

Abstract
Microglia activation is central to the neuroinflammation associated with neurological and neurodegenerative diseases, particularly since activated microglia are often a source of pro-inflammatory cytokines. Despite decades-long research, the molecular cascade of pro-inflammatory transformation of microglia in vivo remains largely elusive. Here, we report increased β–catenin expression, a central intracellular component of WNT signaling, in microglia undergoing a pro-inflammatory morphogenic transformation under pathogenic conditions associated with neuroinflammation, such as Alzheimer’s disease. We substantiate disease-associated β–catenin signaling in microglia in vivo by showing age-dependent β–catenin accumulation in mice with Alzheimer’s-like pathology (APdE9). In cultured mouse microglia expressing the WNT receptors Frizzled (FZD)4, 5, 7, 8 and LDL related protein 5/6 (LRP5/6), we find that WNT-3A can stabilize β–catenin. WNT-3A dose-dependently induces LRP6 phosphorylation with downstream activation of disheveled, β-catenin stabilization and nuclear import. Gene expression profiling reveals that WNT-3A stimulation specifically increases the expression of pro-inflammatory immune response genes in microglia, and exacerbates the release of de novo IL-6, IL-12 and tumor necrosis factor α. In sum, our data suggest that the WNT family of lipoglycoproteins can instruct pro-inflammatory microglia transformation and emphasize the pathogenic significance of β–catenin signaling networks in this cell type.
Keywords: aging, Alzheimer’s disease, Frizzled, neurodegeneration, neuroinflammation
Introduction
Microglia, the immunocompetent cells of the central nervous system, can react to virtually any homeostatic modification by changing their morphology, motility, gene expression and pro-inflammatory cytokine production and release (Hanisch and Kettenmann, 2007). In the inflamed brain, microglia can exert dual actions: besides being the cellular source of pro-inflammatory cytokines, these cells are indispensable for the phagocytic removal of infectious agents and cellular debris (Hanisch and Kettenmann, 2007; Rivest, 2009). Diverse signaling molecules - including growth factors, neurotransmitters, and morphogens - may instruct microglia functions (Hanisch, 2002; Pocock and Kettenmann, 2007) thus emphasizing that the temporal and spatial dynamics of communication between microglia and neurons, astrocytes or oligodendrocytes determine microglia fate.
WNTs bind Frizzled (FZD) family receptors (Schulte and Bryja, 2007; Schulte 2010) whose cooperation with particular co-receptors, such as LDL receptor-related protein 5/6 (LRP5/6), receptor tyrosine kinase-like orphan receptor 1/2 (ROR1/2) or receptor-like tyrosine kinase (RYK), defines downstream signal specificity (Hendrickx and Leyns, 2008). A main WNT signaling branch is the WNT/β-catenin pathway (Fig. S1) progressing though disheveled (DVL), glycogen synthase-3 (GSK-3) and the transcriptional regulator β-catenin (MacDonald et al, 2009). WNT signaling inhibits constitutive β-catenin phosphorylation by GSK-3 and its proteasomal degradation upon FZD-LRP5/6 activation allowing for β-catenin accumulation, nuclear import and the regulation of gene transcription.
The (patho-)physiological significance of WNT/β-catenin signaling in neuronal differentiation, growth as well as dysfunction are broadly accepted (Inestrosa and Arenas, 2010). Particularly, microglia activation is implicated in various neurodegenerative conditions (Morales et al, 2010; Morganti-Kossmann et al, 2007; Perry et al, 2002; Tai et al, 2007; Tansey and Goldberg, 2010; Weinstein et al, 2010). It is broadly accepted that microglia activity exacerbates the underlying, molecularly diverse disease pathologies by releasing pro-inflammatory cytokines to compromise neuronal survival and function (Hanisch and Kettenmann, 2007). Notably, progressive neuronal dysfunction in AD and its transgenic models is associated with decreased WNT/β–catenin signaling (De Ferrari and Moon, 2006; Dinamarca et al, 2008; Inestrosa and Arenas, 2010). Accordingly, GSK3 blockade in a mouse model of AD improves memory performance suggesting that restoring WNT/β–catenin signaling may alleviate the underlying neuronal deficits (Toledo and Inestrosa, 2009).
Although microglia reside and act in an environment where multiple members of the WNT family of lipoglycoproteins are expressed (Inestrosa and Arenas, 2010), it is not known whether WNTs influence microglia functions, particularly their pro-inflammatory transformation. Here, we report that β-catenin levels in active, mobile and macrophage-like microglia are elevated in neurodegenerative conditions associated with chronic neuroinflammation and in the corresponding transgenic models. We show that microglia express appropriate combinations of WNT receptors to activate β-catenin signaling. By using WNT-3A, we show LRP6 and DVL phosphorylation, β-catenin stabilization and nuclear import in cultured microglia, a specific pro-inflammatory gene expression pattern, and pro-inflammatory cytokine release. Therefore, we propose that WNT/β-catenin signaling in microglia is a potent pro-inflammatory regulatory signaling cascade.
Materials and Methods
Histochemistry
Human brains (for human tissue handling see supplement) were blocked, embedded in paraffin, and sectioned at a thickness of 7 µm. Sections were de-waxed in xylene, washed in ethanol, and endogenous peroxidase activity was blocked by 1% H2O2 in 100% methanol (30 min). Antigen retrieval was carried out by incubating sections in 5% urea in 0.1M Tris-HCl (pH9.5) and microwaving (800W, 8 min) followed by incubation in ice-cold NaHBO4 (0.5%, in PB for 5–10 min). Chromogenic detection of β-catenin was performed by employing rabbit anti-β-catenin IgG (1:1,000; Abcam, Cambridge, United Kingdom). Sections were blocked in 0.1% Triton X-100, 5% normal donkey serum (NDS) and 2% BSA in 0.1M phosphate buffer (PB, pH 7.4) for 1h at 22–24 °C. Subsequently sections were exposed to the primary antibody diluted in PB, 0.1% Triton X-100, 1% NDS, 0.1% BSA (pH 7.4) overnight at 4 °C. After repeated rinses in PB, sections were subjected to biotinylated donkey anti-rabbit IgG as secondary antibody (1:600; Jackson ImmunoResearch, Newmarket, UK; 2h at 22–24 °C in PB (pH7.4)) followed by exposure to streptavidin-conjugated horseradish peroxidase (1:300; Jackson ImmunoResearch; 1h at 22–24 °C in PB (pH 7.4)) and development with 3,3’-diaminobenzidine tetrahydrochloride (20 mg/100 ml) and H2O2 (0.05%). Sections were coverslipped with Entellan (in toluene, Merck, Darmstadt, Germany).
