couples metabolic status to the regulation of a wide variety of cellular signaling pathways by acting as a nutrient sensor1. OGT catalyzes the transfer of N-acetyl-glucosamine from UDP-GlcNAc to serines and threonines of cytoplasmic, nuclear and mitochondrial proteins2,3, including numerous transcription factors4, tumor suppressors, kinases5, phosphatases1, and histone-modifying proteins6. Aberrant O-GlcNAcylation by OGT has been linked to insulin resistance7, diabetic complications8, cancer9 and neurodegenerative diseases including Alzheimer’s10. Despite the importance of OGT, the details of how it recognizes and glycosylates its protein substrates are largely unknown. We report here two crystal structures of human OGT, as a binary complex with UDP (2.8 A) and a ternary complex with UDP and a peptide substrate (1.95 A). The structures provide clues to the enzyme mechanism, show how OGT recognizes target peptide sequences, and reveal the fold of the unique domain between the two halves of the catalytic region. This information will accelerate the rational design of biological experiments to investigate OGT’s functions and the design of inhibitors for use as cellular probes and to assess its potential as a therapeutic target.
The ability to sense and respond to nutrient levels is critical for the growth of all living systems. In eukaryotes, a major mechanism for nutrient sensing involves the essential11 protein glycosyltransferase OGT, which senses cellular glucose levels via UDP-GlcNAc concentrations, and responds by dynamically O-GlcNAcylating a wide range of nuclear and cytoplasmic proteins.1,12 These include proteins involved in insulin-like signaling pathways7 and transcriptional activators that regulate glucose levels by controlling gluconeogenesis13. Since many known O-GlcNAcylation sites are also phosphorylation sites, OGT is proposed to play a major role in modulating cellular kinase signaling cascades14. OGT is also involved in widespread transcriptional regulation15–17. Prolonged hyperglycemia, such as occurs in diabetes, or excessive glucose uptake, such as occurs in cancer cells, results in hyper-O-GlcNAcylation of cellular proteins by OGT, and this increased O-GlcNAcylation has been linked to harmful cellular effects18. Thus, strategies to modulate OGT activity may have therapeutic value for treating diabetic complications, cancer, and other diseases13.
The lack of a crystal structure has been a major impediment to investigating OGT’s molecular mechanisms, understanding substrate recognition, and developing inhibitors. OGT comprises two distinct regions: an N-terminal region consisting of a series of tetratricopeptide repeat (TPR) units19,20 and a multidomain catalytic region. The TPR domain is proposed to scaffold interactions with other proteins, which may play a role in determining substrate selectivity21. A crystal structure comprising 11.5 TPR units of human OGT was reported in 200421, but there have been no structures of the catalytic region. From sequence analysis and structures of bacterial glycosyltransferases22–26, including a bacterial homolog of unknown function25,26, OGT was predicted to be a member of the GT-B superfamily of glycosyltransferases (Gtfs)27. However, OGT is unusual because it is the only known member to glycosylate polypeptides and it contains a long uncharacterized intervening sequence (~120 amino acids) in the middle of the catalytic region. It is also proposed to contain a phosphatidylinositol (3,4,5)-trisphosphate (PIP3) binding domain involved in membrane recruitment in response to insulin signaling7.
We report two crystal structures of a human OGT construct (hOGT4.5) containing 4.5 TPR units and the catalytic domain. The catalytic properties of this construct are similar to those of the full-length enzyme (Supplementary Fig. 1)28. One structure (2.8 A, referred to as OGT-UDP) is a complex with UDP; the other structure (1.95 A, referred to as OGT-UDP-peptide) is a complex containing UDP and a well-characterized 14 residue-CKII peptide substrate28. Based on currently available experimental data, we also present a model for the full-length enzyme (see Supplementary Information). Details of structure determination are presented in Methods and Supplementary Tables 1 and 2.
