

Abstract
Previous studies suggested that cleavage of the amyloid precursor protein (APP) at aspartate residue 664 by caspases may play a key role in the pathogenesis of Alzheimer's disease. Mutation of this site (D664A) prevents caspase cleavage and the generation of the C-terminal APP fragments C31 and Jcasp, which have been proposed to mediate amyloid-β (Aβ) neurotoxicity. Here we compared human APP transgenic mice with (B254) and without (J20) the D664A mutation in a battery of tests. Before Aβ deposition, hAPP–B254 and hAPP–J20 mice had comparable hippocampal levels of Aβ1-42. At 2–3 or 5–7 months of age, hAPP–B254 and hAPP–J20 mice had similar abnormalities relative to nontransgenic mice in spatial and nonspatial learning and memory, elevated plus maze performance, electrophysiological measures of synaptic transmission and plasticity, and levels of synaptic activity-related proteins. Thus, caspase cleavage of APP at position D664 and generation of C31 do not play a critical role in the development of these abnormalities.
Introduction
Alzheimer's disease (AD) is the most frequent neurodegenerative disorder and cause of dementia in the elderly. Despite more than 100 years of investigation, no effective treatments are available to prevent, halt, or reverse the disease. A major reason is likely the complex etiology of the condition, which appears to involve a variety of genetic and environmental factors (Blennow et al., 2006). Key among these factors is the amyloid precursor protein (APP). APP is cleaved by at least four types of proteases, α-, β-, γ-secretases, and caspase(s), resulting in metabolites with diverse biological activities (Berger-Sweeney et al., 1999; Gao and Pimplikar, 2001; Müller et al., 2008; Nikolaev et al., 2009). The most studied among these is the amyloid-β (Aβ) peptide, released from APP through sequential cleavage by β- and γ-secretases. Because a compelling body of data supports the hypothesis that Aβ plays a causal role in the pathogenesis of AD, many investigational treatments in clinical trials aim to reduce Aβ production or aggregation or to promote its clearance from the brain (Roberson and Mucke, 2006).
More recent lines of evidence have raised the possibility that APP metabolites other than Aβ may contribute to AD pathogenesis. APP can be cleaved at three sites to generate four proapoptotic peptides: sAPPβ, Aβ, Jcasp, and C31 (Bredesen, 2009). The Jcasp and C31 C-terminal fragments are generated by caspase cleavage at aspartate residue 664 in human APP (D664; hAPP695 numbering) (Gervais et al., 1999; Weidemann et al., 1999; Bertrand et al., 2001). Both fragments can induce cell death in vitro (Lu et al., 2000; Bertrand et al., 2001; McPhie et al., 2001), although recent evidence suggests that C31 is more toxic than Jcasp (Park et al., 2009). Interestingly, Aβ can bind its cognate domain on APP, facilitating homo-oligomerization of APP and subsequent caspase cleavage at D664, producing C31 (Lu et al., 2000; Shaked et al., 2006). These findings raised the intriguing possibility that C31 could be a key mediator of Aβ-dependent deficits.
To assess the importance of caspase cleavage of APP in vivo, transgenic mice expressing hAPP without a functional caspase cleavage site were generated (Galvan et al., 2006). Blocking caspase cleavage of APP did not alter Aβ production but was reported to prevent loss of presynaptic terminals, dentate gyrus (DG) atrophy, reductions in baseline synaptic transmission strength and long-term potentiation (LTP), and deficits in learning and memory in hAPP mice (Galvan et al., 2006, 2008; Saganich et al., 2006).
We aimed to confirm and extend these findings in independent groups of mice from hAPP transgenic lines analyzed in the original studies: line J20 (Mucke et al., 2000) and line B254 (Galvan et al., 2006). The hAPP–B254 transgene encodes the additional D664A mutation, which prevents C31 and Jcasp release by caspases (Weidemann et al., 1999). In regards to AD-related histopathological, electrophysiological, and behavioral abnormalities, hAPP–J20 and hAPP–B254 mice analyzed in the current study were similarly affected in most tests, suggesting that these abnormalities do not require caspase cleavage of hAPP and are not caused by C31 or Jcasp.
Materials and Methods
Animals.
The hAPP transgenic mouse lines J20 (Rockenstein et al., 1995; Mucke et al., 2000) and B254 (Galvan et al., 2006) have been described previously. The presence of the D664A mutation in the B254 line was verified by genomic sequencing. Briefly, a 480 bp PCR product was amplified from genomic DNA with the following primers (5′-3′): forward, AACACAGAAAACGAAGTT and reverse, CCGATGGGTAGTGAAGCA. The same primers were then used to sequence the PCR product (sequencing done by Elim Biopharmaceuticals). Both transgenic lines were on the C57BL/6J background and were maintained by crossing heterozygous transgenic mice with nontransgenic (NTG) C57BL/6J breeders (The Jackson Laboratory). The Institutional Animal Care and Use Committee of the University of California, San Francisco, approved all experiments.
Behavioral tests.
Only male mice were analyzed to reduce variability in behavioral performance. One cohort of mice was behaviorally evaluated at 2–3 months of age. Two additional, independent cohorts of mice were evaluated at 5–7 months. Each cohort contained four groups: hAPP mice and NTG littermates from each transgenic line. For the elevated plus maze, open field, Y-maze, and novel object recognition tests, data from the older cohorts were pooled. For the Morris water maze, data were analyzed separately for each cohort.
Elevated plus maze.
The elevated plus maze consisted of two open (without walls) and two enclosed (with walls) arms elevated 63 cm above the ground (Hamilton-Kinder). Mice were allowed to habituate in the testing room under dim light for 1 h before testing. During testing, mice were placed at the junction between the open and closed arms of the plus maze and allowed to explore for 5 min. The maze was cleaned with 70% alcohol after testing of each mouse. Total distance traveled and time spent in both the open and closed arms were calculated based on infrared photobeam breaks.
Open field.
Spontaneous locomotor activity in an open field arena was measured in an automated Flex-Field/Open Field Photobeam Activity System (San Diego Instruments). Before testing, mice were transferred to the testing room and acclimated for at least 1 h. Mice were tested in a clear plastic chamber (41 × 41 × 30 cm) for 15 min, with two 16 × 16 photobeam arrays detecting horizontal and vertical movements. The apparatus was cleaned with 70% alcohol after testing of each mouse. Total movements in the open field were recorded for additional data analysis.
Novel object recognition.
Mice were transferred to the testing room and acclimated for at least 1 h. Two- to 3-month-old mice were tested in a white round plastic chamber 35 cm in diameter. On the first day, mice were habituated to the testing arena for 30 min. On the second day, two identical objects were placed into the same chamber, and mice were allowed to explore the objects and the chamber for 10 min. Four hours later, the objects were replaced with one exact replica of the original object and a novel object of different shape and texture. Mice were then allowed to explore the objects and the chamber for 10 min. Positions of the familiar and novel objects in the chamber were changed semi-randomly between testing of different mice but were kept constant between training and test sessions for any given mouse. Five- to 7-month-old mice were evaluated in a slightly different paradigm. They were trained and tested in a white rectangular Plexiglas chamber (20 × 40 cm). The entire procedure consisted of four 5-min trials with an intertrial interval of 15 min. During the first three trials, mice were presented with two identical objects that were evenly spaced in the chamber and placed opposite of the mouse drop location. For the fourth trial, one of the two objects was replaced with a novel object that had a different shape but a similar texture. Behavior was recorded with a video tracking system (Noldus). Frequency of interactions with the objects and time spent exploring each object were recorded for subsequent data analysis, and recognition memory was assessed in the final trial. Arenas and objects were cleaned with 70% ethanol after testing of each mouse.
