

Abstract
Focal adhesion kinase (FAK) functions downstream of integrins and growth factor receptors to promote tumor cell motility and invasion. In colorectal cancer, FAK is activated by amidated gastrin, a pro-tumorigenic hormone. However, it is unclear how FAK receives signals from the gastrin receptor or other G-protein-coupled receptors that can promote cell motility and invasion. The Rho guanine-nucleotide exchange factor p190RhoGEF (Rgnef) binds FAK and facilitates fibroblast focal adhesion formation on fibronectin. Here we report that Rgnef mRNA and protein expression is significantly increased during colorectal tumor progression. In human colon carcinoma cells, Rgnef complexes with FAK and upon gastrin stimulation, FAK translocates to newly-forming focal adhesions where it facilitates tyrosine phosphorylation of paxillin. siRNA-mediated knockdown of Rgnef or FAK, or pharmacological inhibition of FAK activity, is sufficient to block gastrin-stimulated paxillin phosphorylation, cell motility, and invadopodia formation in a manner dependent upon upstream cholecystokinin-2 receptor expression. Overexpression of the C-terminal region of Rgnef (Rgnef-C, aa 1279-1582) but not Rgnef-CΔFAK (aa 1302-1582 lacking the FAK binding site) disrupted endogenous Rgnef-FAK interaction and prevented paxillin phosphorylation and cell motility stimulated by gastrin. Rgnef-C-expressing cells formed smaller, less invasive tumors with reduced tyrosine phosphorylation of paxillin upon orthotopic implantation, compared to Rgnef-CΔFAK-expressing cells. Our studies identify Rgnef as a novel regulator of colon carcinoma motility and invasion and they show that a Rgnef-FAK linkage promotes colon carcinoma progression in vivo.
Keywords: gastrin, p190RhoGEF, Rgnef, FAK, invasion, colorectal cancer
Introduction
Colorectal tumor metastasis results in an overall mortality rate of 33% (1). Elucidation of the molecular mechanisms driving tumor invasion may lead to new therapeutic options. During tumor progression, cells can undergo an epithelial to mesenchymal transition associated with increased cell motility (2). This involves the dissolution of cell-cell contacts and the formation of integrin receptor-mediated cell-substratum sites termed focal adhesions (FAs) (3). Tumor progression is associated with increased local tumor cell invasion mediated in part by signals generated at FAs (4, 5). A key protein that localizes to FAs and facilitates tumor cell motility-invasion is focal adhesion kinase (FAK) (6, 7).
FAK is recruited to FAs via interactions with integrin-associated proteins such as talin and paxillin (8). FAK is a cytoplasmic protein-tyrosine kinase that is activated by integrin clustering where FAK auto-phosphorylation at Tyr-397 facilitates the recruitment of Src-family protein-tyrosine kinases into a multi-protein signaling complex (9, 10). Increased paxillin tyrosine phosphorylation by the FAK-Src complex is associated with FA formation and cell movement (11, 12). FAK expression and activity are elevated as a function of tumor progression (13) and FAK signaling promotes tumor metastasis (14).
Activation of the FAK-Src complex can also occur after G protein-coupled receptor (GPCR) stimulation of cells (15). Gastrin is a circulating peptide hormone triggering increased gastric acid secretion (16). Gastrin also promotes the growth of normal gastric mucosa as well as various gastrointestinal cancers (gastric, pancreatic, colorectal) (17, 18). Gastrin binding to the GPCR cholecystokinin receptor (CCK2R) can initiate intracellular signaling events (19, 20). Tumor-associated gastrin and CCK2R expression are associated with malignant characteristics of gastrointestinal tumors (21). How FAK is activated downstream of gastrin-CCKR2 remains undefined.
Current models linking GPCR signaling to FAK involve actin cytoskeletal stress fiber formation, tension generation, and FA formation as key events leading to increased FAK activation (19). This linkage involves RhoA GTPases controlled by in part by guanine nucleotide exchange factors (GEFs) that catalyze the exchange of GDP for GTP on RhoA (22, 23). For GPCR-associated signaling, heterotrimeric Gα13 binding to p115RhoGEF facilitates RhoGTPase activation (24). It is the RGS (regulators of G-protein signaling) domain within p115RhoGEF that connects to GPCRs and is conserved in other GEFs such as PDZ-RhoGEF and LARG (25). Notably, FAK can associate with PDZ-RhoGEF and this connection may influence FA dynamics (26). Additionally, FAK can bind to a non-RGS-containing RhoGEF termed Rgnef (p190RhoGEF) that promotes RhoA activation and FA formation downstream of integrins (27, 28). Rgnef can localize to FAs and this is dependent upon the FAK-binding region (residues 1292-1301) within the Rgnef C-terminal domain. Although over-expression of CCKR2 potentiates gastrin-stimulated FAK activation (29, 30), it remains unknown whether particular GEF interactions with FAK facilitate gastrin-CCK2R stimulated signaling events or colon carcinoma tumor progression.
Herein, we find that Rgnef protein expression becomes elevated during colon tumor progression and that a complex between Rgnef and FAK controls gastrin-stimulated paxillin phosphorylation, cell motility, and matrix degradation. As Rgnef C-terminal domain over-expression prevented orthotopic colon carcinoma tumor growth and local invasion in a FAK binding-dependent manner, our findings establish Rgnef as a novel regulator of FAK signaling downstream of GPCRs involved in promoting tumor progression.