The OGT-UDP complex is shown in Fig. 1. The catalytic region contains three domains: the N-terminal domain (N-Cat), the C-terminal domain (C-Cat), and the intervening domain (Int-D) (Figs. 1a,b). The N-Cat and C-Cat domains have Rossmann-like folds typical of GT-B superfamily members; however, the N-Cat domain is distinctive in containing two additional helices, H1 and H2, which form an essential part of the active site (Fig. 1b). The Int-D domain, which has a novel fold, packs exclusively against the C-Cat domain (Fig. 1c). The UDP moiety binds in a pocket in the C-Cat domain near the interface with the N-Cat domain27. This pocket is lined with conserved residues shown to be important for catalytic activity (Supplementary Table 3)25,26. A transitional helix (H3) links the catalytic region to the TPR repeats, which spiral along the upper surface of the catalytic region from the C-Cat domain to the N-Cat domain. The TPRs and the catalytic region are demarcated by a narrow horizontal cleft.
Figure 1. Overall structure of human OGT complexed to UDP.
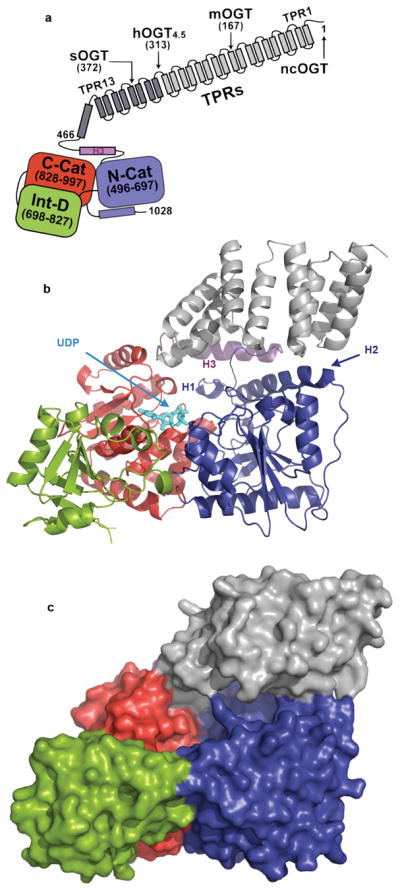
a, Schematic of OGT domain architecture with the TPR units shown in gray, the transitional helix (H3) in purple, the N-Cat domain in blue, the Int-D domain in green, and the C-Cat domain in red. The native isoforms of OGT (sOGT, mOGT, and ncOGT) and the crystallization construct differ only in the number of TPRs, as shown. b, Overall fold of OGT from the OGT-UDP complex in a ribbon representation. The coloring is the same as the schematic in a. The UDP is shown in cyan. The N-Cat domain helices unique to OGT are indicated as H1 and H2. c, Surface representation of the OGT-UDP complex. The coloring scheme is the same as in a and b.
The OGT-UDP-peptide complex (Fig. 2), which crystallized in a different space group from the OGT-UDP complex, has a wider cleft between the TPR domain and the catalytic region than the OGT-UDP complex (Fig. 1c and Fig. 2a), and the CKII peptide binds in this cleft. This peptide, YPGGSTPVS*SANMM, contains three serines and a threonine, but only one serine (underlined; referred to as Ser*) is glycosylated by OGT28. The hydroxyl of Ser* points into the nucleotide-sugar binding site (Fig. 2b). The two residues N-terminal to Ser* lie over the UDP moiety; the residues C-terminal to Ser* traverse towards the back of the cleft along the H2 helix of the N-Cat lobe. Although OGT glycosylates a wide range of target peptides, it prefers sequences in which the residues flanking the glycosylated amino acid enforce an extended conformation (e.g., prolines and β-branched amino acids; see Supplementary Fig. 2 and Supplementary Table 4). Consistent with these preferences, the peptide is anchored mainly by contacts from OGT side chains to the amide backbone, with an additional contact from the UDP moiety to the backbone amide of Ser*. The cleft is also filled with ordered water molecules, enabling it to serve as an adaptable interface to bind a range of polypeptides containing side chains of different sizes and hydrogen bond properties. Since the peptide substrate is anchored by contacts to its backbone, it is reasonable to infer that protein substrates are glycosylated on flexible regions such as loops or termini that can bind in an extended conformation, exposing the amide backbone.