Morris water maze.
The water maze consisted of a pool (122 cm diameter) filled with water (21 ± 1°C) made opaque with nontoxic white tempera paint powder. The pool was located in a room surrounded by distinct extramaze (spatial) cues. Preceding hidden platform training, mice were given four pretraining trials in which they had to swim in a rectangular channel (15 × 122 cm) and mount a platform hidden 1.5 cm below the water surface in the middle of the channel. Mice that did not mount the platform within 90 s were gently guided to it and allowed to sit on it for 10 s before they were removed by the experimenter. The day after pretraining, mice were trained in the circular water maze. For hidden platform training, the platform (14 × 14 cm) was submerged 1.5 cm below the water surface. The platform location remained the same throughout hidden platform training, but the drop location varied semi-randomly between trials. Mice received two training sessions with 3 h intersession intervals for 5 consecutive days. Each session consisted of two (first cohort) or three (second cohort) trials with 10 min intertrial intervals. Mice that did not find the platform within 60 s were guided to it and allowed to sit on it for 10 s. In spatial probe trials, the platform was removed, and mice were allowed to swim for 60 s before they were removed. The drop location was 180° from where the platform was placed during hidden platform training. The same drop location was used for all spatial probe trials. The first cohort of mice was given a probe trial 24 h after completion of the hidden platform training. The second cohort of mice received an additional probe trial 48 h after the beginning of the hidden platform training. After the final probe trial, mice were allowed to rest for 1 d, followed by cued platform training. In this task, the platform was marked with a visible cue (15 cm tall, black and white striped pole placed on top of the platform). Mice received two training sessions per day with 3–4 h intertrial intervals. Each session consisted of two training trials with 10 min intertrial intervals. For each session, the platform was moved to a new location, and the drop location varied semi-randomly between trials. Trials were aborted after 60 s. Behavior was recorded with a video tracking system (Noldus). Escape latencies, distance traveled, swim paths, swim speeds, percentage time spent in each quadrant, and platform crossings were recorded for subsequent analysis.
Immunohistochemistry.
After behavioral testing, mice were anesthetized with Avertin (tribromoethanol, 250 mg/kg) and transcardially perfused with 0.9% saline. One hemibrain was drop-fixed in 4% paraformaldehyde for 48 h, and the other hemibrain was immediately frozen at −70°C. Coronal sections (30 μm) were prepared with a sliding microtome and collected for immunohistochemistry. Primary antibodies used were rabbit anti-calbindin (1:20,000; Swant), rabbit anti-Fos (1:10,000; Ab-5; Oncogene), rabbit anti-neuropeptide Y (NPY) (1:8000; Immunostar), mouse biotinylated anti-Aβ (1:400; 3D6; Elan Pharmaceuticals), mouse anti-synaptophysin (1:1000; Roche Diagnostics), and mouse anti-MAP-2 (1:40; Millipore Corporation). Sections labeled with anti-synaptophysin or anti-MAP-2 were incubated with FITC-conjugated horse anti-mouse IgG secondary antibody (1:75; Vector Laboratories) and mounted onto slides for analysis with a laser-scanning confocal microscope (MRC1024; Bio-Rad). Binding of the other primary antibodies was detected with biotinylated donkey anti-rabbit or anti-mouse (1:1000; Jackson ImmunoResearch), followed by incubation with avidin–biotin complex (Vector Laboratories). Diaminobenzidine was used as the chromagen.
Fos, calbindin, NPY, and Aβ immunoreactivities were quantified as described previously (Palop et al., 2003, 2005, 2007; Chin et al., 2005). For calbindin and NPY immunoreactivities, the integrated optical density (IOD) was determined with the BioQuant Image Analysis system (R & M Biometrics) in two sections of the dentate gyrus 300 μm apart. Relative levels were expressed as the IOD ratio of the molecular layer or mossy fibers over the stratum radiatum in the same section. The average ratio in NTG animals was defined as 1.0. The burden of Aβ deposition (plaque load) was determined by measuring the percentage area of the hippocampus covered with 3D6 immunoreactivity in four sections each 300 μm apart. The relative numbers of Fos-immunoreactive cells was determined by counting positive granule cells in every 10th serial coronal section throughout the entire rostrocaudal axis of the dentate gyrus. Confocal images of synaptophysin and MAP-2 immunoreactivity were analyzed with NIH Image software, essentially as described previously (Mucke et al., 1995; Chin et al., 2004). For each mouse, the frontal cortex and outer molecular layer of the dentate gyrus were analyzed in a total of three sections, and, for each section, an average of 10 fields was examined. The area occupied by fluorescent immunoreactivity of defined signal intensity was quantified and expressed as a percentage of the total image area. All sections were blind coded for analysis.
Aβ ELISAs and immunoblotting.
For Aβ measurements, snap-frozen hippocampal samples were homogenized in 5 m guanidine buffer. Total Aβ1–x and Aβ1-42 were quantified by ELISA as described previously (Johnson-Wood et al., 1997; Mucke et al., 2000). Total hAPP levels were assessed by Western blotting in cortical samples homogenized on ice in buffer containing 320 mm sucrose, 10 mm Tris-HCl, pH 7.4, 10 mm EDTA, 10 mm EGTA, 1% deoxycholate, 1 mm phenylmethylsulfonyl fluoride (PMSF), phosphatase inhibitor cocktails I and II (Sigma), and protease inhibitors (Roche). The samples were sonicated and centrifuged at 10,000 rpm for 10 min. Protein concentrations were determined using the Bradford assay. Equal amounts of protein were separated on 4–12% Bis-Tris gels and transferred to nitrocellulose membranes. Membranes were incubated with anti-hAPP (1:1000; 8E5; Elan Pharmaceuticals) or anti-tubulin (Sigma) overnight at 4°C. Primary antibodies were detected with HRP-conjugated anti-mouse secondary antibody (1:10,000; Calbiochem), and bands were visualized by ECL. Densitometry measurements of the bands were acquired from scanned images with Quantity One software (Bio-Rad).
Measurement of the Aβ*56 oligomer.
Aβ*56 was detected essentially as described previously (Lesné et al., 2006; Cheng et al., 2007). Briefly, cortical samples were homogenized in NP-40 lysis buffer (50 mm Tris-HCl, 150 mm NaCl, 2 mm EDTA, and 0.1% SDS). Protease inhibitor cocktail, phosphatase inhibitor cocktails 1 and 2 (Sigma), 100 mm PMSF in ethanol, and 20 mm phenanthrolene monohydrate in methanol were all added just before use. Homogenized cortical samples were immunoprecipitated overnight at 4°C with the anti-APP/Aβ antibody 6E10 (Signet) and protein G magnetic beads (Thermo Fisher Scientific). Samples were separated on 4–12% Bis-Tris gels and transferred to nitrocellulose membranes. Western blotting was done using biotinylated 6E10 antibody and ExtrAvidin (1:5000; Sigma). Levels of Aβ*56 were normalized to hAPP values on the same membranes.