Materials and Methods
Antibodies and reagents
Antibodies to Hic-5, c-Src, and c-Yes (C-4) were from Santa Cruz Biotechnology. Monoclonal antibody (mAb) to FAK was from Millipore. Paxillin mAb was from BD Biosciences. β-actin mAb was from Sigma. Green fluorescent protein (GFP) mAb was from Covance and polyclonal antibodies to mCherry were from Clontech. FAK Tyr-397 (pY397) and paxillin Tyr-31 (pY31) phosphospecific antibodies were from Invitrogen. Affinity-purified rabbit antibodies to Rgnef were generated using peptides 1247-1265 (EDVHLEPHLLIKPDPGEPP, rabbit 3647 used for immunoblotting) and 1502-1519 (ERLREGQRMVERERQKMR, rabbit 1397 used for immunoprecipitation and immunostaining) coupled to keyhole limpet hemocyanin (OpenBiosystems). Human amidated gastrin (G17 peptide) was from Calbiochem. The PF-228 FAK inhibitor was from Pfizer (31).
Lentiviral constructs
Human FAK, c-Src, paxillin, and a scrambled (Scr) short-hairpin RNA (shRNA) were used as described (32). For knockdown of human Rgnef, combined shRNA and GFP protein expression was achieved using pLentiLox3.7 with the sense 5′TGAATTCTGTTGCCATCATATtcaagagATATGATGGCAACAGAATTCTTTTTTC-3′ primer encompasses then end of the U6 promoter, the shRNA loop (lower case letters), and an added 3′C to generate an Xho1 site. The anti-sense primer was 5′-TCGAGAAAAAAGAATTCTGTTGCCATCATATctcttgaATATGATGGCAACAGAATTCA-3′. For mCherry protein expression, polymerase chain reaction (PCR) was used to add NheI and BamH1 ends and mCherry was cloned into pCDH-CMV-MCS (System Biosciences). The Rgnef C-terminal regions (Rgnef-C, 1279-1582 and Rgnef-CΔFAK 1302-1582) were amplified via PCR from pHM6-Rgnef-C (27) and cloned into pCDH-mCherry for lentivirus production.
Cell culture
DLD-1, Caco-2, HCT-116, HT-29, and SW-480 human colorectal carcinoma cells were obtained from American Type Culture Collection. Early passage cells were used for all experiments and they were not re-authenticated. All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1 mM non-essential amino acids, 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. DLD-1 cells expressing either Scr, anti-FAK, anti-paxillin, anti-c-Src, or anti-Rgnef shRNA and GFP-Rgnef over-expressing cells were obtained by fluorescence activated cell sorting (FACS) and maintained as pool populations. DLD-1 mCherry, DLD-1 mCherry-Rgnef-C, and DLD-1 mCherry-Rgnef-CΔFAK were obtained by FACS. DLD-1 cells contain mutations in K-Ras and p53 (33).
Cell lysis, immunoprecipitation (IP) and blotting
Sub-confluent cells were starved overnight (serum-free media) and dimethyl sulfoxide (DMSO) or gastrin (200 nM) were added for the indicated time. Cells were solubilized in ice-cold modified protein lysis buffer containing 1% Triton X-100, 0.5% sodium deoxycholate and 0.1% SDS (34). For immunoprecipitations, antibodies (2.5 μg) were incubated with lysates (500 to 1 mg total protein) at 4°C and collected by bead binding (Invitrogen). Proteins were resolved by SDS-PAGE and membrane immunoblotting performed as described (34). Relative protein expression levels and phosphospecific antibody reactivity were quantified using Image J (v1.43).
Immunofluorescent staining
Cells were plated at low density onto 1% gelatin-coated glass coverslips until colonies of ~30 cells were formed (48-60 h). After overnight serum starvation, DMSO or gastrin (200 nM) was added for 45 to 60 min. Cells were fixed (3.7% paraformaldehyde in PBS, 10 min), permeabilized (0.1% Triton X-100 in PBS, 10 min) and coverslips were blocked with 2% bovine serum albumin in PBS for 1 h. For staining, coverslips were incubated with anti-FAK or anti-paxillin antibodies at 4°C overnight and visualized using either fluorescein isothiocyanate-conjugated (FITC) goat anti-mouse IgG, Alexa-fluor-488 goat anti-mouse IgG, Texas Red-phalloidin, and nuclei stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were acquired using a spinning disk confocal microscope (IX81, Olympus) and monochrome camera (ORCA ER, Hamamatsu).
Anti-Rgnef tumor staining
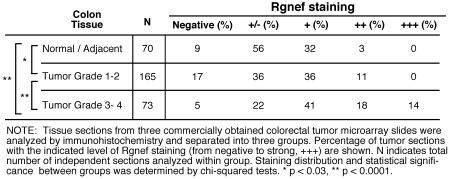
Micro-array slides (US Biomax; CO1002, CO701, CO2085a) with colon tumor (with clinical stage and pathology grade) and adjacent normal tissue sections were incubated at 60°C for 90 min to facilitate tissue core adhesion, paraffin was removed with xylene and tissue cores were rehydrated. Antigen retrieval was performed by boiling in 10 mM sodium citrate pH 6.0 for 5 min. Slides were washed, endogenous peroxidase activity was quenched with 0.3% hydrogen peroxide (10 min), and tissue cores blocked for 45 min (block buffer: PBS with 5% normal goat serum and 0.3% Triton X-100). Affinity-purified rabbit polyclonal Rgnef antibodies (rabbit 1397) were diluted 1:150 in block buffer, incubated overnight at 4°C, and followed by biotinylated goat-anti-rabbit IgG (Vector Labs) at 1:300 for 30 min. Rgnef staining was visualized using the Vectastain ABC Elite reagent, 3,3-diaminobenzidine tetrahydrochloride peroxidase substrate, and counterstained with Hematoxylin QS (Vector Labs). Individual tissue cores were scored blind and recorded as negative (−), +/−, +, ++, +++ to designate levels of Rgnef staining.
Colony scattering-motility assay
Cells were plated at low density onto 1% gelatin-coated glass coverslips until colonies of ~30 cells were formed (48-60 h). After overnight serum starvation, DMSO or gastrin (200 nM) was added and after 18 h, cell colonies (>200 per experiment) were evaluated by phase-contrast microscopy. A colony with <50% cell-cell contacts was considered scattered. At least three independent experiments were performed.