Immunofluorescence histochemistry on human specimens after antigen retrieval and free-floating mouse brain (for information on mouse tissue preparation see supplement) sections was performed according to published protocols (Harkany et al, 2003). Sections were rinsed in PB and pre-incubated with 5% NDS (Jackson ImmunoResearch), 2% BSA and 0.3% Triton X-100 in PB for 1h (22–24 °C). Sections where then exposed to combinations of the following primary antibodies: rabbit anti-β-catenin (1:1,000; Abcam) goat anti-ionized Ca2+-binding adaptor molecule 1 (IBA-1; 1:200; Abcam), mouse anti-glial fibrillary acidic protein (GFAP, 1:1,000, Millipore, Billerica, MA, USA), mouse anti-hyperphosphorylated tau AT8 (1:1,000; Pierce/Thermo Scientific; Rockford, IL, USA) and rabbit anti-CB2 cannabinoid receptor (CB2R; 1:500 (Van Sickle et al, 2005)), diluted in PB, 0.1% Triton X-100, 0.1% BSA, 1% NDS, and incubated overnight at 4 °C. Subsequently, sections were rinsed in PB and incubated with a mixture of carbocyanine (Cy)-conjugated secondary antibodies raised in donkey (IgGH+L; 1:200; Jackson) diluted in PB/ 2% BSA (2h at 22–24 °C). Specimens were counterstained with Hoechst 33,342 (Sigma-Aldrich, St. Louis, USA). Finally, sections were rinsed in PB, dipped in distilled water, mounted on fluorescence-free glass slides, air-dried and coverslipped with Entellan (in toluene, Merck).
Standard control experiments were performed by omission of primary antibodies and yielded the lack of any cellular labeling. We have minimized the likelihood of staining artifacts such as lipofuscin autofluorescence by using Sudan Black B (Schnell et al, 1999) in combination with spectral dye unmixing (Fig. S2).
Images were captured on a Zeiss 710LSM laser-scanning microscope using multi-track consecutive channel capture configuration and spectral unmixing. Image overviews (Fig. 1) were captured using the tile-scanning function and differential interference contrast microscopy. Emission wavelengths for maximum separation of immunofluorescence signals were limited to 407–484 nm (Hoechst 33,342), 510–525 nm (Cy2), 565–620 nm (Cy3) and 650–720 nm (Cy5). Reconstruction of β-catenin+ microglia was carried out by capturing consecutive images at 63× primary magnification. Three-dimensional rendering and maximum intensity projections were prepared using the ZEN2009 software package (Zeiss). Images were processed and figures were assembled in CorelDraw X5 (Corel Corp., Ottawa, ON, Canada).
Fig. 1. β-catenin localization in Alzheimer’s disease (AD).
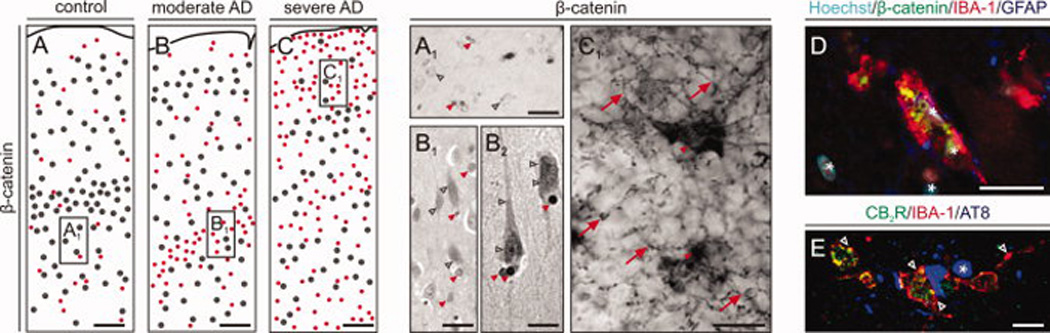
(A–C) Schematic maps of cells clusters (n > 4 cells/circle) with β-catenin immunohistochemistry in temporooccipital cortices from AD patients and age-matched controls. (A1–C1) High-resolution photomicrographs corresponding to fields marked by open rectangels in (A–C) demonstrate differential cellular recruitment of β-catenin during AD progression. Open grey arrowheads mark β-catenin+ neurons, while red filled arrowheads point to β-catenin immunosignal in small diameter nuclei, reminiscent of astro- or microglia. Note that multipolar cells with fine-caliber processes, likely representing microglia, accumulate in severe AD. Scale bars= A–C 250 micrometer, A’–C’ = 100 µm. (D) Indirect fluorescent immunohistochemistry staining for IBA-1 (microglia marker) and β-catenin identify high levels of β-catenin in activated microglia. Scale bar = 50 µm. (E) IBA-1+ microglia associated with AT8+ dystrophic neurons (*) express CB2R (open arrowheads). Scale bar = 10 µm. Asterisks indicate the localization of nuclei as revealed by Hoechst dye staining.
Cell culture
N13 microglia-like cells (Ferrari et al, 1996) were cultured as described (Hammarberg et al, 2003). For biochemical analysis or immunocytochemistry, cells were seeded either directly, or on collagen-coated coverslips, in 24 well plates (density: 5 × 104 cells/well). Serum-starved cells were stimulated with vehicle (0.1% BSA in PBS) with or without recombinant mouse WNT-3A (R&D Systems, Wiesbaden, Germany) as specified. In proliferation assays, N13 cell numbers were determined after stimulation in serum-free medium, trypsination and cell-counting in a Bürker chamber (in triplicate, n = 6). For stimulation with Aβ, lyophilized Aβ(1–42) (from Dr. Botond Penke; University of Szeged, Hungary; Minkeviciene et al, 2009) was dissolved directly in growth medium to yield fibrillar Aβ and added to N13 cells as a 10x solution.
Primary microglia were isolated from newborn mouse brains (Prinz et al, 1999). Cultures typically contained >95% microglia, as validated by cytochemistry using Griffonia simplicifolia isolectin B4 (Sigma-Aldrich). Cultures were challenged 24h after plating.
β-catenin localization in vitro
Cells were fixed in 4% paraformaldehyde (10 min) in PB. β-catenin was detected by using mouse anti-β-catenin antibody (1:1,000; BD Transduction Laboratories). A Cy3-coupled donkey-anti-mouse antibody (1:500; Jackson) was used as secondary antibody in combination with SYTOX Green or DAPI nuclear stains (Molecular Probes/Invitrogen, Eugene, OR, USA). Images were captured on an LSM710 (Zeiss) laser-scanning microscope.
Immunoblotting
Immunoblotting of lysates from cultured cells (Bryja et al, 2007) and mouse cortices (Martín-Ibañez et al, 2006) were performed as described (Table S3). Signals were visualized by the enhanced chemiluminescence method (GE Healthcare, Pollards Wood, UK). Densitometry was done with the Scion Image software.
Cytokine release
Primary mouse microglia were plated in 96 well plates (density: 105 cells/well) and stimulated with WNT-3A (300 ng/ml; R&D Systems) for 24h. Supernatants were analyzed for IL-6, IL-12 (including the IL-12 isoforms p70, p40, and p402) and TNFα using mouse-specific antibody pairs and mouse protein standards designed for ELISA applications (R&D Systems, Minneapolis, USA). Colorimetric reactions were quantified on a microplate reader (Victor, 1420 Multilabel counter, Perkin Wallac, Waltham, MA, USA).