Acute slice preparation.
Two- to 3-month-old hAPP–J20, hAPP–B254, and NTG control mice were anesthetized with Avertin and decapitated. Each brain was quickly removed and placed in ice-cold solution containing the following (in mm): 2.5 KCl, 1.25 NaPO4, 10 MgSO4, 0.5 CaCl2, 26 NaHCO3, 11 glucose, and 234 sucrose, pH ∼7.4 (305 mOsmol), conditions favorable for acute slices from aged mice (Aghajanian and Rasmussen, 1989). A block of brain containing hippocampus was fastened to the stage of a Vibratome-3000 with cyanoacrylate (Krazy Glue), and 400 μm horizontal slices were cut and collected in the above solution. To allow slices to recover from slicing, they were incubated for 30 min in standard artificial CSF (ACSF) (30°C) containing the following (in mm): 2.5 KCl, 126 NaCl, 10 glucose, 1.25 NaH2PO4, 1 MgSO4, 2 CaCl2, and 26 NaHCO3 (290 mOsmol; when gassed with a mixture of 95% O2–5% CO2, the pH was ∼7.4). Subsequently, slices were maintained at room temperature for at least another 30 min before recording. No recordings were made from slices >5 h after dissection. Individual slices were transferred to a submerged recording chamber where they were maintained at 30°C and perfused with ACSF at a rate of 2 ml/min.
Electrophysiological recordings.
Field EPSPs (fEPSPs) were recorded with glass electrodes (∼3 MΩ tip resistance) filled with 1 m NaCl and 25 mm HEPES, pH 7.3, and evoked every 20 s with a bipolar tungsten electrode (FHC). Recordings were filtered at 2 kHz (−3 dB, eight-pole Bessel), digitally sampled at 20 kHz with a Multiclamp 700A amplifier (Molecular Devices), and acquired with a Digidata-1322A digitizer and pClamp 9.2 software. Data were analyzed offline with pClamp9 software and OriginPro 8.0 (OriginLab). For recordings performed in CA1, the stimulating electrode was placed in the stratum radiatum at the border of CA3 and CA1, and the recording electrode was placed ∼150 μm away in CA1 stratum radiatum. For recordings performed in the DG, the stimulating electrode was placed in the medial perforant path (MPP) in the dorsal blade of the DG, and the recording electrode was also placed in the MPP ∼150 μm closer to CA3 than the recording electrode. fEPSPs in the DG were recorded in the presence of 50 μm picrotoxin (Tocris Bioscience). Measures of synaptic strength and plasticity assessed in each slice consisted of input–output (I–O) relationships, paired-pulse ratios, and LTP; these measures were recorded in each brain region in the order listed. Synaptic transmission strength in both the DG and CA1 were assessed by generating I–O curves for fEPSPs; input was the peak amplitude of the fiber volley, and the output was the initial slope of the fEPSP. For each slice, we measured the fiber volley amplitude and initial slope of the fEPSP responses to a range of stimulation from 25 to 800 μA, and a response curve was generated for both values. Stimulus strength was then adjusted to be ∼30% of the maximal fEPSP response for recordings that followed. Paired-pulse ratios were determined by evoking two fEPSPs 50 ms apart and dividing the initial slope of the second fEPSP by the initial slope of the first fEPSP (fEPSP2/fEPSP1). After a 15 min stable baseline was established, LTP was induced in CA1 by high-frequency stimulation (four 100-Hz trains of 100 stimuli every 20 s). After a 15-min stable baseline was established, LTP was induced in the DG by theta burst stimulation (a set of 10 bursts repeated 10 times every 15 s; each burst, consisting of four pulses at 100 Hz, was repeated at 5 Hz).
Statistical analyses.
Statistical analyses were conducted with GraphPad Software Prism version 4.0 or 5.0. Differences between means were analyzed by two-tailed Student's t test or one-way ANOVA with Tukey's post hoc tests. Statistical significance was determined by repeated measures ANOVA with Tukey's post hoc tests for the acquisition curves in the water maze, I–O relationships, and LTP. Data were considered significant when p < 0.05.
Results
Amyloid precursor protein and Aβ levels in hAPP–B254 and hAPP–J20 mice
To determine the extent to which the D664A mutation can prevent the development of AD-related abnormalities in hAPP mice, we compared transgenic and NTG mice from lines hAPP–B254 and hAPP–J20. The presence of the D664A mutation in hAPP–B254 mice was confirmed by sequencing of genomic DNA. In addition to the Indiana and Swedish mutations in both hAPP–B254 and hAPP–J20 lines, we identified an A → C nucleotide substitution in hAPP–B254 mice, resulting in the D → A substitution at the caspase cleavage site at amino acid residue 664 (Fig. 1A). Therefore, the original report of a G-to-C substitution encoding the D664A mutation in this construct (Galvan et al., 2006) was likely in error. However, consistent with the report by Galvan and colleagues, cortical levels of hAPP were ∼20% higher in hAPP–B254 mice than in hAPP–J20 mice (Fig. 1B).
Figure 1.

hAPP levels are higher in hAPP–B254 mice than in hAPP–J20 mice. A, The PDGF–hAPP transgene in the hAPP–J20 line carries the Swedish and Indiana familial AD mutations (Mucke et al., 2000). An additional mutation was introduced into this transgene at the D664 caspase cleavage site to generate the hAPP–B254 line (Galvan et al., 2006). Genomic DNA was amplified and sequenced in both forward and reverse directions to verify that the D664A mutation was indeed present in the hAPP–B254 mice analyzed in this study. An A-to-C nucleotide substitution was identified, resulting in a D-to-A amino acid change at the correct position (data not shown). B, Cortical homogenates from 7- to 10-month-old transgenic mice were analyzed by Western blotting and densitometric analysis of hAPP signals. Tubulin served as a loading control. hAPP levels were ∼1.2-fold higher in hAPP–B254 than hAPP–J20 mice. n = 11 mice per genotype, *p < 0.05 versus J20, t test. Bar graph shows mean ± SEM.
Aβ1–x levels were also ∼20% higher in hAPP–B254 mice than the hAPP–J20 mice when measured at 2 months of age (Fig. 2A), before Aβ deposition is detectable in the J20 line (Mucke et al., 2000). Aβ1-42 levels did not differ significantly between the two lines at 2 months, although hAPP–B254 showed a trend toward higher levels (Fig. 2B). The ratios of Aβ1-42/Aβ1–x were also similar in the two lines at 2 months (Fig. 2C). Aβ deposition in hAPP–J20 mice starts between 4 and 5 months of age (Mucke et al., 2000). Aβ deposition likely begins earlier in hAPP–B254 mice because levels of Aβ1–x, Aβ1-42, and Aβ1-42/Aβ1–x ratios rose markedly in these mice between 2 and 3–4 months (Fig. 2A–C). By 7–10 months, hippocampal Aβ deposition was approximately six times higher in hAPP–B254 mice than hAPP–J20 mice (Fig. 2D,E). Perhaps related to this difference in Aβ deposition, we also observed more astrogliosis in hAPP–B254 mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), consistent with previous findings (Galvan et al., 2008).