In situ zymography
Oregon green® 488-conjugated gelatin (Invitrogen) degradation assays performed as described (35) with minor modifications. Briefly, after gelatin cross-linking and quenching-washing of glass slides, cells were plated in DMEM containing 10% FBS with DMSO or gastrin (200 nM) and incubated at 37°C for 18 h followed by 3.7% paraformaldehyde fixation. Phase and fluorescent images of cells were evaluated for areas of degradation. Positive values are presented as percent of total cells (>200 cells per experimental condition) with at least one degradation patch (independent of size).
Orthotopic colorectal tumor growth
6 week old female nude (nu/nu) mice (Harlan Labs) were housed in pathogen-free conditions under approved institutional animal care and use protocols. One million DLD-1 cells expressing mCherry, mCherry-Rgnef-C, or mCherry-Rgnef-CΔFAK were mixed with growth factor-reduced Matrigel (BD Biosicences) and injected (in 10 μl) sub-mucosally into the distal posterior rectal wall as described (36). After 24 days, the peritoneal cavity was opened and fluorescent tumors visualized (OV100, Olympus). Tumors were resected, weighed, and the posterior colon area was imaged a second time to determine the extent of local tumor invasion. Tumors were divided in half and either homogenized in protein lysis buffer for immunoblotting or imbedded in Optimal Cutting Temperature compound (Tissue Tek), frozen in liquid nitrogen, thin sectioned (7 μM), and mounted onto glass slides. Sections were stained with hematoxylin and eosin (H&E) and images acquired at 10X with a color camera (Infinity1-3C, Lumenera). Detection of mCherry fluorescence and anti-desmin (Thermo Scientific) staining was performed by methods as above. Multi-color images were sequentially captured at 10X (UPLFL objective, 1.3 NA; Olympus) pseudo-colored, overlaid, and merged using Photoshop CS3 (Adobe).
Statistical methods
For cell culture experiments, mean values and standard deviation (SD) of at least triplicate experimental points were calculated. Student’s t-test was used to compare mean value differences. For tumor experiments, differences between groups were determined using one-way ANOVA with Tukey post hoc. Differences between pairs of data were determined using an unpaired two-tailed student’s t-test. The distribution (negative to +++) of Rgnef tumor staining was evaluated using Pearson’s chi-square test. Statistical analyses were performed using GraphPad Prism (version 5.0b).
Results
Elevated expression of Rgnef in colon cancer
Rgnef (p190RhoGEF) is a ubiquitously-expressed protein consisting of a central Dbl/pleckstrin-homology catalytic region associated with GEF activity (37) (Fig. 1A). To determine if Rgnef expression is altered in colon cancer, semi-quantitative reverse transcriptase PCR was performed using RNA samples obtained from colorectal tumor and paired normal colorectal tissue (Supplemental Fig. 1A). Surprisingly, an Rgnef-specific band was detected in only 25% (4 of 16) of cancerous tissues. These positive samples were tumor grade 3-4 whereas Rgnef was below the level of experimental detection in fourteen grade 1-2 tumors and sixteen normal tissue samples (Supplemental Fig. 1A). To extend these observations, polyclonal anti-peptide antibodies were generated to Rgnef (Fig. 1A), affinity-purified, and verified to be specific for Rgnef detection (Supplemental Figs. 1B and C). Anti-Rgnef staining of colon cancer tissue micro-array slides revealed a number of samples with strong (++ to +++) Rgnef staining (Fig. 1B). In the analysis of 308 tissue sections ranging from normal colon to tumor grade 4, moderately increased Rgnef staining was detected in tumor grade 1-2 compared to normal (p<0.03) samples, and further elevated Rgnef levels were detected in tumor grade 3-4 compared to grade 1-2 (p<0.0001) samples (Table I). Together, these results support the conclusion that Rgnef mRNA and protein expression increases as a function of colon cancer tumor progression.
Figure 1.

Elevated Rgnef expression during colon cancer progression. A, Schematic diagram of Rgnef (p190RhoGEF) showing locations of leucine-rich (L-rich), zinc-finger-like (Zn), Dbl/pleckstrin homology (DH/PH), and coiled-coil (C-coil) domains. Indicated is the FAK binding site (1292-1301) and the peptide regions used for polyclonal antibody production. B, Representative colon carcinoma tumor sections stained with antibodies to Rgnef (brown) reveals increased Rgnef expression (from minimal −/+ to strong +++) as a function of tumor stage. Sections were counterstained with hematoxylin (blue). Scale bar is 0.1 mm.
Table 1.
Relative Rgnef protein expression in colon carcinoma microarray slides
|
FAK activation is important for gastrin-stimulated paxillin tyrosine phosphorylation and cell scattering
Immunoblotting analyses revealed ubiquitous Rgnef expression in DLD-1, Caco-2, HCT-116, HT-29, and SW-480 colon carcinoma tumor cells (Fig. 2A). FAK was expressed in all cells except SW-480 wherein FAK-related Pyk2 kinase expression was detected. Previous studies showed that gastrin enhanced FAK and paxillin tyrosine phosphorylation in Colo320 colon carcinoma cells over-expressing the CCK2R receptor (29). As gastrin stimulates signaling within DLD-1 cells (38), we tested whether gastrin enhanced FAK and paxillin tyrosine phosphorylation via endogenous receptors in DLD-1 cells. Phospho-specific blotting analyses revealed enhanced FAK Tyr-397 (pY397), FAK Tyr-576/Tyr-577 (pY576/pY577) as well as paxillin Tyr-31 (pY31) phosphorylation within 40 min of 200 nM gastrin addition to DLD-1 cells (Supplemental Fig. 2A). Increased FAK and paxillin tyrosine phosphorylation by gastrin was dependent on intrinsic FAK activity as shown by pharmacological FAK-specific PF-228 (1 μM) inhibition (Fig. 2B).
Figure 2.