Reverse transcriptase (RT)-PCR and quantitative real-time PCR
RNA was isolated from N13 cells and primary microglia using the RNeasy Mini kit and the RNeasy Micro kit (Qiagen, Hilden, Germany), respectively. cDNA was transcribed using the high-capacity cDNA Archive kit (Applied Biosystems, Foster City, CA, USA). RT-PCR was performed according to a standard protocol: 94°for 5 min followed by 30 cycles of 94 °C for 30s, Tanneal (Table S4) for 45s, 72 °C for 1 min, followed by 72 °C for 10 min (2720 Thermo Cycler, Applied Biosystems). Genomic mouse-tail DNA served as positive control, while non-transcribed (minus reverse transcriptase/-RT) RNA was used as negative control. PCR products were analyzed by 2% agarose/ethidium bromide gel electrophoresis. Quantitative real-time PCR was carried out with the TaqMan gene expression assay (Applied Biosystems) according to the manufacturer’s instructions. Mm00443260_g1 (TNFα), Mm99999064_m (IL6) and Mm01288991_g1 (IL12) primer pairs were from Applied Biosystems. Reactions were performed in triplicates on an ABI Prism 7000 Sequence detector (Applied Biosystems). Results were presented as the difference in the number of cycles to reach the detection threshold (Ct = cycle at threshold), using 18S rRNA (Applied Biosystems/Ambion) as internal reference standard (ΔCt = Ctcytokine−Ct18S). ΔCt was presented as relative fold change in gene expression (mean ± SEM).
Affimetrix expression analysis
Three independent harvests of primary microglia were seeded in 6 well plates and stimulated with 0.1% BSA in PBS (vehicle control) or 300 ng/ml recombinant WNT-3A for 6 h after serum-deprivation overnight. RNA was prepared as described above and analyzed on an Affimetrix Mouse Gene 1.0 ST Array (Bioinformatics and Expression Analysis Core Facility, Department of Biosciences & Nutrition, Karolinska Institutet; www.bea.ki.se). RNA was quality controlled on an Agilent BioAnalyzer (Agilent RNA 6000 Nano assay). Functional grouping was done according to the gene ontology terminology (www.geneontology.org; GO). Data were selected by their p value being <0.05 and their fold change (FC) ≤−2 or ≥2 from the functional grouping GO:0006955 (immune response). Overlap with the following groups is indicated: GO:0006954 (inflammatory response), GO:0035556 (intracellular signal transduction), GO:0001817 (regulation of cytokine production).
Statistical analysis
GraphPad Prism 5.0 (GraphPad Software Inc, San Diego, USA) was used to perform non-linear regression analysis, bar graphs, and group comparisons. Data were analyzed by either one-way ANOVA followed by Bonferroni’s multiple comparison post-hoc test or Student’s t-test (independent group design) as appropriate. *p < 0.05, **p < 0.01, ***p < 0.001.
Results
β-catenin accumulates in microglia in Alzheimer’s disease
We chose to study β–catenin expression by microglia in AD because this neurodegenerative condition presents significant chronic neuroinflammation and microglia activation (Querfurth and LaFerla, 2010). Immunohistochemistry to detect β–catenin was performed on clinicopathologically-verified post-mortem material from AD patients and aged-matched controls. We observed two types of β–catenin immunoreactivity in controls and Braak stage II – IV cases: a) low-to-moderate staining of perikarya, morphologically reminiscent of pyramidal cells (Fig. 1A,A1), and b) intensely-labeled nuclei, (Fig. 1A1,B1) with quasi-random distribution across all cortical laminae in the temporooccipital region (Fig. 1. In contrast, β–catenin immunoreactivity redistributes in cortices from Braak VI subjects: besides being scantly dispersed in large (neuronal) somata, a meshwork of fine-caliber processes of multipolar cells with (peri-)nuclear β–catenin immunoreactivity (Fig. 1C), likely representing micro- or astroglia (Fig. 1C1) emerges. We find closely associated β–catenin and IBA-1 immunoreactivities, the latter being a microglia marker (Fig. 1D) in Braak VI cases. We have verified that IBA-1+ cells around AT8+ dystrophic neurons were activated microglia by their co-expression of CB2Rs (Fig. 1E) (Walter et al, 2003). Thus, our data from a human AD cohort suggest a shift from neuronal towards (micro-)glial β–catenin expression in patients manifesting significant AD-related neuroinflammation.
Surveying microglia undergo phasic transformation to reach a macrophage-like stage with phagocytic capacity (Fig. 2A). Therefore, we have determined whether β–catenin is co-expressed at any particular morphological state(s) of IBA-1+ microglia (Fig 2A1). β–catenin immunoreactivity is not detectable in either surveying/resting microglia with ramified morphology (Fig. 2A1/a2) or ameboid microglia (Fig. 2A1/a1). In contrast, activated, mobile microglia (Fig. 2A1/a3) progressing towards a macrophage-like state are β–catenin+ (2A1/a4). Whilst we acknowledge the resolution limits of light microscopy in rounded-down β–catenin+/IBA-1+ microphage-like cells, our data suggest β–catenin localized in the cytosol, submembraneously or in the nucleus of such cells (Fig. 2A1a4). Colocalization experiments of β–catenin and GFAP show that β–catenin levels are generally low in astrocytes, irrespective of the AD stage analyzed (Fig. 2A1/a2–a4).
Fig. 2. Human microglia morphology in relation to β–catenin levels.
(A) Microglia undergo phenotypical transformation upon activation as schematically shown in a1–a4. Corresponding cell states can be found in human post mortem brain sections (A1), which were stained with β-catenin (green), IBA-1 (red, microglia marker) and GFAP (blue, astrocytic marker). Open arrows point to IBA-1+ but β-catenin− microglia. Filled arrows indicate IBA-1+/β-catenin+ microglia, whereas filled and open arrowheads depict β-catenin+ or β-catenin− astroctyes identified by GFAP staining, respectively. Scale bars in A1(a1) = 15 µm, a2 = 15 µm (inset 5 µm), a3 = 15 µm, a4= 20 µm (inset 5 µm). (B) Human post mortem brain lysates from healthy controls and patients with mild AD (mAD) and severe AD (sAD) were analyzed for β-catenin, GFAP, CB2R and β-actin levels by immunoblotting. After densitometric quantification and normalization of β-catenin, GFAP and CB2R intensities to the corresponding β-actin levels, individual correlation between β-catenin and CB2R (B1) and β-catenin and GFAP was plotted. * p<0,05; ** p<0,01 (unpaired Student's t-test).