Figure 2.

Aβ levels in hAPP–B254 and hAPP–J20 mice. A–C, Aβ1–x and Aβ1-42 levels were measured in cortical lysates from the two transgenic lines at 2 and 3–4 months by ELISA. At 2 months, Aβ1–x (A), but not Aβ1-42 (B), was significantly higher in hAPP–B254 than hAPP–J20 mice. By 3–4 months, both Aβ1–x and Aβ1-42 levels in hAPP–B254 mice were higher than in age-matched hAPP–J20 mice and higher than in 2-month-old hAPP–B254 mice. Aβ1-42/Aβ1–x ratios (C) in hAPP–B254 and hAPP–J20 mice did not differ at 2 months but were significantly higher in hAPP–B254 mice at 3–4 months. n = 4–7 mice per genotype and age; *p < 0.05, **p < 0.005 versus J20, t test; ##p < 0.005 by ANOVA and Tukey's post hoc test. D, Immunostaining for Aβ with the 3D6 antibody at 7–10 months of age revealed a greater level of hippocampal Aβ deposition in hAPP–B254 than hAPP–J20 mice. E, The percentage area covered by 3D6-immunoreactive Aβ deposits was quantified to determine “plaque loads.” n = 11 per genotype; **p < 0.005 versus J20, t test. F, Levels of the Aβ*56 oligomer were measured in cortical homogenates from 7- to 10-month-old transgenic mice by Western blotting. G, Densitometric quantitation of Western blot signals revealed comparable levels of Aβ*56 in the two transgenic lines. H, Aβ*56/hAPP ratios were lower in hAPP–B254 than hAPP–J20 mice. n = 11 per genotype; ***p < 0.0005 versus J20, t test. All bar graphs show mean ± SEM.
Levels of the Aβ*56 oligomer, which are closely related to memory deficits in hAPP mice (Lesné et al., 2006; Cheng et al., 2007), were also approximately similar between the two lines, with hAPP–B254 mice showing a slight (10%) trend toward higher levels (Fig. 2F,G). Because hAPP–B254 mice had higher hAPP levels than hAPP–J20 mice, their ratios of Aβ*56/hAPP were significantly lower than those of hAPP–J20 mice (Fig. 2H).
The D664A mutation may delay but does not prevent hAPP/Aβ-dependent behavioral alterations in the elevated plus maze and the open field
Next, we tested both transgenic lines in a number of behavioral assays that detect abnormalities in hAPP–J20 mice. Behavior in the elevated plus maze is commonly used as a measure of anxiety (Belzung and Griebel, 2001). Several lines of hAPP mice, including hAPP–J20 mice, spend more time in the open arms of this maze than NTG controls, suggesting lower levels of anxiety or disinhibition (Chin et al., 2005; Ognibene et al., 2005; Cheng et al., 2007; Roberson et al., 2007; Meilandt et al., 2009). In agreement with these results, hAPP–J20 mice spent more time in the open arms than NTG controls when tested at 2–3 or 5–7 months of age (Fig. 3A). At 5–7 months, hAPP–B254 mice also spent more time in the open arms than NTG mice, whereas at 2–3 months, they only showed a trend in this direction (Fig. 3A). There were no significant differences between transgenic mice at either age.
Figure 3.

The D664A mutation does not prevent behavioral abnormalities in the elevated plus maze or hyperactivity in an open field arena. Groups of transgenic and NTG mice from lines B254 and J20 were analyzed in the indicated behavioral paradigms at 2–3 or 5–7 months of age. A, In the elevated plus maze, hAPP–B254 mice and hAPP–J20 mice spent more time in the open arms than NTG controls. B, In the open field, hAPP–B254 mice and hAPP–J20 mice were hyperactive compared with their NTG controls. In both paradigms, hAPP–B254 trended toward impairments at 2–3 months of age, but the differences only reached significance in the 5- to 7-month-old group. n = 9 mice per group (at 2–3 months); n = 11–15 J20 mice per group and 22–23 B254 mice per group (at 5–7 months). *p < 0.05, **p < 0.005, ***p < 0.0005 versus NTG, t test. All bar graphs show mean ± SEM.
Another behavioral phenotype shared by several lines of hAPP transgenic mice is hyperactivity in different arenas, including the open field (Chin et al., 2005; Kobayashi and Chen, 2005; Ognibene et al., 2005; Cheng et al., 2007; Roberson et al., 2007; Meilandt et al., 2009). As expected based on previous results, hAPP–J20 mice were hyperactive in the open field at 2–3 and 5–7 months relative to NTG controls (Fig. 3B). hAPP–B254 mice were also significantly more active than NTG controls at the older age but showed only a trend toward hyperactivity at the younger age (Fig. 3B). There were no significant differences in locomotor activity in the open field between transgenic mice at either age. Thus, the D664A mutation does not prevent behavioral abnormalities in the elevated plus maze and the open field but may delay their development.
The D664A mutation does not prevent hAPP/Aβ-dependent deficits in novel object recognition
To test for deficits in nonspatial memory, mice were first allowed to explore two objects during training sessions. Memory for a familiar object was then tested by replacing one of the original objects with a novel object. If mice successfully form memories of the object they previously explored, they will prefer exploring the novel object in the test session (Ennaceur and Delacour, 1988). As expected, NTG mice of both lines and ages spent significantly more time with the novel object than the familiar object during the test session (Fig. 4). In contrast, hAPP–B254 and hAPP–J20 mice at both ages did not show a preference for the novel object, indicating that hAPP mice with or without a functional caspase cleavage site have impaired object recognition learning/memory.
Figure 4.

The D664A mutation does not prevent deficits in nonspatial recognition memory. Groups of transgenic and NTG mice from lines B254 and J20 were analyzed in the novel object recognition test at 2–3 or 5–7 months of age. hAPP–B254 mice and hAPP–J20 mice did not spend more time with a novel than with a familiar object in test sessions, in contrast to NTG controls. n = 9 mice per group (at 2–3 months); n = 11–15 J20 mice per group and 22–23 B254 mice per group (at 5–7 months). *p < 0.05, **p < 0.005 novel versus familiar object or as indicated by brackets, ANOVA with Tukey's post hoc test. All bar graphs show mean ± SEM.
The D664A mutation does not prevent spatial learning and memory deficits in hAPP mice
Spatial learning and memory of mice was examined in the Morris water maze at 5–7 months of age. Mice received two training sessions per day, each consisting of two trials, to learn to use spatial cues outside of the maze to navigate to a hidden platform. The mice were trained in this task for 5 consecutive days. NTG mice from the two different lines did not differ in their ability to learn this task (Fig. 5A). Relative to these controls, hAPP–B254 and hAPP–J20 mice showed comparable deficits in acquiring this task (Fig. 5A) (repeated measures ANOVA, p < 0.0001). Because this result contradicted previous reports (Galvan et al., 2006, 2008), we repeated the test in an independent cohort of age-matched mice. For the second cohort, the training was modified slightly so that mice received three trials per sessions in two sessions per day. hAPP–B254 and hAPP–J20 mice again showed similar learning deficits relative to NTG controls (Fig. 5B) (repeated measures ANOVA, p < 0.0001). Swim speeds during the hidden platform training were not different among all groups of mice, and all groups performed equally well in the cued version of the task (data not shown).