Gastrin stimulates FA formation, translocation of FAK to FAs, and the formation of an Rgnef-FAK-paxillin complex. A, Rgnef, FAK, and Pyk2 expression compared to β-actin in human colon carcinoma cell lines as determined by immunoblotting. B, Pharmacological FAK inhibitor addition (PF-228, 1 μM) blocks gastrin-stimulated FAK and paxillin tyrosine phosphorylation as determined by immunoprecipitation (IP) and immunoblotting. C, Serum-starved DLD-1 colonies were stimulated by gastrin or DMSO (control) addition and after 60 min, processed for immunostaining. Paxillin (FA marker) was in a punctate cytoplasmic distribution in DMSO-treated cells and upon gastrin stimulation, increased paxillin staining at peripheral cell projections and at cell-cell junctions (arrows) was observed that were also sites of filamentous actin accumulation. Cell nuclei were visualized with DAPI. Accumulation of FAK staining at peripheral FAs upon gastrin addition to DLD-1 cells. Scale bar is 10 μm. D, Rgnef-FAK complex formation in DLD-1 cells as determined by co-IP with antibodies to FAK and Rgnef and reciprocal immunoblotting. Gastrin stimulates increased FAK-paxillin association. Antibodies to FAK co-IP Rgnef and paxillin after gastrin addition.
Interestingly, gastrin addition to DLD-1 cells increased FA formation as detected by vinculin staining (Supplemental Fig. 2B, arrows). Gastrin also triggered the dissolution of cell-cell contacts associated with the loss of E-cadherin surface expression (Supplemental Fig. 2C). Increased paxillin staining at sites resembling FAs was detected both within the center of DLD-1 cell colonies and at peripheral sites of enhanced membrane ruffling stimulated by gastrin addition (Fig. 2C, arrows). Notably, FAK was localized in the cytoplasm of starved DLD-1 cells and exhibited enhanced FA localization upon gastrin addition (Fig. 2C). PF-228 FAK inhibition prevented gastrin-initiated FA formation (data not shown) and blocked gastrin-triggered cell scattering-motility (Supplemental Fig. 2D). Importantly, lentiviral-mediated shRNA stable knockdown of CCK2R in DLD-1 cells prevented both gastrin-triggered cell scattering and elevated paxillin tyrosine phosphorylation compared to Scr shRNA and parental DLD-1 cells (Supplemental Fig. 3). These results support the notion that gastrin promotes CCK2R-dependent FAK catalytic activation and paxillin tyrosine phosphorylation in DLD-1 cells associated with increased FA formation and the acquisition of a motile cell phenotype.
Rgnef is in a signaling complex with FAK in colon carcinoma cells
In fibroblasts, cell replating onto fibronectin facilitates FA formation and the recruitment of Rgnef into a signaling complex with FAK at these sites (28). In DLD-1 cells, Rgnef co-immunoprecipitates (co-IPs) with antibodies to FAK in starved cells and FAK co-IPs with antibodies to Rgnef (Fig. 2D). Rgnef-FAK complex formation is not altered by gastrin addition. The identity of the smaller Rgnef-immunoreactive bands associated with FAK IPs after gastrin stimulation was not determined. Notably, increased paxillin association was detected with FAK by co-IP within 30 to 60 min after gastrin addition (Fig. 2D). This was verified by paxillin co-IP analyses and is consistent with the notion that FAK and paxillin are recruited to newly-forming FAs upon gastrin addition. Although paxillin co-IPs with over-expressed Rgnef upon fibronectin-mediated fibroblast adhesion (28), endogenous Rgnef did not detectably co-IP with paxillin in lysates of gastrin-stimulated DLD-1 cells (Fig. 2D). These results support a model-hypothesis that gastrin triggers FA formation, FAK recruitment to these sites, and FAK-dependent paxillin tyrosine phosphorylation potentially involved in FA maturation events (39).
Rgnef knockdown reveals its importance in gastrin-stimulated paxillin tyrosine phosphorylation, cell scattering, and invasion
Although we could not detect Rgnef localization to FAs upon gastrin stimulation of DLD-1 cells (data not shown), a key role for Rgnef in gastrin-stimulated signaling was revealed through the analysis of stable knockdown DLD-1 cells (Fig. 3). Lentiviral-mediated shRNA expression was used to individually knockdown Rgnef, FAK, c-Src, and paxillin in DLD-1 cells and >80% stable reduction in target protein expression was achieved (Fig. 3A). Interestingly, in FAK, c-Src, and paxillin shRNA-expressing cells, elevated expression of c-Yes tyrosine kinase was detected (Fig. 3A) and increased Hic-5 expression was detected in paxillin shRNA cells (data not shown). Despite potential compensatory changes associated with stable shRNA expression, Rgnef and FAK knockdown significantly prevented cell scattering upon gastrin addition compared to scrambled (Scr) shRNA DLD-1 controls (Fig. 3B and C). Paxillin and c-Src knockdown did not significantly affect gastrin-stimulated cell scattering and it was unclear whether this was associated with compensatory cell alterations. Importantly, knockdown of either Rgnef or FAK prevented gastrin-stimulated paxillin tyrosine phosphorylation, thus reinforcing the importance of the Rgnef-FAK signaling complex (Fig. 3D).
Figure 3.

Knockdown of Rgnef or FAK prevent gastrin-stimulated DLD-1 cell scattering and paxillin tyrosine phosphorylation. A, Lysates from parental DLD-1 or stable scrambled (Scr) or c-Src, FAK, paxillin, or Rgnef short-hairpin RNA (shRNA)-expressing DLD-1 cells were evaluated by immunoblotting with β-actin expression used as a control. B, Representative images of the indicated starved DLD-1 shRNA-expressing cell colonies treated with DMSO or gastrin (200 nM) for 24 h. Scattered cells exhibit loss of cell-cell contacts. Scale bar is 200 μm. C, FAK or Rgnef knockdown prevent gastrin-stimulated DLD-1 scattering. Colony scattering percentage was determined and values are means +/− SD from 3 experiments. Statistical significance is compared to Scr shRNA DLD-1 cell results. D, Time course of gastrin-stimulated paxillin tyrosine phosphorylation and Image J analyses of the immunoblots (right) reveals that Rgnef or FAK knockdown prevent gastrin-stimulated paxillin phosphorylation in DLD-1 cells.