We have performed regression analysis to assess whether β–catenin levels could correlate with CB2R or GFAP in microglia and astroglia, respectively in the AD brain (Fig. 2B,B1). We find significantly increased CB2R levels in severe AD, as compared to age-matched controls (p < 0.05) or moderate AD subjects (p < 0.01). We find a quasi-random relationship between β–catenin and CB2R levels in control and moderate AD subjects. However, our data support a close positive association between β–catenin and CB2R protein expression in severe AD. Although a similar relationship between β–catenin and GFAP contents appears in both moderate and severe AD cohorts (Fig. 2B1,C), the considerable population spread of GFAP expression negates any association. Analysis of SNAP-25 protein levels, a marker of glutamatergic synapses in human brain (Garbelli et al, 2008; Matteoli et al, 2009), revealed significantly reduced protein concentrations in moderate but not severe AD (Fig. S3D,D1), and a lack of expressional relationship with β–catenin. Collectively, our neuroanatomical and biochemical analysis suggest that β–catenin expression is significantly increased in microglia or astrocytes in AD.
β-catenin accumulation in microglia is age-dependent but unrelated to acute β-amyloid toxicity
We confirmed our above data in APdE9 mice (Jankowsky et al, 2004; Minkeviciene et al, 2009) exhibiting considerable microgliosis and astrogliosis coinciding progressive β-amyloid production. We provide added support for chronic neuroinflammation in APdE9 mice by showing increased tumor necrosis factor α (TNFα) mRNA expression (Fig. 3) in brains of 7 and 12 months old APdE9 mice (p < 0.05 vs. control (7 months); p < 0.005 vs. control (12 months).
Fig. 3. Expression of pro-inflammatory TNFα in APdE9 mice indicates chronic neuroinflammation.

cDNA (6 animals per group) from age-matched control and APdE9 mouse brains was analyzed for the expression of TNFα by QPCR. Error bars give standard error of the mean. Groups were compared with unpaired Student's t-test. * p < 0.05, ** p < 0.01.
β–catenin levels were found increased with age in IBA-1+ microglia in the polymorph layer of the dentate gyrus (Fig. 4A) in wild-type mice (Fig. 4A1,A2). The density of both β–catenin+ microglia and β–catenin− astroglia in the dentate gyrus of aged APdE9 mice (Fig. 4A3), as well as total β–catenin levels in hippocampus (129.2 ± 13.1%, n = 3/group; p < 0.05; Fig. 4C,C1) exceeded that of age-matched littermate controls (Fig. 4A2). High-resolution microscopy suggests perinuclear β–catenin in microglia (Fig. 4B,B1) but not astrocytes. Although our analysis focused on the hippocampus, the brain region primarily affected by β-amyloid (Aβ) overexpression in APdE9 mice, β–catenin+ microglia were also found in the cerebral cortex, striatum and basal forebrain (data not shown). Cumulatively, our findings in post-mortem AD brains and APdE9 mouse hippocampi reveal progressive and significant β-catenin stabilization in pro-inflammatory microglia.
Fig. 4. Increased β–catenin levels in microglia in APdE9 mice.

(A) Fluorescent immunohistochemistry was performed on brain sections from wild type and APdE9 mice. (A1–A3) show cellular distribution of β-catenin (green), IBA-1 (red, microglia marker) and GFAP (blue, astrocyte marker) in the dentate gyrus and the granular cell layer (gc) according to the schematic overview provided in A (hl, hilus; fh, hippocampal fissure; mo, molecular layer; yellow circles with red outline symbolize the increase in β-catenin+/IBA-1+ microglia). Open arrowheads point to IBA-1+ microglia devoid of β-catenin. Filled arrowheads pinpoint IBA-1+/β-catenin+ microglia. Asterisks mark β-catenin− astrocytes. (B) High-resolution photomicrograph showing a microglial cell harboring β-catenin immunoreactivity. (B1) Three-dimensional reconstruction of an IBA-1+ microglia reveals intracellular β-catenin accumulation in this cell type. (C, C1) Immunoblot analysis confirms increased β-catenin levels in APdE9 mice at 14 months of age. * p < 0.05 (Student’s t-test, n = 3/group) (D) Proto-fibrillar Aβ (20 and 100 µM, dissolved in medium) fails to increase β-catenin in N13 cells but increases expression of inducible nitric oxide synthase (iNOS). Representative data are shown from n = 3 independent experiments. Scale bars in A1– A3 = 20 µm, B, B1 = 10 µm.
Considering that Aβ can bind FZDs, such as e g FZD5 (Magdesian et al, 2008), we hypothesized that Aβ may be critical to stabilize β–catenin in microglia. Therefore, we exposed N13 microglia-like cells to Aβ(1–42) for 6h (data not shown) and 24h (Fig. 4D) and monitored β–catenin levels. We find that Aβ elevates inducible nitric oxide synthase (iNOS) expression, a marker of microglia activation (Hanisch and Kettenmann, 2007). However, β-catenin levels remained unchanged suggesting that Aβ per se may not regulate β-catenin levels in microglia.
WNT-3A increases microglial β-catenin levels
We have performed a ligand array coupled to Western analysis of β-catenin and LRP6 phosphorylation (P-LRP6) to identify factor(s) inducing β-catenin stabilization and signaling in microglia. We included ligands (Table S2) that have previously been reported to affect β-catenin stabilization or are otherwise important for microglia function (Pocock and Kettenmann, 2007; Jin et al, 2008). As Fig. 5A shows, WNT-3A and LiCl, the latter used as a positive control given its inhibitory effect on GSK-3β, but not lipopolysaccaride (LPS), thrombin, interferon α/β, T NFα, insulin, glutamate, ATP/UTP, N-ethylcarboxiamidoadenosine, isoproterenol or apomorphine induced β-catenin accumulation in N13 microglia-like cells by 60 min. Only WNT-3A exposure led to LRP6 phosphorylation.
Fig. 5. WNT-3A selectively induces β–catenin stabilization in microglia cells.

(A) Serum-starved N13 microglia-like cells were stimulated with WNT-3A, LiCl, LPS, insulin, IFNα/β, TNFα, ATP, UTP, NECA, isoproterenol, apomorphine, thrombin or glutamate (Table S2) for 60 min, lysed and analyzed by immunoblotting to determine P-LRP6 and β-catenin levels. β-actin served as loading control. Results were plotted (from duplicates) with values normalized to unstimulated control. (B) RT-PCR was used to investigate expression of WNT-1, -2, -3 and -7A mRNA in adult brain and to profile the presence of WNT receptor mRNAs in N13 cells and primary mouse microglia (C). Abbreviations: −, without reverse transcriptase; +, with reverse transcriptase; G, genomic DNA.
WNTs are expressed in the adult brain (Malaterre et al, 2007). Here, we confirmed WNT-1, WNT-2, WNT-3 and WNT-7A expression, all of which can activate WNT/β-catenin signaling (Shimizu et al, 1997; Hwang et al, 2004), in adult mouse brain (Fig. 5B). Therefore, we hypothesize that WNTs may represent a novel class of signaling molecules modulating microglia functions.