Figure 5.

The D664A mutation does not prevent deficits in spatial learning and memory. Two independent cohorts of 5- to 7-month-old hAPP–B254 mice, hAPP–J20 mice, and NTG controls were trained in the Morris water maze for 5 d. A, In the first cohort, hAPP–B254 and hAPP–J20 mice both had longer latencies to find a hidden platform than NTG controls (p < 0.0001, repeated measures ANOVA). Total distances of the swim paths to reach the platform were also analyzed and showed similar results as latency to platform (data not shown). B, Similar to the first cohort, a second, independent cohort of hAPP–B254 and hAPP–J20 mice also had impaired learning in the hidden platform component of the task (p < 0.0001, repeated measures ANOVA). C, In the first cohort of mice, a probe trial 24 h after the hidden task training was completed showed that all groups of mice remembered the location of the platform, spending more time in the target quadrant compared with the other quadrants of the pool. D, In the second cohort, an earlier probe trial was given 2 d after the start of hidden platform training. This probe trial revealed memory deficits in both hAPP–B254 and hAPP–J20 mice, whereas the NTG littermates from both lines already showed a significant preference for the target quadrant. n = 6–9 mice per group (cohort 1; A, C); n = 5–13 mice per group (cohort 2; B, D). *p < 0.05, **p < 0.005, ***p < 0.0005, percentage time in target quadrant versus other quadrants by t test. Graphs show mean ± SEM.
Memory retention was assessed by removing the platform for a probe trial. In probe trials conducted 24 h after the last day of hidden platform training, all groups of mice spent more time in the target quadrant of the water maze in which the platform was previously located than in the other quadrants, indicating good memory retention (Fig. 5C and data not shown). Because prolonged training can obscure differences in memory retention, we administered an earlier probe trial after 2 d of hidden platform training in the second cohort of mice. In this probe trial, both groups of NTG controls already showed significant preference for the target quadrant, whereas hAPP–B254 and hAPP–J20 did not, although there was a trend toward better memory retention in hAPP–B254 mice (Fig. 5D).
The D664A mutation does not prevent alterations in calcium- and synaptic activity-related proteins in the dentate gyrus of hAPP mice
Deficits in learning and memory in hAPP–J20 mice strongly correlate with depletions of calbindin-D28K and Fos in granule cells of the dentate gyrus (Palop et al., 2003; Chin et al., 2005; Cheng et al., 2007; Roberson et al., 2007; Meilandt et al., 2008). Ectopic expression of NPY in mossy fibers and increased levels of NPY in the molecular layer of the dentate gyrus are also observed in hAPP–J20 mice and other hAPP transgenic mice and likely represent a response to Aβ-induced aberrant network excitability (Palop et al., 2007). To determine whether caspase cleavage of hAPP is required for the development of these molecular alterations, we analyzed all three biomarkers in mice that previously underwent behavioral testing. Both hAPP–B254 and hAPP–J20 mice had significant depletions of calbindin and Fos relative to NTG controls (Fig. 6A,B,E,F). In addition, both hAPP–B254 and hAPP–J20 mice displayed increased expression of NPY in the molecular layer and ectopic expression of NPY in mossy fibers (Fig. 6C,D,G,H). Of note, calbindin levels were significantly worse in hAPP–B254 mice when directly compared with hAPP–J20 mice (Fig. 6E). All other molecular alterations were not significantly different between hAPP–B254 and hAPP–J20 mice.
Figure 6.
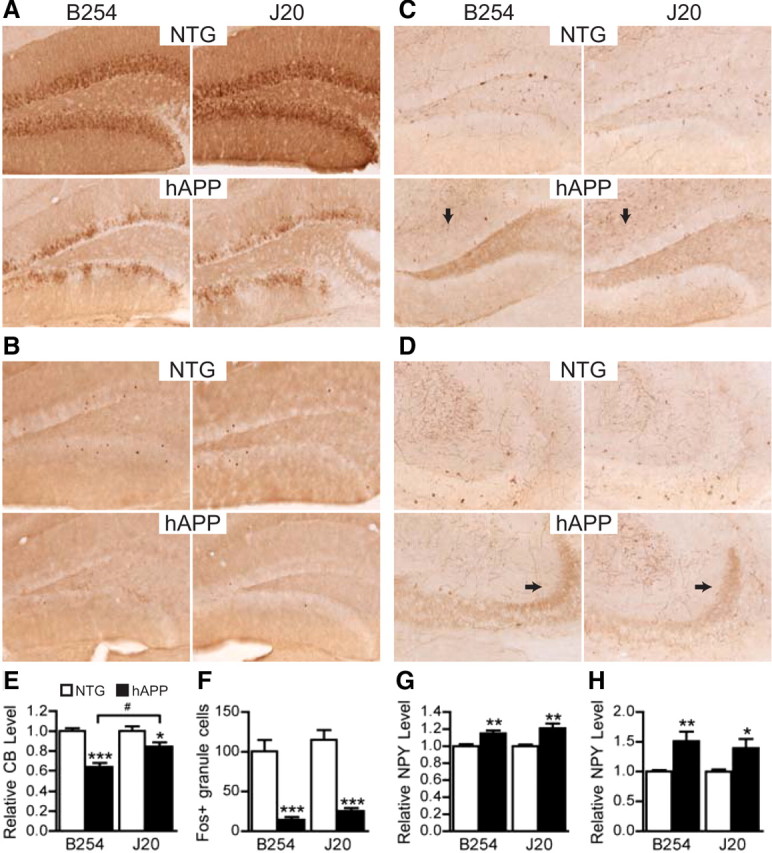
The D664A mutation does not prevent alterations in synaptic activity-related proteins in the dentate gyrus. A–D, Representative photomicrographs show brain sections from 7- to 10-month-old mice immunostained for calbindin (A), Fos (B), or NPY (C, D). Arrows indicate the molecular layer (C) and mossy fiber terminals in CA3 (D). E–H, Protein levels of calbindin (E) and NPY (G, H) were quantified by densitometry of immunoreactivities. Fos expression (F) was quantified by counting granule cells immunoreactive for Fos. Compared with NTG controls, both hAPP–B254 and hAPP–J20 mice had significant reductions in calbindin (E) and Fos (F). hAPP–B254 mice had significantly less calbindin expression than hAPP–J20 mice. Transgenic mice of both lines had comparable increases in NPY in the molecular layer of the dentate gyrus (G) and the mossy fiber pathway (H). n = 11 per genotype; *p < 0.05, **p < 0.005, ***p < 0.0005 versus NTG from the same line, t test; #p < 0.005 by ANOVA and Tukey's post hoc test. Graphs show mean ± SEM.