As previous studies showed that gastrin promoted colon carcinoma protease secretion and invasion (40, 41) and that FA formation can be a precursor to invadopodia formation (42), Rgnef and FAK shRNA-expressing DLD-1 cells were evaluated in an in situ zymography-cell invasion activity assay (Fig. 4). Knockdown of FAK or Rgnef significantly reduced gastrin-induced gelatin degradation activity (visualized as cell-associated dark spots) compared to Scr shRNA-expressing DLD-1 cells (Fig. 4A and B). Therefore, gastrin-stimulated cell spreading was correlated with the transition to an invasive cell phenotype. To establish a direct link between Rgnef and cell invasion, a GFP-Rgnef fusion protein was stably-overexpressed in DLD-1 cells (Fig. 4C). GFP-Rgnef over-expression significantly increased cell scattering-motility and gelatin degradation activity compared to GFP-DLD-1 cells (Fig. 4C and D). Together, these results show that both Rgnef and FAK expression are required for gastrin-stimulated DLD-1 cell motility and the generation of an invasive cell phenotype.
Figure 4.

Rgnef and FAK facilitate gastrin-stimulated DLD-1 matrix degradation. A, In situ gelatin zymography analyses after DMSO (control) or gastrin addition were performed with parental DLD-1 cells or the indicated shRNA-expressing DLD-1 cells and analyzed by microscopy. Values represent percent of cells with associated gelatin degradation patches. Values are the mean +/− SD from 3 experiments. Statistical significance is compared to Scr shRNA DLD-1 cell results. B, Representative phase and fluorescent images of Scr- or Rgnef shRNA-expressing DLD-1 cells plated on Oregon green gelatin under DMSO (control) or gastrin-treated conditions. Scale bar is 50 μm. C, Lysates from DLD-1 cells expressing GFP-(vector) or GFP-Rgnef were analyzed by anti-GFP, -Rgnef, and β-actin blotting. Colony scattering percentage was determined and values are means +/− SD from four experiments. D, Gastrin-stimulated gelatin degradation. Values represent percent of cells with degradation patches and are the mean +/− SD from 3 experiments.
Blocking Rgnef-FAK interaction prevents gastrin-stimulated paxillin tyrosine phosphorylation and cell scattering
To test the importance of Rgnef-FAK signaling complex in mediating gastrin signaling, the Rgnef C-terminal domain (Rgnef-C, residues 1279-1582) or Rgnef-C lacking the FAK binding site (Rgnef-CΔFAK, residues 1302-1582) were stably over-expressed in DLD-1 cells as mCherry fusion proteins (Fig. 5A). Rgnef-C levels were markedly elevated compared to endogenous Rgnef expression (Fig. 5A). Rgnef-C constitutively bound to FAK and disrupted the association with endogenous Rgnef whereas Rgnef-CΔFAK expression did not effect FAK-Rgnef association (Fig. 5A). Importantly, Rgnef-C but not Rgnef-CΔFAK prevented gastrin-stimulated paxillin tyrosine phosphorylation (Fig. 5B) and blocked gastrin-initiated cell scattering (Fig. 5C and D). These results support the notion that Rgnef-C acts as a dominant-negative inhibitor of the endogenous Rgnef-FAK signaling complex.
Figure 5.

Over-expression of the C-terminal Rgnef region (Rgnef-C) binds FAK and acts in a dominant-negative manner to block gastrin-stimulated cell scattering and paxillin tyrosine phosphorylation. A, Stable expression of mCherry, mCherry Rgnef-C (67 kDa), or mCherry Rgnef-CΔFAK (65 kDa) in DLD-1 cells. Anti-Rgnef blotting shows Rgnef-C over-expression compared to endogenous Rgnef. Co-IP analyses reveal that FAK-Rgnef-C association and that endogenous Rgnef does not co-IP with FAK in cells expressing Rgnef-C. B, Inhibition of gastrin-stimulated paxillin tyrosine phosphorylation in Rgnef-C-expressing cells and quantified by Image J analysis (below). C, Representative images of starved DLD-1 mCherry, Rgnef-C, or Rgnef-CΔFAK cell colonies treated with DMSO or gastrin (200 nM) for 24 h. Scale bar is 100 μm. D, Rgnef-C prevents gastrin-stimulated DLD-1 scattering. Colony scattering percentage was determined and values are means +/− SD from 2 experiments.
Rgnef-C inhibits DLD-1 orthotopic tumor growth and invasion
Prior to evaluating the tumor growth characteristics of Rgnef-C DLD-1 cells, equivalent expression of mCherry Rgnef-C and mCherry Rgnef-CΔFAK was confirmed by flow cytometry and no significant cell growth differences either under adherent or suspended conditions were observed (Supplemental Fig. 4). One million mCherry-labeled DLD-1 cells were injected into the distal posterior rectum of nude mice and allowed to grow as orthotopic tumors (Fig. 6). After 24 days, the peritoneal cavity was opened and fluorescent tumors visualized prior to and after surgical resection (Supplemental Fig. 5). Interestingly, mCherry DLD-1 and mCherry Rgnef-CΔFAK tumors were difficult to completely remove as they had become locally invasive into the distal posterior musculature of the mice. In contrast, mCherry Rgnef-C DLD-1 tumors were primarily localized to the colon surface and were readily surgically removed (Supplemental Fig. 5).
Figure 6.