Figure 2. Structure of the OGT-UDP-Peptide Complex.

a, Surface rendering of the OGT complex with UDP and the CKII peptide substrate28. The view and the coloring is the same as in Figure 1. The peptide, shown in yellow, lies over the UDP moiety, which is not visible in this orientation. b, Close up surface rendering of the OGT active site (gray) containing the CKII peptide in a stick representation (carbon atoms shown in yellow) with the UDP (purple) in a space filling representation lying directly underneath it. The peptide binds in the cleft between the TPR region and the catalytic region and extends along the interface between the C-Cat and N-Cat domains. Protein residues implicated in catalytic activity are colored orange, green, or blue in decreasing order of importance based on residual activity after mutation (Supplementary Table 3). Lysine 842 (orange) lies underneath UDP in this view. c, View of UDP (carbon atoms shown in cyan) and part of the CKII peptide (carbon atoms shown in yellow) with selected OGT side chains shown. Dashed lines indicate inferred hydrogen bonds based on distances in the OGT-UDP-peptide complex. The 2Fo-Fc omit map is contoured at the 1σ level. d, Proposed mechanism of OGT. The ordered sequential bi-bi kinetic mechanism shown is based on the structure of the ternary complex and supporting kinetic experiments (Supplementary Fig. 4). The peptide is depicted in yellow with only the reactive serine hydroxyl shown. H498A is the proposed catalytic base. Lys842, also shown to be essential for activity25,26, stabilizes the UDP moiety. e, Histogram showing the relative activities of the H498A and H558A mutants compared to the wildtype protein (average ± s.d., n=3).
The closed conformation of the substrate-binding cleft in the OGT-UDP structure is stabilized by a ‘latch’ comprising contacts between TPRs 10/11 and the H2 helix of the catalytic domain (Fig. 2a and Supplementary Fig. 3). Opening of the cleft in the OGT-UDP-peptide complex occurs due to a hinge-like motion around a pivot point between TPRs 12 and 13. The two structures suggest that glycosylation substrates enter the active site from the face of the enzyme shown in Fig. 2a, with the TPR domain restricting or allowing access depending on its conformation and its interactions with the catalytic domain. Molecular dynamics simulations indicate that the ‘hinge’ between the catalytic domain and the TPR domain is capable of large motions that fully expose the active site, which would allow protein substrates to approach closely enough for surface loops to enter (Supplemental Movie 1). The molecular mechanisms that facilitate or stabilize opening of the cleft to allow access of protein substrates remain to be determined, but may involve interactions between protein substrates or adapter proteins and the other regions of OGT.
The OGT-UDP-peptide complex, in addition to revealing how peptide substrates bind, provides unexpected insights into the kinetic mechanism. OGT was previously proposed to have a random sequential bi-bi mechanism.28 The structure, however, indicates that the peptide substrate binds over the nucleotide-sugar binding pocket, blocking access to it. Moreover, the α-phosphate of the UDP moiety contacts the backbone amide of Ser* (Fig. 2c), which helps orient the peptide. The peptide complex suggests an ordered mechanism in which UDP-GlcNAc binds prior to the polypeptide substrate. To assess the order of substrate binding, we analyzed the product inhibition patterns for UDP. At saturating peptide concentrations, a competitive inhibition pattern was obtained for UDP with respect to UDP-GlcNAc, which is inconsistent with a random mechanism, but supports the ordered sequential bi-bi mechanism implied by the crystal structure (Supplementary Fig. 4).