Microglia express WNT receptors
A prerequisite of functional WNT signaling is the expression of WNT receptors. By RT-PCR we identify that both N13 cells and primary mouse microglia express a variety of WNT receptors invariably dominated by FZD4, FZD5, FZD7, FZD8, LRP5 and LRP6 (Fig. 5C). Neither cell type expressed ROR1, ROR2 or RYK, atypical receptor tyrosine kinases that can cooperate with FZDs or act as cell-autonomous WNT receptors. Although minor differences in WNT receptor expression exist between N13 and primary microglia exist (Fig. 5C), our data validate the use and responsiveness to WNT-3A of both models in functional assays in vitro.
WNT/β-catenin signaling in microglia
Cytosolic, perinuclear and nuclear β-catenin localization in microglia suggests that WNTs could signal through β-catenin in this cell type. To test this possibility, we explored the potential of purified, recombinant WNT-3A to stimulate β-catenin signaling in microglia. We emphasize that WNT-3A was used as a model ligand to establish signaling mechanisms in proof-of-principle studies. Yet, the molecular identity of WNTs activating microglia in vivo may vary and might be specific to certain brain areas or particular disease conditions.
First, we verified that recombinant WNT-3A induces β-catenin stabilization and nuclear import in N13 cells as well as primary microglia (Fig. 6A). We find increased β-catenin immunoreactivity in microglia when stimulated with recombinant WNT-3A (Fig. 5A). Importantly, β-catenin underwent nuclear translocation as revealed by β-catenin, SYTOX green/DAPI co-localization (Fig. 6A).
Fig. 6. WNT-3A induces WNT/β–catenin signaling in microglia cells.

(A) β-catenin localization in N13 and primary mouse microglia cells in combination with SYTOX Green (N13) or DAPI (primary microglia) as nuclear counterstain. Confocal laser-scanning photomicrographs were captured in control and WNT-3A-stimulated (100 ng/ml, 2h) cells. Scale bar = 20 µm. (B) WNT-3A induces LRP6 phosphorylation (P-LRP6) and β-catenin stabilization over time in N13 cells and (C) with increasing concentrations (2 h). P-LRP6, β-catenin and β-actin levels were monitored by immunoblotting. (C’) A dose-response curve was constructed where each data-point represents the mean ± SEM from n=4 – 6 independent observations/data point. P-LRP6 and β–catenin levels in control (ctrl) were set to zero while response to 300 ng/ml of WNT-3A was considered 100%. (D) WNT-3A stimulation for 2h activates DVL1, 2, 3 isoforms visualized by electrophoretic mobility shift (PS-DVL is indicated by open triangle; non-shifted DVL form is marked with filled triangle). (E) Dose- and time-dependence of P-LRP6 and β-catenin levels in primary mouse microglia with β-actin as loading control.
Next, we determined WNT/β–catenin signaling kinetics by detecting P-LRP6 and β–catenin upon WNT-3A stimulation for up to 6 h (Fig. 6B). In agreement with previous studies in other cell types (Bryja et al, 2007), WNT-3A induced β-catenin stabilization and P-LRP6 accummulation. WNT-3A-induced changes in P-LRP6 and β-catenin levels occurred simultaneously with increases detected from 15 min post-stimulation and reaching saturation by 120 min. Therefore, we used 2-h stimulation to establish dose-response relationships upon WNT-3A stimulation in N13 cells (Fig. 6C,C'). Non-linear regression analysis of WNT-3A-induced P-LRP6 or β-catenin stabilization estimated EC50 values at 205 ng/ml (115–367 ng/ml) and 147 ng/ml (84–256 ng/ml), respectively.
An upstream regulator of β-catenin is the phosphoprotein disheveled (DVL), which becomes phosphorylated and shifted (PS-DVL) upon WNT stimulation (Cong et al, 2004; Bryja et al, 2007). WNT-3A at concentrations significantly stabilizing β-catenin progressively induced PS-DVL3 formation (from 30 min post-stimulation; Fig. 6D). All three mammalian DVL isoforms responded to WNT-3A stimuli (2h) with PS-DVL formation (Fig. 6D').
We have excluded the possibility that the above WNT-3A-induced signaling events were specific to N13 cells by stimulating primary mouse microglia with WNT-3A. WNT-3A increased both P-LRP6 and β-catenin levels at 30 and 100 ng/ml (Fig. 6E), concentrations previously shown to be effective in N13 cells. Similarly, the temporal induction of P-LRP6 and β-catenin were similar to those seen in N13 cells (Fig. 6E). Collectively, these data suggest that WNT-3A signaling through P-LRP6 activates DVL, and results in β-catenin stabilization and nuclear translocation in microglia.
WNT-3A modulates microglia activity
Our findings show that microglia express a repertoire of receptors suited to mediate WNT-3A responses. However, the cellular consequence(s) and physiological response(s) WNT-3A imposes on microglia, including cell proliferation and pro-inflammatory cytokine (IL-6, IL-12 and TNFα) expression and release remain to be determined
Since enhanced proliferation is a hallmark of generalized microglia activation (Hanisch and Kettenmann, 2007), and β-catenin-dependent signaling pathways can exert mitogenic effects in select cellular systems (Castelo-Branco et al, 2003; Willert et al, 2003; Boland et al, 2004), we studied whether WNT-3A affects N13 cell growth. WNT-3A (300 ng/ml) stimulation did not affect the rate of N13 cell proliferation, as compared to serum-starved control cells (Fig. 7A). Treatment with fetal calf serum (FCS, 10%), used as positive control (Kloss et al, 1997), provided the expected increase in N13 cell proliferation (p < 0.001, n = 6 in triplicate).
Fig. 7. WNT-3A induces pro-inflammatory cytokines but does not affect microglia proliferation.

(A) Serum-deprived N13 microglia-like cells were stimulated with WNT-3A (300 ng/ml) for 24h, cells were counted and compared to unstimulated controls (ctrl). Fetal calf serum (FCS, 10%) served as a positive control. ***p < 0.001 (control vs. FCS-treated N13; one-way ANOVA). Data were expressed as means ± SEM. (B) cDNA was prepared from primary microglia cells, which were stimulated with WNT-3A (300 ng/ml, 24h) to determine cytokine expression levels by QPCR. (B’) supernatants from these cells were analyzed for released IL-6, IL-12 and TNFα. Data are from three or more independent experiments and are expressed as means ± SEM. ***p < 0.001, **p < 0.01, *p < 0.05 (WNT-3A vs. control; one-way ANOVA).
Pro-inflammatory cytokine release is a hallmark of activated microglia, and contributes to chronic neuroinflammation (Farfara et al, 2008). Therefore, we tested whether exposure of primary mouse microglia to WNT-3A affects the expression and release of the pro-inflammatory cytokines IL-6, IL-12 or TNFα. WNT-3A (300 ng/ml, 24h) increased mRNA levels, and facilitated release of corresponding cytokines in the medium (Fig. 7B,B'). Thus, WNT-3A can potently exacerbate neuroinflammatory responses.