Rgnef-C inhibits DLD-1 orthotopic tumor growth and invasion associated with reduced paxillin tyrosine phosphorylation in vivo. A, Orthotopic DLD-1 mCherry, Rgnef-C, or Rgnef-CΔFAK tumor size in nude mice as box-whisker diagrams. B, Tumor lysates were evaluated by paxillin phospho-specific (pY31) and total paxillin immunoblotting. Top, representative analyses from four different DLD-1, DLD-1 Rgnef-C, or DLD-1 Rgnef-CΔFAK tumors. Bottom, ratio of pY31 paxillin to total paxillin reveals that the FAK binding region of Rgnef-C inhibits paxillin phosphorylation in vivo. C, Representative tumor sections were stained with H&E and reveal surface-associated growth of DLD-1 Rgnef-C tumors (T) on the muscularis propria (MP) of the colon (C) and the interface of DLD-1 Rgnef-CΔFAK tumors with the posterior musculature (M). Scale bar is 1 mm. D, Tumor sections were stained for desmin (green), cell nuclei (blue), and tumor-associated mCherry (red) expression. DLD-1 Rgnef-C tumors are encapsulated whereas DLD-1 Rgnef-CΔFAK are locally invasive within the musculature. Scale is 1 mm. Differences between groups were evaluated using ANOVA followed by the Tukey post hoc test. Box-whisker diagrams show the distribution of the data: square, mean; bottom line, 25th percentile; middle line, median; top line, 75th percentile; and whiskers, 5th or 95th percentiles.
Rgnef-C but not Rgnef-CΔFAK expression significantly inhibited DLD-1 tumor growth in vivo (Fig. 6A), despite under-estimating excised tumor size due to local tumor invasion. Analysis of tumor cell lysates by immunoblotting showed that paxillin tyrosine phosphorylation was significantly inhibited in Rgnef-C but not Rgnef-CΔFAK tumors compared to DLD-1 controls (Fig. 6B). H&E staining of tumor sections confirmed that DLD-1 Rgnef-C tumors were at the surface of the colon muscularis propria and Rgnef-CΔFAK tumors were connected to the posterior musculature (Fig. 6C). Combined immunofluorescent staining of tumor sections for the muscle intermediate filament protein desmin (green), intrinsic mCherry fluorescence (red) for tumor cell detection, and DAPI staining (blue) for cell nuclei revealed that Rgnef-C tumors were encapsulated by a host-associated cells and not detectably invasive into the muscularis propria at the colon surface (Fig. 6D). Conversely, Rgnef-CΔFAK tumor cells were extensively invading into the surrounding musculature (Fig. 6D). Together, our results support the conclusion that Rgnef binding to FAK plays important roles in promoting both gastrin-stimulated DLD-1 cell motility in vitro and tumor progression in vivo associated with the regulation of paxillin tyrosine phosphorylation.
Discussion
Epithelial cancer cells metastasize in a series of linked, sequential steps initiated by extracellular matrix remodeling followed by local tumor invasion. Elucidation of the molecular processes contributing to an invasive cell phenotype is critical to understanding tumor cell metastasis. In this study, we have identified a new role for Rgnef within colon cancer cells in facilitating FAK-associated paxillin tyrosine phosphorylation initiated by gastrin and dependent upon CCK2R expression.
There is a growing body of literature implicating FAK signaling in cancer (13). In colon cancer, transcription factors such as Eps8 elevate FAK expression (43) and tyrosine phosphorylation of proteins such as FAK and paxillin are correlated with an invasive cell phenotype (44, 45). The molecular pathways connecting GPCRs to FAK activation remain undefined. A canonical pathway for gastrin and GPCR-mediated FAK activation is likely to involve Gα12/Gα13 proteins leading to RhoA GTP binding, Rho-kinase activation, myosin light chain phosphorylation, actin stress fiber formation, and FA assembly triggering integrin clustering leading to FAK-associated paxillin phosphorylation (19). Where Rgnef fits into this pathway is unclear. However, our results support the conclusion that Rgnef may be a key RhoGEF controlling RhoA GTP binding as shRNA knockdown (Figs. 3 and 4) or dominant-negative Rgnef expression (Fig. 5) prevented gastrin-initiated cell-cell dissociation, FA formation (data not shown), and FAK-associated signaling as measured by paxillin tyrosine phosphorylation. The fact that Rgnef and FAK form a complex in quiescent DLD-1 colon carcinoma cells prior to gastrin-initiated FA formation (Fig. 2) suggests that an Rgnef-FAK signaling complex may coordinate or localize RhoA activation and FA formation in response to gastrin. This role is consistent with Rgnef function in normal fibroblasts as an important regulator of RhoA, FA formation, and motility downstream of integrins (28).
While RhoA activation has been implicated in colorectal tumor progression (46) and active RhoA is localized to podosomes or invadopodia (35), very little is known about the key regulators contributing to elevated RhoA signaling in colon cancer. Our results using the Rgnef C-terminal domain as a dominant-negative inhibitor blocking the formation of a Rgnef-FAK signaling complex reveal that a direct binding interaction with FAK is important in regulating gastrin-stimulated paxillin phosphorylation, cell motility, and DLD-1 tumor progression (Figs. 5 and 6). Interestingly, Rgnef-C expression did not affect tumor cell proliferation in vitro, but inhibited orthotopic tumor growth in nude mice implicating a role for modulation of the tumor microenvironment. This dominant-negative effect was lost upon deletion of the FAK binding site in Rgnef-C. Although how Rgnef-C limits tumor growth in vivo remains unclear, Rgnef-C expression resulted in lower levels of tumor-associated paxillin tyrosine phosphorylation and less invasive tumors (Fig. 6). As paxillin tyrosine phosphorylation is increased upon epithelial to mesenchymal transitions in normal cells (47), involved in the assembly of FAs (12, 39), and has been correlated with tumor invasion (48), our in vitro and in vivo results are consistent with the hypothesis that Rgnef-C blocks a FAK-paxillin motility and invasion signaling pathway.