Another insight from the crystal structure is the identity of the catalytic base. Based on analyses of other GT-B family members, including the bacterial OGT homolog, it was proposed that His558 is the catalytic base. Although we have verified that this residue is critical for catalytic activity, the peptide complex shows that it is more than 5 A away from the reactive serine hydroxyl and makes an apparent hydrogen bond with the backbone carbonyl of the preceding residue. In contrast, His498, which is invariant in metazoan OGTs but absent in the homologous bacterial enzyme, protrudes from helix H1 into the active site within 3.5 A of the Ser* hydroxyl. Since His498 is critical for activity and is located between the reactive serine hydroxyl and the GlcNAc binding pocket, it is the probable catalytic base in OGT.
We were unable to obtain a crystal of the OGT-UDP-GlcNAc complex due to hydrolysis of the substrate, but according to computational docking experiments the GlcNAc is oriented in a manner that exposes its β face to the overlying peptide (Supplementary Fig. 5) and places the anomeric carbon near the reactive serine. This conformation is similar to the UDP-GlcNAc conformation observed in a complex of another GT-B family member23, and its relevance is supported by evidence that the C2 N-acetyl moiety projects up from the OGT sugar binding pocket29. Furthermore, it is consistent with the enzymatic reaction, which involves displacement of the α-UDP group to yield an inverted product. Based on the accumulated biochemical and structural data, we propose a general mechanism for the reaction (Fig. 2d).
The most unusual feature of OGT is the intervening domain between the catalytic lobes, which is only found in metazoans (Supplementary Figs. 6 and 7). This polypeptide adopts a topologically novel fold with a seven-stranded beta sheet core stabilized by flanking alpha helices (Fig. 3a). There are two long unstructured loops for which electron density is missing. An electrostatic surface rendering shows that the intervening domain and an adjacent helix of the C-Cat domain form a large basic surface comprising ten lysine residues (Figs. 3a and 3b), including K981 and K982, which were previously reported to constitute part of a PIP3 binding motif that recruits OGT to membranes7. We mutated eight of these ten lysines in various combinations (Supplementary Table 3). All mutants were catalytically active (Supplementary Figure 8), but we were unable to identify a role for the Int-D domain in PIP binding (Supplementary Table 5). We suggest this domain is involved in other functions in vivo. These functions may include substrate selection, cellular localization, or interactions with regulatory factors or receptors. The reported structures and mutant data provide a crucial starting point for investigating the possible roles of the intervening domain.
Figure 3. Structure of the intervening domain and full length models of human OGT.

a, Ribbon representation of the intervening domain rendered in light green with missing loops represented by dotted lines. Lysine sidechains that form an extensive positive surface (see b) are displayed in a “ball-and-stick” representation. Shown in coral is a helix from the C-cat domain containing four basic residues that contribute to the positively charged surface7. b, Surface representation of OGT colored according to electrostatic potential with blue representing areas of positive charge and red representing areas of negative charge. The protein is rotated 90 degrees around the X-axis from the representation shown in Figures 1, 2, and 3c, exposing the bottom surface of the catalytic region. c, Model of full-length human OGT, shown as a surface rendering and colored as in Figure 1a, based on the hOGT4.5 structures and the previously reported TPR domain structure. The TPRs preceding the boundary of hOGT4.5 are shown in light grey. The model is shown as a monomer but OGT may exist in different oligomerization states in cells.21,28 Hinge and latch regions are indicated by arrows. d, Model of full-length OGT opening to accommodate larger substrates. The “open” conformation is based on MD simulations (Supplementary Movie 1), as described in Methods. (PDB coordinates for full-length models are available for download. See Supplementary Information.)
The structures reported here show how OGT recognizes peptide sequences and provide new information on the enzymatic mechanism as well as a view of the intervening domain. Models of full-length human OGT in its open and closed states, constructed based on crystal structures and MD simulations, highlight the conformational changes that may regulate access of substrates to the active site (Figs. 3c and d). Our structures may assist in the development of inhibitors with possible therapeutic value for treating diseases associated with excessive O-GlcNAcylation.