The D664A mutation does not prevent presynaptic and dendritic loss in the frontal cortex and dentate gyrus
Loss of synaptic terminals is a characteristic feature of AD pathology and strongly correlates with the degree of cognitive decline in humans (Terry et al., 1991; Sze et al., 1997). Reduction in synaptophysin-immunoreactive presynaptic terminals (SIPTs) is also found in transgenic mouse models of AD, including hAPP–J20 mice (Mucke et al., 2000; Chin et al., 2004; Galvan et al., 2006). The percentage area covered by SIPTs in the frontal cortex (Fig. 7A) and outer molecular layer of the dentate gyrus (Fig. 7B) was significantly less in hAPP–B254 and hAPP–J20 mice than in NTG controls. In addition to presynaptic degeneration, hAPP mice and patients with AD also show loss of dendrites and dendritic spines (Games et al., 1995; Moolman et al., 2004; Jacobsen et al., 2006; Rockenstein et al., 2007; Knobloch and Mansuy, 2008). The percentage area occupied by the dendritic marker MAP-2 in the frontal cortex (Fig. 7C) and dentate gyrus (Fig. 7D) was also significantly lower in both lines of hAPP mice.
Figure 7.

The D664A mutation does not prevent loss of synaptophysin and MAP-2. A–D, The percentage area of immunostained sections covered by synaptophysin (A, B) or MAP2 (C, D) immunoreactivity was quantified in the frontal cortex and outer molecular layer of the dentate gyrus. hAPP–B254 mice and hAPP–J20 mice showed comparable reductions in synaptophysin and MAP-2 in both regions. n = 9–11 mice per group. **p < 0.005, ***p < 0.0005 versus NTG from the same line, t test. Graphs show mean ± SEM.
The D664A mutation does not prevent electrophysiological deficits in CA1 and the dentate gyrus
We previously identified brain-region-specific electrophysiological deficits in hAPP–J20 mice (Palop et al., 2007). Here, we confirmed differences between synapses in CA1 and the dentate gyrus in hAPP–J20 mice and found them to be present also in hAPP–B254 mice. Extracellular recordings of fEPSPs at the Schaffer collateral to CA1 pyramidal cell synapse in acute hippocampal slices revealed reductions in baseline synaptic transmission strength in 3-month-old hAPP–B254 and hAPP–J20 mice (Fig. 8A,B). Baseline synaptic transmission strength in the dentate gyrus along the medial perforant pathway to granule cell synapse was normal in both hAPP lines (Fig. 8C,D). Induction of LTP and paired-pulse ratios in CA1 were also normal in both groups of transgenic mice and NTG controls (Fig. 9A–C). In contrast, LTP at the medial perforant pathway synapse on dentate gyrus granule cells was depressed in both hAPP–B254 and hAPP–J20 mice relative to NTG controls (Fig. 9D,E). At this synapse, paired-pulse ratios, which reflect short-term plasticity, were also depressed in both hAPP lines compared with NTG controls (Fig. 9F). Thus, the pattern and severity of electrophysiological synaptic alterations in the hippocampus of hAPP mice were not altered by the D664A mutation.
Figure 8.

The D664A mutation does not prevent deficits in baseline synaptic transmission strength in CA1. Acute hippocampal slices were obtained from 2- to 3-month-old mice and analyzed electrophysiologically. A, B, The input–output relationship along the Schaffer collateral–CA1 synapse was impaired in both hAPP–B254 mice and hAPP–J20 mice relative to NTG controls (p < 0.005, Tukey's post hoc analysis on repeated measures ANOVA; n = 6 slices from 3 mice per genotype). C, D, In contrast, synaptic strength at the medial perforant path synapses onto granule cells of the dentate gyrus was not affected in either hAPP–B254 or hAPP–J20 mice (n = 6 slices from 3 mice per genotype). Insets show example fEPSP traces for different levels of stimulation. Calibration: 10 ms, 0.5 mV.
Figure 9.

The D664A mutation does not prevent deficits in synaptic plasticity in the dentate gyrus. Acute hippocampal slices were obtained from 2- to 3-month-old mice and analyzed electrophysiologically. LTP and paired-pulse ratios were analyzed at synapses in CA1 and the DG. A, B, LTP was not impaired at the Schaffer collateral–CA1 synapse in either hAPP–B254 (A) or hAPP–J20 (B) mice compared with NTG controls (n = 6–7 slices from 3 mice per genotype). C, Paired-pulse ratios at the Schaffer collateral–CA1 synapse were also unimpaired in both hAPP transgenic lines relative to NTG controls (n = 6 slices from 3 mice per genotype). D, E, LTP at the medial perforant path synapse on granule cells of the dentate gyrus was depressed in both hAPP–B254 (D) and hAPP–J20 (E) mice relative to NTG controls (p < 0.005, Tukey's post hoc analysis on repeated measures ANOVA; n = 6–7 slices from 3 mice per genotype). F, Paired-pulse ratios at the medial perforant pathway were also reduced in both hAPP–B254 and hAPP–J20 mice relative to NTG controls (***p < 0.0005 vs NTG, t test; n = 6–7 slices from 3 mice per genotype). Insets show example fEPSP traces before (black line) and after (gray line) LTP induction stimuli. Calibration: 10 ms, 0.5 mV.
Discussion
Our study demonstrates that caspase cleavage at D664 in hAPP is not required for the development of deficits in learning and memory, related alterations in calcium- and synaptic activity-dependent proteins, synaptic dysfunction, and synaptodendritic loss in hAPP transgenic mice. Because many of these abnormalities are absent or much less severe in transgenic mice expressing wild-type hAPP at approximately comparable levels (Mucke et al., 2000; Palop et al., 2003, 2007), it is unlikely that they are caused by the hAPP holoprotein itself. In addition, a substantial body of positive evidence supports the conclusion that these abnormalities are caused by pathogenic assemblies of Aβ (Walsh and Selkoe, 2007; Selkoe, 2008; Sun et al., 2008). Although contributions by several other APP metabolites, or caspase cleavage of different substrate molecules, have not yet been rigorously excluded, our results make it unlikely that C31 or Jcasp plays a critical role in the pathogenesis of the AD-related abnormalities examined here.
Because several of our findings are at odds with previously published reports (Galvan et al., 2006, 2008; Saganich et al., 2006), it is important to discuss potential methodological differences and to highlight the steps we took to ensure the reliability of our data. To exclude the possibility that the transgenic mice in our hAPP–B254 colony had lost the D664A mutation, we confirmed its presence by genomic sequencing. We also confirmed that expression levels of hAPP and Aβ had not inadvertently changed in either of the lines we analyzed. This confirmation is important because the biological effects of hAPP/Aβ are clearly dose dependent. For example, transgenic mice from line hAPP–J9 carry the same transgene construct as hAPP–J20 mice, but their expression levels of hAPP and Aβ are approximately half of those found in hAPP–J20 mice (Mucke et al., 2000), likely reflecting differences in genomic integration sites and related positional effects on transgene expression. Consistent with their lower levels of hAPP/Aβ, behavioral and histopathological abnormalities are much more subtle in hAPP–J9 mice than in hAPP–J20 mice (Chin et al., 2005).