To date, the best characterized GEFs involved in colon cancer are the adenomatous poyposis coli-associated proteins Asef1-Asef2 involved in promoting Rac1 and Cdc42 activation and knockout studies have revealed their importance in promoting tumor progression (49). RhoA regulation is also important in promoting tumor spread (46), but the GEFs controlling this pathway remain undefined. Our findings that Rgnef mRNA and protein levels are elevated as a function of colon carcinoma tumor progression add important biological relevance to our mechanistic studies characterizing the importance of Rgnef-FAK interactions. Future studies will explore whether changes in Rgnef expression occur within other cancers and whether this parallels elevated expression of FAK/Pyk2 within tumors. In summary, our results support the importance of Rgnef-FAK signaling in promoting colorectal cancer progression and complement mouse genetic studies showing that loss of FAK expression prevents colon cancer tumorigenesis downstream of Wnt/c-Myc signaling (50). FAK inhibitors may serve to control the invasive and metastatic properties of advanced colorectal cancers.
Precis (edited).
Findings reveal a mechanism through which the pro-tumorigenic gastrointestinal hormone gastrin acts to promote invadopodia formation and invasive motility of colorectal carcinoma cells.
Supplementary Material
Acknowledgements
Supported by NIH CA102310 and an American Heart Association Established Investigator Award (0540115N) to David Schlaepfer. Ju-Ock Nam was partially supported by a fellowship from the Korean Science Foundation (KRF-2008-357-E00007). Alok Tomar was supported by an American Heart Association postdoctoral fellowship (0825166F).
Abbreviations
- CCK2R
cholecystokinin 2 receptor
- DMEM
Dulbecco’s modified eagles medium
- DMSO
dimethyl sulfoxide
- FACS
fluorescence activated cell sorting
- FA
focal adhesion
- FAK
focal adhesion kinase
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- GEF
guanine-nucleotide exchange factor
- GFP
green fluorescent protein
- H&E
hematoxylin and eosin
- IP
immunoprecipitation
- mAb
monoclonal antibody
- PCR
polymerase chain reaction
- Scr
scramble shRNA
- shRNA
short-hairpin RNA
References
- 1.Cunningham D, Atkin W, Lenz HJ, et al. Colorectal cancer. Lancet. 2010;375:1030–47. doi: 10.1016/S0140-6736(10)60353-4. [DOI] [PubMed] [Google Scholar]
- 2.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Tilghman RW, Parsons JT. Focal adhesion kinase as a regulator of cell tension in the progression of cancer. Semin Cancer Biol. 2008;18:45–52. doi: 10.1016/j.semcancer.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rathinam R, Alahari SK. Important role of integrins in the cancer biology. Cancer Metastasis Rev. 2010;29:223–37. doi: 10.1007/s10555-010-9211-x. [DOI] [PubMed] [Google Scholar]
- 5.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–23. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 7.Chan KT, Cortesio CL, Huttenlocher A. FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion. J Cell Biol. 2009 doi: 10.1083/jcb.200809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–78. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 9.Schlaepfer DD, Mitra SK, Ilic D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim Biophys Acta. 2004;1692:77–102. doi: 10.1016/j.bbamcr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 10.Schaller MD. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci. 2010;123:1007–13. doi: 10.1242/jcs.045112. [DOI] [PubMed] [Google Scholar]
- 11.Deakin NO, Turner CE. Paxillin comes of age. J Cell Sci. 2008;121:2435–44. doi: 10.1242/jcs.018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaidel-Bar R, Milo R, Kam Z, Geiger B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J Cell Sci. 2007;120:137–48. doi: 10.1242/jcs.03314. [DOI] [PubMed] [Google Scholar]
- 13.Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- 14.Walsh C, Tanjoni I, Uryu S, et al. Oral delivery of PND-1186 FAK inhibitor decreases spontaneous breast to lung metastasis in pre-clinical tumor models. Cancer Biology & Therapy. 2010;9:776–88. doi: 10.4161/cbt.9.10.11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rozengurt E. Mitogenic signaling pathways induced by G protein-coupled receptors. J Cell Physiol. 2007;213:589–602. doi: 10.1002/jcp.21246. [DOI] [PubMed] [Google Scholar]
- 16.Dufresne M, Seva C, Fourmy D. Cholecystokinin and gastrin receptors. Physiol Rev. 2006;86:805–47. doi: 10.1152/physrev.00014.2005. [DOI] [PubMed] [Google Scholar]
- 17.Lee HK, Lee HJ, Hur K, et al. Growth effect of gastrin on gastric cancer and its clinical implications for gastric cancer surgery. Oncol Rep. 2005;14:383–8. [PubMed] [Google Scholar]
- 18.Grabowska AM, Watson SA. Role of gastrin peptides in carcinogenesis. Cancer Lett. 2007;257:1–15. doi: 10.1016/j.canlet.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 19.Rozengurt E, Walsh JH, Gastrin CCK. signaling, and cancer. Annu Rev Physiol. 2001;63:49–76. doi: 10.1146/annurev.physiol.63.1.49. [DOI] [PubMed] [Google Scholar]
- 20.Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- 21.Jin G, Ramanathan V, Quante M, et al. Inactivating cholecystokinin-2 receptor inhibits progastrin-dependent colonic crypt fission, proliferation, and colorectal cancer in mice. J Clin Invest. 2009;119:2691–701. doi: 10.1172/JCI38918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–5. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- 23.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–80. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki N, Nakamura S, Mano H, Kozasa T. Galpha 12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc Natl Acad Sci U S A. 2003;100:733–8. doi: 10.1073/pnas.0234057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siehler S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br J Pharmacol. 2009;158:41–9. doi: 10.1111/j.1476-5381.2009.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwanicki MP, Vomastek T, Tilghman RW, et al. FAK, PDZ-RhoGEF and ROCKII cooperate to regulate adhesion movement and trailing-edge retraction in fibroblasts. J Cell Sci. 2008;121:895–905. doi: 10.1242/jcs.020941. [DOI] [PubMed] [Google Scholar]