Methods Summary
Human OGT residues 313-1031 (CPTH…KPVE) was expressed in E. coli and purified by nickel affinity chromatography and gel filtration. Protein was then incubated with UDP or UDP and a 17-residue substrate peptide (KKKYPGGSTPVSSANMM), which was cleaved to YPGGSTPVSSANMM in the crystallization drop (confirmed by mass spectrometry). The OGT-UDP structure was determined using MIRAS (Supplementary Table 2). The OGT-UDP-peptide complex structure was solved by molecular replacement using the refined OGT-UDP structure. The crystal packing for the two complexes is described in Supplementary Fig. 9. Kinetic analysis was performed using UDP-14C-GlcNAc and a lysine tagged CKII peptide using our previously described filter binding assay29. The molecular dynamics simulation was performed by using the program Desmond30 on an optimized 64-node Linux-based Infiniband cluster.
Supplementary Material
Acknowledgments
We thank B. Gross and C. Drennan for helpful advice. We also thank the National Institutes of Health, National Science Foundation, and Harvard Biomedical Accelerator Fund for financial support. This work is based upon research conducted at the Advanced Photon Source (Northeastern Collaborative Access Team beamlines) and Brookhaven National Laboratory (X25 and X29 beamlines).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions S.W. conceived the project. M.B.L. obtained the crystallization construct and initial diffracting crystals. M.B.L., Y.N., and P.S. determined and refined the crystal structures. J.J. and M.B.L. performed the enzymatic assays. M.B.L., Y.N., J.J., P.S. and S.W. designed experiments, discussed results, and prepared the manuscript.
Author Information The structures of the OGT-UDP complex and the OGT-UDP-peptide complex have been submitted to the Protein Data Bank under accession numbers 3PE3 and 3PE4. PDB Coordinates for the full-length models of OGT as well as the docked UDP-GlcNAc structure are available for download from the Walker Lab web site (See Supplementary Information). Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
References
- 1.Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–22. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 2.Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem. 1984;259:3308–17. [PubMed] [Google Scholar]
- 3.Haltiwanger RS, Holt GD, Hart GW. Enzymatic addition of O-GlcNAc to nuclear and cytoplasmic proteins. Identification of a uridine diphospho-N-acetylglucosamine:peptide beta-N-acetylglucosaminyltransferase. J Biol Chem. 1990;265:2563–8. [PubMed] [Google Scholar]
- 4.Yang X, Zhang F, Kudlow JE. Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell. 2002;110:69–80. doi: 10.1016/s0092-8674(02)00810-3. [DOI] [PubMed] [Google Scholar]
- 5.Dias WB, Cheung WD, Wang Z, Hart GW. Regulation of calcium/calmodulin-dependent kinase IV by O-GlcNAc modification. J Biol Chem. 2009;284:21327–37. doi: 10.1074/jbc.M109.007310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujiki R, et al. GlcNAcylation of a histone methyltransferase in retinoic-acid-induced granulopoiesis. Nature. 2009;459:455–9. doi: 10.1038/nature07954. [DOI] [PubMed] [Google Scholar]
- 7.Yang X, et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature. 2008;451:964–9. doi: 10.1038/nature06668. [DOI] [PubMed] [Google Scholar]
- 8.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 9.Caldwell SA, et al. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene. 2010;29:2831–42. doi: 10.1038/onc.2010.41. [DOI] [PubMed] [Google Scholar]
- 10.Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:10804–9. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shafi R, et al. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci U S A. 2000;97:5735–9. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Love DC, Hanover JA. The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Sci STKE. 2005;2005:re13. doi: 10.1126/stke.3122005re13. [DOI] [PubMed] [Google Scholar]