While we reveal select changes upon WNT-3A challenge, pro-inflammatory microglia transformation is likely to trigger widespread changes in complex cytokine signaling networks. Therefore, we determined genome-wide expressional changes upon WNT-3A exposure (300 ng/ml, 6h) in primary mouse microglia. Affimetrix gene expression profiling shows (Fig. 8) WNT-3A-induced upregulation of immune response genes, particularly IL-6 and TNFα. IL-12 mRNA also increased by 6h; however, sample variability rendered the changes statistically non-significant (+7.73 fold increase, p = 0.32). Since quantitative PCR confirmed our expression analysis, we employed the gene expression microarray approach for in-depth analysis of WNT-3A’s inflammatory profile. We grouped WNT-3A-regulated genes according to GO:"immune response" (Fig. 8). Soluble pro-inflammatory factors, such as IL-6, IL-1α, IL-15, oncostatin M, chemokines (CXCL11, CCL7, CXCL2, 10, CCL2, CCL4, 5) and complement factor 3 were upregulated. Members of the TNF-superfamily (TNFSF9, 10, 15, TNF) and factors involved in immune signaling or function (MX1 and 2, TLR3, ICAM1, SP110, NFKB2, IRGM1, MYD88, IRAK3) were also induced (Fig. 8). Furthermore, we find that WNT-3A robustly induces iNOS/NOS2), CD69, matrix metalloproteases (MMP13 and 14), prostaglandin-endoperoxide synthase 2 or cyclooxygenase 2 (PTGS2/COX-2; Table S5). Thus, our microarray analysis supports our hypothesis that WNT-3A can trigger pro-inflammatory microglia transformation.
Fig. 8. The pro-inflammatory fingerprint of WNT-3A on primary mouse microglia.

For Affimetrix gene expression analysis, primary mouse microglia were treated with ctrl (0.1 % BSA in PBS) or 300 ng/ml WNT-3A for 6 h (n=3). Genes were clustered indicating a correlation between gene expression patterns in ctrl and stimulated samples. Data were grouped according to gene ontology term # GO:0006955 "immune response" indicated in green (selected GO terms). Overlap with other selected GO terms is indicated in orange: inflammatory response, blue: regulation of cytokine production and purple: intracellular signal transduction. Hits with fold changes larger than 2 or smaller than −2 and a p value <0.05 were plotted in a heat map. The heatmap coloring is individually created for each gene with light green as the lowest expression level measured, black is the mean and bright red represents the highest detected level.
Discussion
Microglia serve as a crucial point of convergence for innate immune responses in the brain (Rivest, 2009). Our results suggest that β–catenin signaling is active in microglia in the inflamed brain, and implicate enhanced cytosolic β–catenin stabilization and nuclear import in pro-inflammatory microglia transformation under conditions of chronic neuroinflammation. Previous studies have shown that peripheral macrophages can employ WNT-based communication to modulate inflammatory responses (Pereira et al, 2009) suggesting that brain microglia could also process WNT signals. A prerequisite of relaying WNT signals intracellularly is the expression of WNT receptors. We show that both N13 and primary microglia express such receptors, including various FZDs and LRP5/6. However, we did not explore the identity of specific WNT receptor complexes in microglia. Instead, we have focused on identifying cellular response patterns to WNT-3A, thus providing primary data on the cellular functions of WNT//β–catenin signaling in this particular cell type. WNT-3A is the prototypic WNT that activates WNT/β–catenin signaling. In vitro, WNT-3A induces WNT/β–catenin signaling (LRP6 phosphorylation, PS-DVL formation, β–catenin stabilization/nuclear translocation in both N13 cells and primary microglia with expected kinetics and dose-response relationships (Willert et al, 2003; Bryja et al, 2007; MacDonald et al, 2009).
Neuroinflammation in AD
AD is accompanied by a severe inflammatory reaction with microglia implicated to act in both detrimental and beneficial fashions (Querfurth and LaFerla, 2010). Our data show increased β-catenin levels in microglia transiting from a surveying to an active macrophage-like stage in post-mortem AD brains. This notion suggests that enhanced β-catenin signaling is an intricate part of the inflammatory transformation cascade in microglia, and participates in exacerbating AD-related neuroinflammation. Further, β-catenin stabilization with disease progression from moderate to severe AD correlates with an increased expression of CB2Rs, expressed in activated (IBA1+) microglia rather than with astrocytic or neuronal markers, emphasizing the selective elevation of β-catenin levels in microglia.
APdE9 mice and microglial β-catenin
Compatible with the human AD pathology, APdE9 mice show important features of chronic inflammation, such as microgliosis and astrogliosis, increased production of inflammatory cytokines, and upregulation of the complement system (Akiyama et al, 2000). Important differences exist between various models of chronic neuroinflammation, regarding, e.g. the extent of tissue damage, gliosis, or blood-brain-barrier permeability. Interestingly, aging is also associated with a progressive inflammatory reaction and a transformation in microglia morphology and function (Miller and Streit, 2007). In aged wild-type mice we also observed an increased density of IBA+ microglia accumulating β-catenin. Excess density of β-catenin+/IBA-1+ microglia in the APdE9 phenotype prompts us to speculate that β–catenin and also WNT/β–catenin communication could serve as a fundamental means of microglia communication in neuroinflammation.
The inflammatory fingerprint of WNT-3A on microglia
Enhanced β–catenin signaling in microglia in the diseased brain could affect the immunocompetence of these cells by mediating either an anti-inflammatory response to compensate ongoing inflammation or a pro-inflammatory reaction exacerbating chronic inflammation. Our in vitro data show that WNT-3A-induced β–catenin in cultured microglia parallels a strong pro-inflammatory response manifesting in the increased expression of microglia ‘activity markers’: IL6, IL12 and TNFα expression and release (Fig. 7). Gene expression analysis (Fig. 8) corroborates these observations by identifying an immuno response network provoked by WNT-3A, including cytokines, chemokines, and innate immune response factors. WNT-3A-evoked reprogramming may allow microglia cells to increase their activation status (e g CD40, ICAM1), intercellular communication with other microglia, astrocytes, T- and B-cells (CD40, CD28, chemokines, cytokines), and phagocytic activity (TLR3, MYD88) (Lynch, 2009). In fact, CCL2 is recognized as one of the driving forces of the acute inflammatory response in the brain (Lu et al, 1998).
Upregulation of IL6, a destructive, pro-inflammatory cytokine, is particularly intriguing in an AD-context as IL-6 may link downstream neurotoxicity ubiquitously following microglia activation. In summary, the overall shift towards a pro-inflammatory phenotype upon WNT-3A stimulation suggests that WNTs can affect essential immune functions in the central nervous system, and orchestrate the infiltration of peripheral immune cells.