- 27.Zhai J, Lin H, Nie Z, et al. Direct interaction of focal adhesion kinase with p190RhoGEF. J Biol Chem. 2003;278:24865–73. doi: 10.1074/jbc.M302381200. [DOI] [PubMed] [Google Scholar]
- 28.Lim Y, Lim ST, Tomar A, et al. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol. 2008;180:187–203. doi: 10.1083/jcb.200708194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu HG, Schrader H, Otte JM, Schmidt WE, Schmitz F. Rapid tyrosine phosphorylation of focal adhesion kinase, paxillin, and p130Cas by gastrin in human colon cancer cells. Biochem Pharmacol. 2004;67:135–46. doi: 10.1016/j.bcp.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 30.Yu HG, Tong SL, Ding YM, et al. Enhanced expression of cholecystokinin-2 receptor promotes the progression of colon cancer through activation of focal adhesion kinase. Int J Cancer. 2006;119:2724–32. doi: 10.1002/ijc.22207. [DOI] [PubMed] [Google Scholar]
- 31.Slack-Davis JK, Martin KH, Tilghman RW, et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282:14845–52. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- 32.Wu L, Bernard-Trifilo JA, Lim Y, et al. Distinct FAK-Src activation events promote alpha5beta1 and alpha4beta1 integrin-stimulated neuroblastoma cell motility. Oncogene. 2008;27:1439–48. doi: 10.1038/sj.onc.1210770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260:85–8. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- 34.Lim ST, Miller NL, Nam JO, Chen XL, Lim Y, Schlaepfer DD. PYK2 inhibition of p53 as an adaptive and intrinsic mechanism facilitating cell proliferation and survival. J Biol Chem. 2010;285:1743–53. doi: 10.1074/jbc.M109.064212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berdeaux RL, Diaz B, Kim L, Martin GS. Active Rho is localized to podosomes induced by oncogenic Src and is required for their assembly and function. J Cell Biol. 2004;166:317–23. doi: 10.1083/jcb.200312168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawakami M, Yanai Y, Hata F, Hirata K. Vascular endothelial growth factor C promotes lymph node metastasis in a rectal cancer orthotopic model. Surg Today. 2005;35:131–8. doi: 10.1007/s00595-004-2896-0. [DOI] [PubMed] [Google Scholar]
- 37.Gebbink MF, Kranenburg O, Poland M, van Horck FP, Houssa B, Moolenaar WH. Identification of a novel, putative Rho-specific GDP/GTP exchange factor and a RhoA-binding protein: control of neuronal morphology. J Cell Biol. 1997;137:1603–13. doi: 10.1083/jcb.137.7.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang AJ, Song DH, Wolfe MM. Attenuation of peroxisome proliferator-activated receptor gamma (PPARgamma) mediates gastrin-stimulated colorectal cancer cell proliferation. J Biol Chem. 2006;281:14700–10. doi: 10.1074/jbc.M602623200. [DOI] [PubMed] [Google Scholar]
- 39.Pasapera AM, Schneider IC, Rericha E, Schlaepfer DD, Waterman CM. Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J Cell Biol. 2010;188:877–90. doi: 10.1083/jcb.200906012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kermorgant S, Lehy T. Glycine-extended gastrin promotes the invasiveness of human colon cancer cells. Biochem Biophys Res Commun. 2001;285:136–41. doi: 10.1006/bbrc.2001.5132. [DOI] [PubMed] [Google Scholar]
- 41.Wroblewski LE, Pritchard DM, Carter S, Varro A. Gastrin-stimulated gastric epithelial cell invasion: the role and mechanism of increased matrix metalloproteinase 9 expression. Biochem J. 2002;365:873–9. doi: 10.1042/BJ20020068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis. 2006;23:97–105. doi: 10.1007/s10585-006-9014-1. [DOI] [PubMed] [Google Scholar]
- 43.Maa MC, Lee JC, Chen YJ, et al. Eps8 facilitates cellular growth and motility of colon cancer cells by increasing the expression and activity of focal adhesion kinase. J Biol Chem. 2007;282:19399–409. doi: 10.1074/jbc.M610280200. [DOI] [PubMed] [Google Scholar]
- 44.Leroy C, Fialin C, Sirvent A, et al. Quantitative phosphoproteomics reveals a cluster of tyrosine kinases that mediates SRC invasive activity in advanced colon carcinoma cells. Cancer Res. 2009;69:2279–86. doi: 10.1158/0008-5472.CAN-08-2354. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y, Zhang X, Guda K, et al. Identification and functional characterization of paxillin as a target of protein tyrosine phosphatase receptor T. Proc Natl Acad Sci U S A. 2010;107:2592–7. doi: 10.1073/pnas.0914884107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2:133–42. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 47.Tumbarello DA, Brown MC, Hetey SE, Turner CE. Regulation of paxillin family members during epithelial-mesenchymal transformation: a putative role for paxillin delta. J Cell Sci. 2005;118:4849–63. doi: 10.1242/jcs.02615. [DOI] [PubMed] [Google Scholar]
- 48.Jagadeeswaran R, Surawska H, Krishnaswamy S, et al. Paxillin is a target for somatic mutations in lung cancer: implications for cell growth and invasion. Cancer Res. 2008;68:132–42. doi: 10.1158/0008-5472.CAN-07-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kawasaki Y, Tsuji S, Muroya K, et al. The adenomatous polyposis coli-associated exchange factors Asef and Asef2 are required for adenoma formation in Apc(Min/+)mice. EMBO Rep. 2009;10:1355–62. doi: 10.1038/embor.2009.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ashton GH, Morton JP, Myant K, et al. Focal adhesion kinase is required for intestinal regeneration and tumorigenesis downstream of Wnt/c-Myc signaling. Dev Cell. 2010;19:259–69. doi: 10.1016/j.devcel.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.