- 13.Dentin R, Hedrick S, Xie J, Yates J, 3rd, Montminy M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science. 2008;319:1402–5. doi: 10.1126/science.1151363. [DOI] [PubMed] [Google Scholar]
- 14.Wells L, Vosseller K, Hart GW. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science. 2001;291:2376–8. doi: 10.1126/science.1058714. [DOI] [PubMed] [Google Scholar]
- 15.Gambetta MC, Oktaba K, Muller J. Essential role of the glycosyltransferase sxc/Ogt in polycomb repression. Science. 2009;325:93–6. doi: 10.1126/science.1169727. [DOI] [PubMed] [Google Scholar]
- 16.Sinclair DA, et al. Drosophila O-GlcNAc transferase (OGT) is encoded by the Polycomb group (PcG) gene, super sex combs (sxc) Proc Natl Acad Sci U S A. 2009;106:13427–32. doi: 10.1073/pnas.0904638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Love DC, et al. Dynamic O-GlcNAc cycling at promoters of Caenorhabditis elegans genes regulating longevity, stress, and immunity. Proc Natl Acad Sci U S A. 2010;107:7413–8. doi: 10.1073/pnas.0911857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldberg HJ, Whiteside CI, Hart GW, Fantus IG. Posttranslational, reversible O-glycosylation is stimulated by high glucose and mediates plasminogen activator inhibitor-1 gene expression and Sp1 transcriptional activity in glomerular mesangial cells. Endocrinology. 2006;147:222–31. doi: 10.1210/en.2005-0523. [DOI] [PubMed] [Google Scholar]
- 19.Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem. 1997;272:9308–15. doi: 10.1074/jbc.272.14.9308. [DOI] [PubMed] [Google Scholar]
- 20.Lubas WA, Frank DW, Krause M, Hanover JA. O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J Biol Chem. 1997;272:9316–24. doi: 10.1074/jbc.272.14.9316. [DOI] [PubMed] [Google Scholar]
- 21.Jinek M, et al. The superhelical TPR-repeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin alpha. Nat Struct Mol Biol. 2004;11 :1001–7. doi: 10.1038/nsmb833. [DOI] [PubMed] [Google Scholar]
- 22.Ha S, Walker D, Shi Y, Walker S. The 1.9 A crystal structure of Escherichia coli MurG, a membrane-associated glycosyltransferase involved in peptidoglycan biosynthesis. Protein Sci. 2000;9:1045–52. doi: 10.1110/ps.9.6.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Y, et al. Crystal structure of the MurG:UDP-GlcNAc complex reveals common structural principles of a superfamily of glycosyltransferases. Proc Natl Acad Sci U S A. 2003;100:845–9. doi: 10.1073/pnas.0235749100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wrabl JO, Grishin NV. Homology between O-linked GlcNAc transferases and proteins of the glycogen phosphorylase superfamily. J Mol Biol. 2001;314:365–74. doi: 10.1006/jmbi.2001.5151. [DOI] [PubMed] [Google Scholar]
- 25.Martinez-Fleites C, et al. Structure of an O-GlcNAc transferase homolog provides insight into intracellular glycosylation. Nat Struct Mol Biol. 2008;15:764–5. doi: 10.1038/nsmb.1443. [DOI] [PubMed] [Google Scholar]
- 26.Clarke AJ, et al. Structural insights into mechanism and specificity of O-GlcNAc transferase. Embo J. 2008;27:2780–8. doi: 10.1038/emboj.2008.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–55. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 28.Kreppel LK, Hart GW. Regulation of a cytosolic and nuclear O-GlcNAc transferase. Role of the tetratricopeptide repeats. J Biol Chem. 1999;274:32015–22. doi: 10.1074/jbc.274.45.32015. [DOI] [PubMed] [Google Scholar]
- 29.Gross BJ, Kraybill BC, Walker S. Discovery of O-GlcNAc transferase inhibitors. J Am Chem Soc. 2005;127:14588–9. doi: 10.1021/ja0555217. [DOI] [PubMed] [Google Scholar]
- 30.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06) ACM Press; 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.