β-catenin and AD
Several molecular mechanisms regulate β-catenin levels (e.g., GSK-3), and many become impaired in AD (Hooper et al, 2008; Pei et al, 1999). Thus, it is of importance to characterize the signaling pathways as well as their cell type-specificities that impact β-catenin signaling in AD. Recent studies concentrated on establishing the molecular complexity of WNT/β-catenin signaling in neurons (Inestrosa and Arenas, 2010), and propose that WNT-induced inhibition of GSK-3 diminishes Aβ neurotoxicity by reducing tau hyperphosphorylation (Inestrosa et al, 2007). This concept is supported by findings demonstrating that WNT-3A engaging FZD1 decreases Aβ neurotoxicity (Alvarez et al, 2004; Chacón et al, 2008), inhibition of GSK-3 abrogates Aβ processing (Phiel et al, 2003), and LiCl treatment improves behavioral outcome in an AD mouse model (Toledo and Inestrosa, 2009). The association of Dickkopf-1, a WNT-inhibitor, with neuronal degeneration reinforces the beneficial effects of WNT signaling on neuronal survival (Caricasole et al, 2004). Thus, decreased neuronal β-catenin is detrimental for nerve cell survival. Compensation for the loss of neuronal β-catenin by enhanced WNT signaling or pharmacological inhibition of GSK3 appears therefore to have neuroprotective potential (De Ferrari et al, 2003; Alvarez et al, 2004; Chacón et al, 2008).
Our studies point to an increase in β-catenin levels in microglia, thus raising an important question: Is enhanced β–catenin signaling in microglia in brain inflammation beneficial or detrimental for disease outcome? On one hand microglia activation is - particularly in the case of AD - associated with positive effects based on the reduction of the Aβ load by phagocytosing microglia. If this function turns out to be prominently affected by treatment with GSK3 inhibitors, there is the potential for beneficial therapeutic effects through support of pro-inflammatory microglia. On the other hand WNT-3A inflicts a robust pro-inflammatory phenotype in cultured microglia suggests that this pathway might enhance the ongoing inflammation with a negative outcome. Therefore, we conclude that selective blockade of WNT signaling in microglia could be of therapeutic significance. Current drug development, however, promotes GSK3 inhibitors for the treatment of AD to counteract the decrease of neuronal β–catenin and the increase in GSK3-mediated tau phosphorylation (Hooper et al, 2008; Hu et al, 2008). However, treatment with a GSK3 inhibitor would inevitably enhance microglial WNT signaling and could thus exacerbate pro-inflammatory responses, possibly imposing enhanced neurotoxicity as long-term side effect. Thus, it is necessary to evaluate the net effect of GSK3 inhibition by weighing in both neuronal tau hyperphosphorylation and microglia-dependent inflammatory processes.
Supplementary Material
Acknowledgments
Dr. Botond Penke (University of Szeged, Hungary) is acknowledged for providing β-amyloid(1–42) peptide. This work was supported by grants from Karolinska Institutet (Department of Physiology & Pharmacology; G.S.), Tore Nilsson Foundation (G.S.), Åke-Wiberg Foundation (G.S.), Jeansson Foundation (G.S.), Åhlén Foundation (G.S.), Knut & Alice Wallenberg Foundation (KAW2008.0149), the Foundations of the National Board of Health and Welfare of Sweden (G.S.), the Swedish Medical Research Council K2008-68P-20810-01-4, K2008-68X-20805-01-4 (G.S.) and K2008-66X-20762-01-3 (T.Ha.), Scottish Universities Life Science Alliance (SULSA, T.Ha.), Alzheimer’s Association (K.M., T.Ha.), European Molecular Biology Organization Young Investigator Programme (T.Ha.), European Commission (HEALTH-F2-2007-201159, T.Ha., H.T.), National Institutes of Health Grants DA023214 (T.Ha.), DA11322 (K.M.), DA21969 (K.M.), the National Institute of Health Research (NIHR) Specialist Biomedical Research Centre for Mental Health award to South London and Maudsley NHS Foundation Trust (SLaM) and the Institute of Psychiatry at King’s College London (T. Ho), and the Alzheimer’s Research Trust (ART) UK (T.Ha.). J.M. is recipient of a research fellowship from ART UK.
Footnotes
The authors declare no conflict of interest.
References
- Alvarez AR, Godoy JA, Mullendorff K, Olivares GH, Bronfman M, Inestrosa NC. Wnt-3a overcomes β-amyloid toxicity in rat hippocampal neurons. Exp Cell Res. 2004;297:186–196. doi: 10.1016/j.yexcr.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Boland GM, Perkins G, Hall DJ, Tuan RS. Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J Cell Biochem. 2004;93:1210–1230. doi: 10.1002/jcb.20284. [DOI] [PubMed] [Google Scholar]
- Bryja V, Schulte G, Arenas E. Wnt-3a utilizes a novel low dose and rapid pathway that does not require casein kinase 1-mediated phosphorylation of Dvl to activate β-catenin. Cell Signal. 2007;19:610–616. doi: 10.1016/j.cellsig.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Caricasole A, Copani A, Caraci F, Aronica E, Rozemuller AJ, Caruso A, Storto M, Gaviraghi G, Terstappen GC, Nicoletti F. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer's brain. J Neurosci. 2004;24:6021–6027. doi: 10.1523/JNEUROSCI.1381-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelo-Branco G, Wagner J, Rodriguez FJ, Kele J, Sousa K, Rawal N, Pasolli HA, Fuchs E, Kitajewski J, Arenas E. Differential regulation of midbrain dopaminergic neuron development by Wnt-1, Wnt-3a, and Wnt-5a. Proc Natl Acad Sci U S A. 2003;100:12747–12752. doi: 10.1073/pnas.1534900100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacón MA, Varela-Nallar L, Inestrosa NC. Frizzled-1 is involved in the neuroprotective effect of Wnt3a against Aβ oligomers. J Cell Physiol. 2008;217:215–227. doi: 10.1002/jcp.21497. [DOI] [PubMed] [Google Scholar]
- Cong F, Schweizer L, Varmus H. Wnt signals across the plasma membrane to activate the β-catenin pathway by forming oligomers containing its receptors, Frizzled and LRP. Development. 2004;131:5103–5115. doi: 10.1242/dev.01318. [DOI] [PubMed] [Google Scholar]
- De Ferrari GV, Chacón MA, Barría MI, Garrido JL, Godoy JA, Olivares G, Reyes AE, Alvarez A, Bronfman M, Inestrosa NC. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by β-amyloid fibrils. Mol Psychiatry. 2003;8:195–208. doi: 10.1038/sj.mp.4001208. [DOI] [PubMed] [Google Scholar]
- De Ferrari GV, Moon RT. The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene. 2006;25:7545–7553. doi: 10.1038/sj.onc.1210064. [DOI] [PubMed] [Google Scholar]
- Dinamarca MC, Colombres M, Cerpa W, Bonansco C, Inestrosa NC. β-amyloid oligomers affect the structure and function of the postsynaptic region: role of the Wnt signaling pathway. Neurodegener Dis. 2008;5:149–152. doi: 10.1159/000113687. [DOI] [PubMed] [Google Scholar]
- Farfara D, Lifshitz V, Frenkel D. Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer's disease. J Cell Mol Med. 2008;12:762–780. doi: 10.1111/j.1582-4934.2008.00314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D, Villalba M, Chiozzi P, Falzoni S, Ricciardi-Castagnoli P, Di Virgilio F. Mouse microglial cells express a plasma membrane pore gated by extracellular ATP. J Immunol. 1996;156:1531–1539. [PubMed] [Google Scholar]