In agreement with previous reports (Galvan et al., 2006; Saganich et al., 2006), the levels of hAPP, Aβ1–x, and plaque loads were higher in hAPP–B254 than hAPP–J20 mice. Because of the dose dependence of hAPP/Aβ effects (see above), this difference might lead one to predict that abnormalities should be worse in hAPP–B254 mice than in hAPP–J20 mice, if the D664A mutation had no beneficial effects. However, behavioral, histopathological, and electrophysiological abnormalities in hAPP–B254 and hAPP–J20 mice were quite similar. At least three interpretations are possible. (1) The D664A mutation may have had a protective effect after all, making hAPP–B254 mice relatively resistant against higher levels of hAPP/Aβ. (2) The dose–response curve of Aβ-induced abnormalities may plateau above a certain level, and this level may have already been reached in the hAPP–J20 line. (3) Other measurements may be more relevant to phenotypic abnormalities than plaque loads and levels of hAPP and Aβ1–x. Indeed, abnormalities in hAPP mice are more closely related to levels of soluble Aβ1-42 and specific Aβ oligomers than to plaque loads and levels of hAPP and Aβ1–x (Hsia et al., 1999; Mucke et al., 2000; Lesné et al., 2006; Cheng et al., 2007; Meilandt et al., 2009). Consistent with the similarity in their phenotypes, hAPP–B254 and hAPP–J20 mice showed no significant differences in their levels of Aβ1-42 before plaque deposition or of the Aβ*56 oligomer, which causes memory deficits in rodents (Lesné et al., 2006). Thus, the D664A mutation does not appear to protect hAPP mice against adverse effects of soluble Aβ1-42 and Aβ*56.
Did we obtain any evidence for a protective effect of the D664A mutation? We only observed trends in this direction that did not reach statistical significance. hAPP–B254 and hAPP–J20 mice showed mostly similar abnormalities at 5–7 months of age, whereas some differences between transgenic and NTG mice reached statistical significance already at 2 months of age in hAPP–J20 mice but not in hAPP–B254 mice. This included disinhibition in the elevated plus maze and hyperactivity in an open field. Thus, it remains possible that the D664A mutation has subtle effects on the speed with which Aβ-dependent abnormalities develop in hAPP mice. However, it should be noted that there were no statistically significant differences between hAPP mice from the two transgenic lines at 2 months. We also observed a trend toward better memory retention in hAPP–B254 mice probed at an early stage during training to a hidden platform in the Morris water maze.
Can methodological differences explain the discrepancies between our study and the previous reports? In the previous studies, D664A-mutant hAPP mice performed as well as NTG controls during training to locate a hidden platform in a Morris water maze. Both the moderate expressor hAPP–B21 line at 3–4 and 7–12 months (Galvan et al., 2006; Saganich et al., 2006) and the high expressor hAPP–B254 line at 7–13 months did not differ significantly from NTG controls, although a trend toward poorer learning was noted in hAPP–B254 mice at 13 months (Galvan et al., 2006, 2008; Saganich et al., 2006). In these studies, cued platform training preceded hidden platform training, whereas the opposite sequence was used in our study. Our protocol is likely to be more challenging and, thus, may be more sensitive to spatial learning deficits, which were evident and of similar severity in hAPP–B254 and hAPP–J20 mice at 5–7 months of age. At the same time, a more challenging protocol could be less sensitive to subtle improvements in spatial learning that might occur in hAPP–B254 mice (Zhang et al., 2009).
We also used a more widely used paradigm to assess nonspatial learning and memory than that used by Galvan et al. (2006), who used unfamiliar mouse pups instead of inanimate objects. Their version of this task likely explores social behavioral domains, whereas the novel object recognition task we used focuses more squarely on nonspatial learning and memory, which did not differ between hAPP–B254 and hAPP–J20 mice. To examine potential differences in their social behaviors, we also compared the amount of time hAPP–B254 and hAPP–J20 mice spent with an unfamiliar pup; in our hands, both transgenic groups interacted with the pup as much as NTG controls (data not shown).
Synaptic function is clearly affected in diverse lines of hAPP transgenic mice (Chapman et al., 1999; Hsia et al., 1999; Moechars et al., 1999; Fitzjohn et al., 2001; Dewachter et al., 2002; Oddo et al., 2003; Palop et al., 2007). Interestingly, whether basal synaptic transmission strength, LTP, or short-term plasticity (as reflected in paired-pulse ratio measurements) is disrupted in hAPP mice depends, in part, on the particular synapse analyzed. At the perforant pathway to granule cell synapse, both LTP and paired-pulse ratios were impaired in hAPP–B254 and hAPP–J20 mice, whereas baseline synaptic transmission strength was unaffected. At the Schaffer collateral to CA1 pyramidal cell synapse, baseline synaptic transmission strength was reduced in hAPP–B254 and hAPP–J20 mice, whereas LTP and paired-pulse ratios were unaffected. These results are consistent with those obtained by us (Hsia et al., 1999; Palop et al., 2007; Sun et al., 2008) and others (Fitzjohn et al., 2001) in hAPP–J20 mice and other hAPP lines lacking the D664A mutation. Some groups identified LTP deficits at the Schaffer collateral synapse in hAPP transgenic lines, including line J20 (Moechars et al., 1999; Dewachter et al., 2002; Saganich et al., 2006). The reasons for this discrepancy in the literature remain unclear. We were unable to confirm the LTP rescue observed by Saganich et al. (2006) at the Schaffer collateral synapse in hAPP–B254 mice because we elicited robust LTP at this synapse in both hAPP–B254 and hAPP–J20 lines.
In addition to the electrophysiological analyses of synaptic functions, we also measured biochemical markers that reflect synaptic activity and the density of synaptic elements. Alterations in neuronal expression of Fos, calbindin, and NPY in hAPP mice likely result from imbalances between excitatory and inhibitory synaptic activity (Palop et al., 2003, 2007; Roberson et al., 2007; Palop and Mucke, 2009) and were either comparable between hAPP–B254 and hAPP–J20 mice or worse in the D664A-mutant group. Losses in the somatodendritic marker MAP-2 and the presynaptic terminal marker synaptophysin were also comparable between both transgenic lines.
In conclusion, our study demonstrates that most behavioral, histopathological, biochemical, and electrophysiological abnormalities in hAPP mice with high levels of Aβ1-42 are independent of caspase cleavage of hAPP at D664 and, thus, are not likely mediated by C31 or Jcasp. Inhibiting this cleavage of hAPP may therefore be of lesser therapeutic benefit than inhibiting the generation or accumulation of pathogenic Aβ1-42 assemblies.
Footnotes
This work was supported by National Institutes of Health Grants AG011385 and NS041787 (L.M.). L.M. is a consultant for Merck, serves on the scientific advisory board of iPierian, and has received research funding from Elan. We thank H. Solanoy and X. Wang for expert technical support, G. Howard for editorial review, and E. Juarez for administrative assistance.