- Garbelli R, Inverardi F, Medici V, Amadeo A, Verderio C, Matteoli M, Frassoni C. Heterogeneous expression of SNAP-25 in rat and human brain. J Comp Neurol. 2008;506:373–386. doi: 10.1002/cne.21505. [DOI] [PubMed] [Google Scholar]
- Hammarberg C, Schulte G, Fredholm BB. Evidence for functional adenosine A3 receptors in microglia cells. J Neurochem. 2003;86:1051–1054. doi: 10.1046/j.1471-4159.2003.01919.x. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Harkany T, Härtig W, Berghuis P, Dobszay MB, Zilberter Y, Edwards RH, Mackie K, Ernfors P. Complementary distribution of type 1 cannabinoid receptors and vesicular glutamate transporter 3 in basal forebrain suggests input-specific retrograde signalling by cholinergic neurons. Eur J Neurosci. 2003;18:1979–1992. doi: 10.1046/j.1460-9568.2003.02898.x. [DOI] [PubMed] [Google Scholar]
- Hendrickx M, Leyns L. Non-conventional Frizzled ligands and Wnt receptors. Dev Growth Differ. 2008;50:229–243. doi: 10.1111/j.1440-169X.2008.01016.x. [DOI] [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Begum AN, Jones MR, Oh MS, Beech WK, Beech BH, Yang F, Chen P, Ubeda OJ, Kim PC, Davies P, Ma Q, Cole GM, Frautschy SA. GSK3 inhibitors show benefits in an Alzheimer's disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis. 2008;33:192–206. doi: 10.1016/j.nbd.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SG, Ryu JH, Kim IC, Jho EH, Jung HC, Kim K, Kim SJ, Chun JS. Wnt-7a causes loss of differentiated phenotype and inhibits apoptosis of articular chondrocytes via different mechanisms. J Biol Chem. 2004;279:26597–26604. doi: 10.1074/jbc.M401401200. [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci. 2010;11:77–86. doi: 10.1038/nrn2755. [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Varela-Nallar L, Grabowski CP, Colombres M. Synaptotoxicity in Alzheimer's disease: the Wnt signaling pathway as a molecular target. IUBMB Life. 2007;59:316–321. doi: 10.1080/15216540701242490. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Jin T, George Fantus I, Sun J. Wnt and beyond Wnt: multiple mechanisms control the transcriptional property of beta-catenin. Cell Signal. 2008;20:1697–1704. doi: 10.1016/j.cellsig.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Kloss CU, Kreutzberg GW, Raivich G. Proliferation of ramified microglia on an astrocyte monolayer: characterization of stimulatory and inhibitory cytokines. J Neurosci Res. 1997;49:248–254. [PubMed] [Google Scholar]
- Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. The multifaceted profile of activated microglia. Mol Neurobiol. 2009;40:139–156. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdesian MH, Carvalho MM, Mendes FA, Saraiva LM, Juliano MA, Juliano L, Garcia-Abreu J, Ferreira ST. Amyloid-β binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/β-catenin signaling. J Biol Chem. 2008;283:9359–9368. doi: 10.1074/jbc.M707108200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaterre J, Ramsay RG, Mantamadiotis T. Wnt-Frizzled signalling and the many paths to neural development and adult brain homeostasis. Front Biosci. 2007;12:492–506. doi: 10.2741/2077. [DOI] [PubMed] [Google Scholar]
- Martín-Ibañez R, Jenstad M, Berghuis P, Edwards RH, Hioki H, Kaneko T, Mulder J, Canals JM, Ernfors P, Chaudhry FA, Harkany T. Vesicular glutamate transporter 3 (VGLUT3) identifies spatially segregated excitatory terminals in the rat substantia nigra. Eur J Neurosci. 2006;23:1063–1070. doi: 10.1111/j.1460-9568.2006.04601.x. [DOI] [PubMed] [Google Scholar]
- Matteoli M, Pozzi D, Grumelli C, Condliffe SB, Frassoni C, Harkany T, Verderio C. The synaptic split of SNAP-25: different roles in glutamatergic and GABAergic neurons? Neuroscience. 2009;158:223–230. doi: 10.1016/j.neuroscience.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Miller KR, Streit WJ. The effects of aging, injury and disease on microglial function: a case for cellular senescence. Neuron Glia Biol. 2007;3:245–253. doi: 10.1017/S1740925X08000136. [DOI] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales I, Farías G, Maccioni RB. Neuroimmunomodulation in the pathogenesis of Alzheimer's disease. Neuroimmunomodulation. 2010;17:202–204. doi: 10.1159/000258724. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury. 2007;38:1392–1400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Distribution of active glycogen synthase kinase 3β (GSK-3β) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- Pereira CP, Bachli EB, Schoedon G. The wnt pathway: a macrophage effector molecule that triggers inflammation. Curr Atheroscler Rep. 2009;11:236–242. doi: 10.1007/s11883-009-0036-4. [DOI] [PubMed] [Google Scholar]
- Perry VH, Cunningham C, Boche D. Atypical inflammation in the central nervous system in prion disease. Curr Opin Neurol. 2002;15:349–354. doi: 10.1097/00019052-200206000-00020. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3α regulates production of Alzheimer's disease amyloid-β peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30:527–535. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Prinz M, Kann O, Draheim HJ, Schumann RR, Kettenmann H, Weber JR, Hanisch UK. Microglial activation by components of gram-positive and -negative bacteria: distinct and common routes to the induction of ion channels and cytokines. J Neuropathol Exp Neurol. 1999;58:1078–1089. doi: 10.1097/00005072-199910000-00006. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9:429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J Histochem Cytochem. 1999;47:719–730. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- Schulte G. IUPHAR Nomenclature Report: The Class Frizzled Receptors. Pharmacol Rev. 2010 doi: 10.1124/pr.110.002931. in press. [DOI] [PubMed] [Google Scholar]
- Schulte G, Bryja V. The Frizzled family of unconventional G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:518–525. doi: 10.1016/j.tips.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Julius MA, Giarré M, Zheng Z, Brown AM, Kitajewski J. Transformation by Wnt family proteins correlates with regulation of β-catenin. Cell Growth Differ. 1997;8:1349–1358. [PubMed] [Google Scholar]
- Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, Piccini P. Imaging microglial activation in Huntington's disease. Brain Res Bull. 2007;72:148–151. doi: 10.1016/j.brainresbull.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo EM, Inestrosa NC. Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer's disease. Mol Psychiatry. 2009;15:272–285. doi: 10.1038/mp.2009.72. [DOI] [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD, Sharkey KA. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, Mackie K, Stella N. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci. 2003;23:1398–1405. doi: 10.1523/JNEUROSCI.23-04-01398.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JR, Koerner IP, Möller T. Microglia in ischemic brain injury. Future Neurol. 2010;5:227–246. doi: 10.2217/fnl.10.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.