References
- Aghajanian GK, Rasmussen K. Intracellular studies in the facial nucleus illustrating a simple new method for obtaining viable motoneurons in adult rat brain slices. Synapse. 1989;3:331–338. doi: 10.1002/syn.890030406. [DOI] [PubMed] [Google Scholar]
- Belzung C, Griebel G. Measuring normal and pathological anxiety-like behaviour in mice: a review. Behav Brain Res. 2001;125:141–149. doi: 10.1016/s0166-4328(01)00291-1. [DOI] [PubMed] [Google Scholar]
- Berger-Sweeney J, McPhie DL, Arters JA, Greenan J, Oster-Granite ML, Neve RL. Impairments in learning and memory accompanied by neurodegeneration in mice transgenic for the carboxyl-terminus of the amyloid precursor protein. Mol Brain Res. 1999;66:150–162. doi: 10.1016/s0169-328x(99)00014-5. [DOI] [PubMed] [Google Scholar]
- Bertrand E, Brouillet E, Caillé I, Bouillot C, Cole GM, Prochiantz A, Allinquant B. A short cytoplasmic domain of the amyloid precursor protein induces apoptosis in vitro and in vivo. Mol Cell Neurosci. 2001;18:503–511. doi: 10.1006/mcne.2001.1030. [DOI] [PubMed] [Google Scholar]
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Bredesen DE. Neurodegeneration in Alzheimer's disease: caspases and synaptic element interdependence. Mol Neurodegener. 2009;4:27. doi: 10.1186/1750-1326-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puoliväli J, Lesné S, Ashe KH, Muchowski PJ, Mucke L. Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Yu GQ, Kojima N, Masliah E, Mucke L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci. 2004;24:4692–4697. doi: 10.1523/JNEUROSCI.0277-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewachter I, Reversé D, Caluwaerts N, Ris L, Kuipéri C, Van den Haute C, Spittaels K, Umans L, Serneels L, Thiry E, Moechars D, Mercken M, Godaux E, Van Leuven F. Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci. 2002;22:3445–3453. doi: 10.1523/JNEUROSCI.22-09-03445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1. Behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Morton RA, Kuenzi F, Rosahl TW, Shearman M, Lewis H, Smith D, Reynolds DS, Davies CH, Collingridge GL, Seabrook GR. Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J Neurosci. 2001;21:4691–4698. doi: 10.1523/JNEUROSCI.21-13-04691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, Greenberg DA, Bredesen DE. Reversal of Alzheimer's-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl Acad Sci U S A. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan V, Zhang J, Gorostiza OF, Banwait S, Huang W, Ataie M, Tang H, Bredesen DE. Long-term prevention of Alzheimer's disease-like behavioral deficits in PDAPP mice carrying a mutation in Asp664. Behav Brain Res. 2008;191:246–255. doi: 10.1016/j.bbr.2008.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Gao Y, Pimplikar SW. The γ-secretase-cleaved C-terminal fragment of amyloid precursor protein mediates signaling to the nucleus. Proc Natl Acad Sci U S A. 2001;98:14979–14984. doi: 10.1073/pnas.261463298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-β precursor protein and amyloidogenic Ab peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Mansuy IM. Dendritic spine loss and synaptic alterations in Alzheimer's disease. Mol Neurobiol. 2008;37:73–82. doi: 10.1007/s12035-008-8018-z. [DOI] [PubMed] [Google Scholar]
- Kobayashi DT, Chen KS. Behavioral phenotypes of amyloid-based genetically modified mouse models of Alzheimer's disease. Genes Brain Behav. 2005;4:173–196. doi: 10.1111/j.1601-183X.2005.00124.x. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid β-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- McPhie DL, Golde T, Eckman CB, Yager D, Brant JB, Neve RL. β-Secretase cleavage of the amyloid precursor protein mediates neuronal apoptosis caused by familial Alzheimer's disease mutations. Mol Brain Res. 2001;97:103–113. doi: 10.1016/s0169-328x(01)00294-7. [DOI] [PubMed] [Google Scholar]
- Meilandt WJ, Yu GQ, Chin J, Roberson ED, Palop JJ, Wu T, Scearce-Levie K, Mucke L. Enkephalin elevations contribute to neuronal and behavioral impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2008;28:5007–5017. doi: 10.1523/JNEUROSCI.0590-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilandt WJ, Cisse M, Ho K, Wu T, Esposito LA, Scearce-Levie K, Cheng IH, Yu GQ, Mucke L. Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Aβ oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J Neurosci. 2009;29:1977–1986. doi: 10.1523/JNEUROSCI.2984-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moechars D, Dewachter I, Lorent K, Reversé D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML. Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol. 2004;33:377–387. doi: 10.1023/B:NEUR.0000044197.83514.64. [DOI] [PubMed] [Google Scholar]
- Mucke L, Abraham CR, Ruppe MD, Rockenstein EM, Toggas SM, Mallory M, Alford M, Masliah E. Protection against HIV-1 gp120-induced brain damage by neuronal expression of human amyloid precursor protein. J Exp Med. 1995;181:1551–1556. doi: 10.1084/jem.181.4.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller T, Meyer HE, Egensperger R, Marcus K. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics-relevance for Alzheimer's disease. Prog Neurobiol. 2008;85:393–406. doi: 10.1016/j.pneurobio.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Ognibene E, Middei S, Daniele S, Adriani W, Ghirardi O, Caprioli A, Laviola G. Aspects of spatial memory and behavioral disinhibition in Tg2576 transgenic mice as a model of Alzheimer's disease. Behav Brain Res. 2005;156:225–232. doi: 10.1016/j.bbr.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer's disease. Arch Neurol. 2009;66:435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Bien-Ly N, Massaro C, Yeung BZ, Yu GQ, Mucke L. Vulnerability of dentate granule cells to disruption of Arc expression in human amyloid precursor protein transgenic mice. J Neurosci. 2005;25:9686–9693. doi: 10.1523/JNEUROSCI.2829-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SA, Shaked GM, Bredesen DE, Koo EH. Mechanism of cytotoxicity mediated by the C31 fragment of the amyloid precursor protein. Biochem Biophys Res Commun. 2009;388:450–455. doi: 10.1016/j.bbrc.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Mucke L. 100 years and counting: prospects for defeating Alzheimer's disease. Science. 2006;314:781–784. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rockenstein EM, McConlogue L, Tan H, Power M, Masliah E, Mucke L. Levels and alternative splicing of amyloid β protein precursor (APP) transcripts in brains of transgenic mice and humans with Alzheimer's disease. J Biol Chem. 1995;270:28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB, Crews L, Masliah E. Neuroprotective effects of regulators of the glycogen synthase kinase-3β signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci. 2007;27:1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saganich MJ, Schroeder BE, Galvan V, Bredesen DE, Koo EH, Heinemann SF. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J Neurosci. 2006;26:13428–13436. doi: 10.1523/JNEUROSCI.4180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked GM, Kummer MP, Lu DC, Galvan V, Bredesen DE, Koo EH. Aβ induces cell death by direct interaction with its cognate extracellular domain on APP (APP 597–624) FASEB J. 2006;20:1254–1256. doi: 10.1096/fj.05-5032fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Zhou Y, Halabisky B, Lo I, Cho SH, Mueller-Steiner S, Devidze N, Wang X, Grubb A, Gan L. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron. 2008;60:247–257. doi: 10.1016/j.neuron.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Aβ oligomers: a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Weidemann A, Paliga K, Dürrwang U, Reinhard FB, Schuckert O, Evin G, Masters CL. Proteolytic processing of the Alzheimer's disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J Biol Chem. 1999;274:5823–5829. doi: 10.1074/jbc.274.9.5823. [DOI] [PubMed] [Google Scholar]
- Zhang J, Gorostiza O, Tang H, Bredesen DE, Galvan V. Reversal of learning deficits in hAPP transgenic mice carrying a mutation at Asp664: a role for early experience. Behav Brain Res. 2009;206:202–207. doi: 10.1016/j.bbr.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]